

Abstract
It has long been suspected that chronic stress can exacerbate, or even cause, disease. We now propose that the RCAN1 gene, which can generate several RCAN1 protein isoforms, may be at least partially responsible for this phenomenon. We review data showing that RCAN1 proteins can be induced by multiple stresses, and present new data also implicating psychosocial/emotional stress in RCAN1 induction. We further show that transgenic mice overexpressing the RCAN1–1L protein exhibit accumulation of hyperphosphorylated tau protein (AT8 antibody), an early precursor to the formation of neurofibrillary tangles and neurodegeneration of the kind seen in Alzheimer disease. We propose that, although transient induction of the RCAN1 gene might protect cells against acute stress, persistent stress may cause chronic RCAN1 overexpression, resulting in serious side effects. Chronically elevated levels of RCAN1 proteins may promote or exacerbate various diseases, including tauopathies such as Alzheimer disease. We propose that the mechanism by which stress can lead to these diseases involves the inhibition of calcineurin and the induction of GSK-3β by RCAN1 proteins. Both inhibition of calcineurin and induction of GSK-3β contribute to accumulation of phosphorylated tau, formation of neurofibrillary tangles, and eventual neurodegeneration.—Ermak, G., Pritchard, M. A., Dronjak, S., Niu, B., Davies, K. J. A. Do RCAN1 proteins link chronic stress with neurodegeneration?
Keywords: glucocorticoids, tau
The RCAN1 gene was originally discovered in the mid-1990s (1, 2). The gene was “rediscovered” several times, resulting in a confusing plethora of unrelated names, including DSCR1, Adapt78, MCIP1, RCN1, and others. In 2007, sufficient agreement was achieved to rename the gene RCAN1 to reflect one of its main functions: regulation of the serine/threonine phosphatase, commonly known as calcineurin (3). There is also evidence that RCAN1 proteins may have other important properties, including regulating the mitochondrial ADP/ATP translocator (ANT; ref. 4) and binding, and possibly modulating the activity of, MAP3K kinase (5).
The RCAN1 gene consists of 7 exons, 3 of which (exons 5, 6, and 7) appear to be invariant in all RCAN1 isoforms. The remaining 4 exons (exons 1–4) can be alternatively spliced to produce a number of different mRNA isoforms (6). In human brains, RCAN1 is expressed at the highest levels in neurons, and at least 3 RCAN1 proteins (RCAN1s) are expressed: 2 RCAN1–1s (1–1L and 1–1S) and RCAN1–4 (3, 7). Of these three, RCAN1–1L is the most abundant isoform in adult human brain.
In addition to important roles in normal physiology, RCAN1 proteins are also connected to tau protein pathology and Aβ pathology. On one hand, overexpression of RCAN1s can lead to accumulation of phosphorylated tau (ref. 7 and more details below). On the other hand, Aβ, which is responsible for the formation of amyloid plaques, can directly induce overexpression of RCAN1s (8) that may, subsequently, lead to tau hyperphosphorylation and the formation of neurofibrillary tangles. Thus, RCAN1s may explain why both amyloid plaques and neurofibrillary tangles are formed in Alzheimer disease and Down syndrome. It is currently accepted that the production of Aβ, which then leads to tau hyperphosphorylation, plays a causal role in both diseases. In the current work, we review evidence and provide new data to suggest that many types of stress, including psychosocial/emotional stress, can cause chronic RCAN1 overexpression. Thus, chronic stress may contribute to accumulation of phosphorylated tau, formation of neurofibrillary tangles, and eventual neurodegeneration.
RCAN1 GENE CAN BE INDUCED BY MULTIPLE STRESSES
In our laboratory, RCAN1 (originally called Adapt78) was discovered as a gene that is induced during transient adaptation to oxidative stress (1). It has, subsequently, become clear that RCAN1 is also induced by a variety of other stresses. This is potentially very important, because many human diseases are associated with, or exacerbated by, increased stress. The fact that RCAN1 can be induced biomechanically by traumatic injuries is of particular interest in regard to neurodegeneration. For example, in vitro studies have demonstrated direct biomechanical induction of RCAN1 in cardiac myocytes (9). We have also observed similar phenomena in cultured neuronal-like PC-12 cells, in which RCAN1 can be induced by scraping adherently grown cells (unpublished results).
Recent studies and new data suggest that RCAN1 gene expression may also be induced by psychological stress. It has been shown that expression of RCAN1–1 mRNA, which encodes the RCAN1–1L and RCAN1–1S proteins, is induced by glucocorticoids, but RCAN1–4 mRNA is not induced (10, 11). Interestingly, expression of RCAN1–1 mRNA was induced in the absence of protein synthesis, indicating that it was induced directly by glucocorticoids rather than through the synthesis of signaling proteins or peptides. It is also important to note that glucocorticoid levels can be increased by biomechanical stress (12), suggesting that elevated levels of RCAN1–1L and RCAN1–1S after biomechanical stress may result from production of glucocorticoids.
Glucocorticoids carry out a number of important functions, including adaptation to stressful conditions, particularly, psychosocial/emotional stresses of the kind with which most humans must contend in their everyday lives. This, and the fact that RCAN1–1L is the isoform predominantly expressed in human brain, makes RCAN1–1L a particularly interesting protein for further study in stress regulation and adaptation in human brain, and in various brain diseases. Therefore, we performed studies to address whether psychological stress can actually lead to RCAN1–1L induction, using a rat model. In these studies, rats were exposed either to immobilization or isolation stress, and the levels of RCAN1–1L were analyzed (Fig. 1). The efficacy of stress was evaluated by measuring hormonal levels in plasma, and all brain samples were obtained from previously described animals (13). We analyzed the hippocampus, since it contains high levels of glucocorticoid receptors, which make it more vulnerable to long-term stress than other parts of the brain (14, 15). Our results demonstrate that RCAN1–1L levels approximately doubled in rat hippocampus within 2 h after either isolation or immobilization stress. Thus, it does appear that psychosocial/emotional stress can induce RCAN1–1L. Similarly, RCAN1–1S levels were also approximately doubled after both stresses. However, levels of RCAN1–4 were not significantly elevated (data not shown). This may be due to the fact that the RCAN1–1 and RCAN1–4 isoforms are regulated by different promoters: RCAN1–4 is regulated by the proximal promoter, while both RCAN1–1s are regulated by the distal promoter (3).
Figure 1.

RCAN1–1L levels are elevated in hippocampus of rats exposed to immobilization or isolation stress. Adult rats were exposed to stress. Eleven-week-old male Wistar rats were maintained and manipulated as fully described in Gavrilovic et al. (13). All experiments described were performed in accordance with the U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication 80-23). Animals were sacrificed at 2 h after stress was terminated, and the hippocampus from the right hemisphere of each brain was isolated for analysis. A) Example of Western blot analysis of RCAN1–1L expression; 5 control and 6 stressed animals per group were analyzed. β-Tubulin was used to control sample loading. B) Summary of Western analyses. RCAN1–1L levels are reported in arbitrary units, which were set as follows: absorption of the first RCAN1–1L control sample was set as 1.0 in the group of immobilized rats; absorption of the first control sample was set as 1.0 in the group of isolated rats. All values were then adjusted according to the loading levels, as measured by β-tubulin absorbance. All values are shown as means ± se. The experiments were repeated using β3-tubulin as the loading control, and we obtained essentially identical results (confirmatory data not shown). In both the immobilized and isolated groups, hippocampal RCAN1–1L levels were significantly increased (2.0-fold after immobilization stress and 1.7-fold after isolation stress), as evaluated by Student's t test (P<0.05).
STRESS CAN CAUSE NEURODEGENERATION
A variety of stresses have been associated with neurodegeneration. For example, epidemiological studies suggest that mechanical stress caused by head injury or trauma (16, 17), or by contact sport exposures, such as soccer (18) or boxing (19), can cause neurodegenerative diseases. It has been estimated, for example, that the risk of Alzheimer disease may be increased by as much as 15% as a result of head trauma (20). Interestingly, data indicating that psychological stress can also cause neurodegeneration and Alzheimer disease have been published. For example, it has been shown that elderly individuals subjected to psychological stress are more likely to develop Alzheimer disease than are age-matched, nondistressed individuals (21). Moreover, it has been shown, using the Alzheimer disease triple-transgenic mouse model, that chronic stress diminishes learning and memory, while increasing amyloid deposition (22), one of the hallmarks of Alzheimer disease. It has been also demonstrated, using mouse models, that stress can induce the accumulation of phosphorylated tau (23–25). In turn, the accumulation of phosphorylated tau can lead to the formation of neurofibrillary tangles, another hallmark of Alzheimer disease, and other forms of neurodegeneration.
RCAN1 GENE OVEREXPRESSION IS ASSOCIATED WITH NEURODEGENERATION
It was observed that RCAN1 is overexpressed in Alzheimer disease (8, 26, 27), which is characterized by progressive neurodegeneration. Alzheimer disease belongs to a group of ailments called “tauopathies,” one of the hallmarks of which is neurodegeneration that involves the formation of neurofibrillary tangles due to accumulation of hyperphosphorylated tau protein. RCAN1 is also overexpressed in Down syndrome (8, 28, 29), and it should be noted that patients with Down syndrome typically experience an early-onset and aggressive form of Alzheimer disease. It has also been found that both loss-of-function and overexpression of the Drosophila ortholog of the RCAN1, called nebula, leads to severe learning defects (30). Finally, it has been reported that RCAN1 overexpression diminishes mitochondrial respiratory activity (MTT assay) in cultured murine neurons (31), which would be expected to cause neuronal damage.
MECHANISMS BY WHICH RCAN1 INDUCTION MAY LEAD TO NEURODEGENERATION
One mechanism by which RCAN1 may harm neurons might involve calcineurin, which dephosphorylates the tau protein. The activity of calcineurin can be down-regulated by RCAN1 proteins, and calcineurin activity levels have been reported to decrease in Alzheimer disease (32). Notably, the RCAN1 gene and calcineurin are expressed at similar times, in similar regions of the brain, and in similar (neuronal) cell types (8, 33, 34). Calcineurin inhibition has been associated with tau phosphorylation at threonine 181 and 231 (T181 and T231). These same phosphorylated residues are seen in paired helical filament preparations from Alzheimer disease brains (35). Other investigations have shown that calcineurin inhibitors cause tau phosphorylation on both serine and threonine residues, both of which are phosphorylated in Alzheimer disease (36). A separate study reported that calcineurin inhibition prevented the proteolysis of tau by calpain (37), and we have also shown that calcineurin inhibition can decrease the degradation of tau by the proteasome (38). Finally, it has also been demonstrated that elevated levels of T181 and T231 tau, and total tau in the cerebrospinal fluid, together constitute a biomarker test with quite good accuracy for predicting incipient Alzheimer disease (39).
Calcineurin may not, however, play a crucial role in Alzheimer disease. It has been claimed, for example, that other phosphatases, such as PP2A, might exert more important effects (40). Therefore, the finding that RCAN1s also induce GSK-3β synthesis (resulting in increased tau phosphorylation) may be even more interesting (8) because there is a general agreement that activation of GSK-3β is an important factor in neurodegeneration and Alzheimer disease (reviewed in refs. 40, 41). Induction of GSK-3β by RCAN1 was discovered in PC-12 neuronal-like cells, and more studies using more relevant models would be extremely helpful in properly exploring this phenomenon.
Regardless of whether RCAN1s operate mostly by inhibition of calcineurin or by induction of GSK-3β (or both), RCAN1–1L induction may lead to accumulation of hyperphosphorylated tau, which is an important step in the formation of neurofibrillary tangles. To begin to test this hypothesis, we have now performed preliminary studies using an RCAN1-overexpressing transgenic mouse that was recently described (42) as a model. In this model, the isoform 1 splice variant, which encodes the RCAN1–1L and RCAN1–1S proteins, was placed under the control of the endogenous promoter to mirror, as closely as possible, the situation in Down syndrome and Alzheimer disease. Our data indicate that hyperphosphorylated tau accumulates in hippocampal neurons in mice overexpressing RCAN1–1L (Fig. 2). This accumulation is only found using the AT8 antibody, which detects early stages of neurofibrillary tangle formation, but not with AT100 or PFH6 antibodies, which detect later stages of neurofibrillary tangle maturation. These data indicate that the accumulation of hyperphosphorylated tau, after RCAN1–1L overexpression, is a slow and gradual process, which corresponds to the slow process (spanning many years) of tangle formation in Alzheimer disease. It is estimated, for example, that just the process of forming mature neurofibrillary tangles from the pretangle stage may take ∼20 yr (43). This is entirely consistent with the observation that acute RCAN1 overexpression is protective against transient stress, with no expected negative long-term effects on tau, because such side effects would require years of chronic RCAN1 overexpression. Similarly, since RCAN1 is chronically overexpressed in the brains of patients affected by Down syndrome from birth (8, 28, 29), this hypothesis is also consistent with the fact that patients with Down syndrome typically develop neurofibrillary tangles and experience onset of Alzheimer disease at ∼40 yr of age.
Figure 2.

Tau phosphorylation is increased in the hippocampus of RCAN1 transgenic mice. Mice (CBA/Bl6 males) were aged to ∼16 mo, and wild-type mice were compared to transgenic mice that constitutively overexpress RCAN1–1L (fully described in ref. 42). A) Representative photographs of hippocampus from wild-type and RCAN1 transgenic mice stained for the AT8 antibody. More darkly stained cells were observed in sections from transgenic mice (arrows) compared with controls. B) Summary of immunohistochemical analysis. Hippocampal sections from 4 transgenic and 4 wild-type mice were probed with AT8, AT100, and PHF6 antibodies; 8 sections/animal were analyzed. Sections were photographed and signal intensities in whole fields were quantified using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA). Results are means ± se. Tau phosphorylation levels were significantly (1.97-fold) increased in sections stained with the AT8 antibody, as evaluated by Student's t test (P<0.05).
In summary, we propose that stress can cause neurodegeneration by inducing RCAN1 gene overexpression. While transient induction of RCAN1 (e.g., transient oxidative stress), typically lasts for just a few hours and can protect cells against stresses, chronic overexpression will cause serious side effects and cell damage. At least one such side effect seems to be increased phosphorylation of the tau protein, which may result both from induction of GSK-3β and from inhibition of calcineurin. The consequent accumulation of phosphorylated tau may ultimately lead to the formation of neurofibrillary tangles, which then contribute to the neurodegenerative process observed in many tauopathies (Fig. 3).
Figure 3.
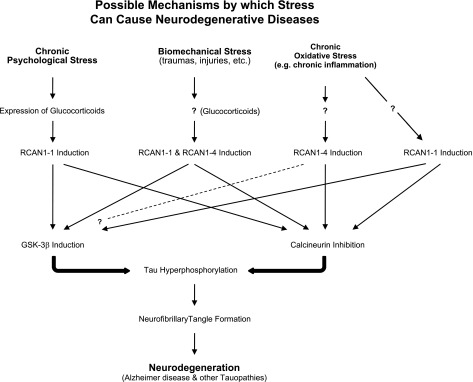
Possible mechanisms by which stress may cause neurodegenerative diseases via chronic RCAN1 induction. Multiple forms of chronic stress can induce expression of RCAN1 proteins. Although both RCAN1–1 and RCAN1–4 protein levels are induced by stress, the mechanisms of induction appear to be different: RCAN1–4 is induced by mRNA transcription (transcriptional activation), whereas RCAN1–1 proteins seem to be induced by posttranscriptional mechanisms (translational activation) (31, 44). Chronically elevated levels of RCAN1 proteins can inhibit calcineurin and activate GSK-3β, both of which can lead to the accumulation of hyperphosphorylated tau protein. The accumulation of hyperphosphorylated tau can (eventually) cause the formation of neurofibrillary tangles, and lead to neurodegeneration.
Finally, it is noteworthy that RCAN1–1L production can be also induced by glucocorticoids. Expression of glucocorticoids is induced by several stresses, including psychosocial/emotional stresses of the kind we experience in everyday life. Since exposure to glucocorticoids can induce the accumulation of phosphorylated tau (23–25), it is possible that mechanisms by which psychological stress contributes to neuronal damage might also involve RCAN1s: i.e., chronic psychosocial/emotional stress induces chronic production of glucocorticoids, which then cause overexpression of RCAN1–1L, leading to accumulation of phosphorylated tau and eventual cell degeneration (Fig. 3). It has long been suspected that emotional and psychological stresses can cause neurodegeneration. We now propose that RCAN1 proteins may be at least a partial explanation for this phenomenon.
Acknowledgments
The authors thank their colleagues, Prof. C. Finch and Prof. C. Pike (Davis School of Gerontology, University of Southern California, Los Angeles, CA, USA) as well as Prof. E. L. Sabban (Department of Biochemistry and Molecular Biology, New York Medical College, New York, NY, USA) for critical reading and helpful suggestions.
REFERENCES
- 1. Crawford D. R., Leahy K. P., Abramova N., Lan L., Wang Y., Davies K. J. (1997) Hamster adapt78 mRNA is a Down syndrome critical region homologue that is inducible by oxidative stress. Arch. Biochem. Biophys. 342, 6–12 [DOI] [PubMed] [Google Scholar]
- 2. Fuentes J. J., Pritchard M. A., Planas A. M., Bosch A., Ferrer I., Estivill X. (1995) A new human gene from the Down syndrome critical region encodes a proline-rich protein highly expressed in fetal brain and heart. Hum. Mol. Gen. 4, 1935–1944 [DOI] [PubMed] [Google Scholar]
- 3. Davies K. J., Ermak G., Rothermel B. A., Pritchard M., Heitman J., Ahnn J., Henrique-Silva F., Crawford D., Canaider S., Strippoli P., Carinci P., Min K. T., Fox D. S., Cunningham K. W., Bassel-Duby R., Olson E. N., Zhang Z., Williams R. S., Gerber H. P., Perez-Riba M., Seo H., Cao X., Klee C. B., Redondo J. M., Maltais L. J., Bruford E. A., Povey S., Molkentin J. D., McKeon F. D., Duh E. J., Crabtree G. R., Cyert M. S., de la Luna S., Estivill X. (2007) Renaming the DSCR1/Adapt78 gene family as RCAN: regulators of calcineurin. FASEB J. 21, 3023–3028 [DOI] [PubMed] [Google Scholar]
- 4. Chang K. T., Min K. T. (2005) Drosophila melanogaster homolog of Down syndrome critical region 1 is critical for mitochondrial function. Nat. Neurosci. 8, 1577–1585 [DOI] [PubMed] [Google Scholar]
- 5. Cho Y. J., Abe M., Kim S. Y., Sato Y. (2005) Raf-1 is a binding partner of DSCR1. Arch. Biochem. Biophys. 439, 121–128 [DOI] [PubMed] [Google Scholar]
- 6. Ermak G., Harris C. D., Davies K. J. (2002) The DSCR1 (Adapt78) isoform 1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. FASEB J. 16, 814–824 [DOI] [PubMed] [Google Scholar]
- 7. Ermak G., Harris C. D., Battocchio D., Davies K. J. (2006) RCAN1 (DSCR1 or Adapt78) stimulates expression of GSK-3beta. FEBS J. 273, 2100–2109 [DOI] [PubMed] [Google Scholar]
- 8. Ermak G., Morgan T. E., Davies K. J. (2001) Chronic overexpression of the calcineurin inhibitory gene DSCR1 (Adapt78) is associated with Alzheimer's disease. J. Biol. Chem. 276, 38787–38794 [DOI] [PubMed] [Google Scholar]
- 9. Wang Y., De Keulenaer G. W., Weinberg E. O., Muangman S., Gualberto A., Landschulz K. T., Turi T. G., Thompson J. F., Lee R. T. (2002) Direct biomechanical induction of endogenous calcineurin inhibitor Down syndrome critical region-1 in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 283, H533–H539 [DOI] [PubMed] [Google Scholar]
- 10. Shen M. U. L., Oshida T., Miyauchi J., Yamada M., Miyashita T. (2004) Identification of novel direct transcriptional targets of glucocorticoid receptor. Leukemia 18, 1850–1856 [DOI] [PubMed] [Google Scholar]
- 11. Hirakawa Y., Nary L. J., Medh R. D. (2009) Glucocorticoid evoked upregulation of RCAN1–1 in human leukemic CEM cells susceptible to apoptosis. J. Mol. Signal. 4, 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goldkuhl R., Klockars A., Carlsson H. E., Hau J., Abelson K. S. Impact of surgical severity and analgesic treatment on plasma corticosterone in rats during surgery. Eur. Surg. Res. 44, 117–123 [DOI] [PubMed] [Google Scholar]
- 13. Gavrilovic L., Spasojevic N., Dronjak S. (2010) Chronic individual housing-induced stress decreased expression of catecholamine biosynthetic enzyme genes and proteins in spleen of adult rats. Neuroimmunomodulation 17, 265–269 [DOI] [PubMed] [Google Scholar]
- 14. Joels M. (2008) Functional actions of corticosteroids in the hippocampus. Eur. J. Pharmacol. 583, 312–321 [DOI] [PubMed] [Google Scholar]
- 15. Woon F. L., Sood S., Hedges D. W. Hippocampal volume deficits associated with exposure to psychological trauma and posttraumatic stress disorder in adults: a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatr. 34, 1181–1188 [DOI] [PubMed] [Google Scholar]
- 16. Piazza O., Siren A. L., Ehrenreich H. (2004) Soccer, neurotrauma and amyotrophic lateral sclerosis: is there a connection? Curr. Med. Res. Opin. 20, 505–508 [DOI] [PubMed] [Google Scholar]
- 17. Guo Z., Cupples L. A., Kurz A., Auerbach S. H., Volicer L., Chui H., Green R. C., Sadovnick A. D., Duara R., DeCarli C., Johnson K., Go R. C., Growdon J. H., Haines J. L., Kukull W. A., Farrer L. A. (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54, 1316–1323 [DOI] [PubMed] [Google Scholar]
- 18. Chio A., Benzi G., Dossena M., Mutani R., Mora G. (2005) Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 128, 472–476 [DOI] [PubMed] [Google Scholar]
- 19. Clausen H., McCrory P., Anderson V. (2005) The risk of chronic traumatic brain injury in professional boxing: change in exposure variables over the past century. Br. J. Sports Med. 39, 661–664; discussion 664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Graves A. B., White E., Koepsell T. D., Reifler B. V., van Belle G., Larson E. B., Raskind M. (1990) The association between head trauma and Alzheimer's disease. Am. J. Epidemiol. 131, 491–501 [DOI] [PubMed] [Google Scholar]
- 21. Wilson R. S., Barnes L. L., Bennett D. A., Li Y., Bienias J. L., Mendes de Leon C. F., Evans D. A. (2005) Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology 64, 380–382 [DOI] [PubMed] [Google Scholar]
- 22. Jeong Y. H., Park C. H., Yoo J., Shin K. Y., Ahn S. M., Kim H. S., Lee S. H., Emson P. C., Suh Y. H. (2006) Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer's disease model. FASEB J. 20, 729–731 [DOI] [PubMed] [Google Scholar]
- 23. Korneyev A., Binder L., Bernardis J. (1995) Rapid reversible phosphorylation of rat brain tau proteins in response to cold water stress. Neurosci. Lett. 191, 19–22 [DOI] [PubMed] [Google Scholar]
- 24. Papasozomenos S. C. (1996) Heat shock induces rapid dephosphorylation of tau in both female and male rats followed by hyperphosphorylation only in female rats: implications for Alzheimer's disease. J. Neurochem. 66, 1140–1149 [DOI] [PubMed] [Google Scholar]
- 25. Yanagisawa M., Planel E., Ishiguro K., Fujita S. C. (1999) Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer's disease. FEBS Lett. 461, 329–333 [DOI] [PubMed] [Google Scholar]
- 26. Cook C. N., Hejna M. J., Magnuson D. J., Lee J. M. (2005) Expression of calcipressin1, an inhibitor of the phosphatase calcineurin, is altered with aging and Alzheimer's disease. J. Alzheimers Dis. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 27. Harris C. D., Ermak G., Davies K. J. (2007) RCAN1–1L is overexpressed in neurons of Alzheimer's disease patients. FEBS J. 274, 1715–1724 [DOI] [PubMed] [Google Scholar]
- 28. Fuentes J. J., Genesca L., Kingsbury T. J., Cunningham K. W., Perez-Riba M., Estivill X., de la Luna S. (2000) DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 9, 1681–1690 [DOI] [PubMed] [Google Scholar]
- 29. Baek K. H., Zaslavsky A., Lynch R. C., Britt C., Okada Y., Siarey R. J., Lensch M. W., Park I. H., Yoon S. S., Minami T., Korenberg J. R., Folkman J., Daley G. Q., Aird W. C., Galdzicki Z., Ryeom S. (2009) Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459, 1126–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chang K. T., Shi Y. J., Min K. T. (2003) The Drosophila homolog of Down's syndrome critical region 1 gene regulates learning: implications for mental retardation. Proc. Natl. Acad. Sci. U. S. A. 100, 15794–15799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Porta S., Serra S. A., Huch M., Valverde M. A., Llorens F., Estivill X., Arbones M. L., Marti E. (2007) RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Hum. Mol. Genet. 16, 1039–1050 [DOI] [PubMed] [Google Scholar]
- 32. Ladner C. J., Czech J., Maurice J., Lorens S. A., Lee J. M. (1996) Reduction of calcineurin enzymatic activity in Alzheimer's disease: correlation with neuropathologic changes. J. Neuropathol. Exp. Neurol. 55, 924–931 [DOI] [PubMed] [Google Scholar]
- 33. Goto S., Matsukado Y., Mihara Y., Inoue N., Miyamoto E. (1986) The distribution of calcineurin in rat brain by light and electron microscopic immunohistochemistry and enzyme-immunoassay. Brain Res. 397, 161–172 [DOI] [PubMed] [Google Scholar]
- 34. Kuno T., Mukai H., Ito A., Chang C. D., Kishima K., Saito N., Tanaka C. (1992) Distinct cellular expression of calcineurin A alpha and A beta in rat brain. J. Neurochem. 58, 1643–1651 [DOI] [PubMed] [Google Scholar]
- 35. Garver T. D., Kincaid R. L., Conn R. A., Billingsley M. L. (1999) Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231. Mol. Pharmacol. 55, 632–641 [PubMed] [Google Scholar]
- 36. Kayyali U. S., Zhang W., Yee A. G., Seidman J. G., Potter H. (1997) Cytoskeletal changes in the brains of mice lacking calcineurin A alpha. J. Neurochem. 68, 1668–1678 [DOI] [PubMed] [Google Scholar]
- 37. Xie H. Q., Johnson G. V. (1998) Calcineurin inhibition prevents calpain-mediated proteolysis of tau in differentiated PC12 cells. J. Neurosci. Res. 53, 153–164 [DOI] [PubMed] [Google Scholar]
- 38. Poppek D., Keck S., Ermak G., Jung T., Stolzing A., Ullrich O., Davies K. J., Grune T. (2006) Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochem. J. 400, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mattsson N., Zetterberg H., Hansson O., Andreasen N., Parnetti L., Jonsson M., Herukka S. K., van der Flier W. M., Blankenstein M. A., Ewers M., Rich K., Kaiser E., Verbeek M., Tsolaki M., Mulugeta E., Rosen E., Aarsland D., Visser P. J., Schroder J., Marcusson J., de Leon M., Hampel H., Scheltens P., Pirttila T., Wallin A., Jonhagen M. E., Minthon L., Winblad B., Blennow K. (2009) CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 302, 385–393 [DOI] [PubMed] [Google Scholar]
- 40. Iqbal K., Grundke-Iqbal I. (2008) Alzheimer neurofibrillary degeneration: significance, etiopathogenesis, therapeutics and prevention. J. Cell. Mol. Med. 12, 38–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martinez A., Perez D. I. (2008) GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer's disease? J. Alzheimers Dis. 15, 181–191 [DOI] [PubMed] [Google Scholar]
- 42. Keating D. J., Dubach D., Zanin M. P., Yu Y., Martin K., Zhao Y. F., Chen C., Porta S., Arbones M. L., Mittaz L., Pritchard M. A. (2008) DSCR1/RCAN1 regulates vesicle exocytosis and fusion pore kinetics: implications for Down syndrome and Alzheimer's disease. Hum. Mol. Genet. 17, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 43. Morsch R., Simon W., Coleman P. D. (1999) Neurons may live for decades with neurofibrillary tangles. J. Neuropathol. Exp. Neurol. 58, 188–197 [DOI] [PubMed] [Google Scholar]
- 44. Michtalik H. J., Narayan A. V., Bhatt N., Lin H. Y., Mulligan M. T., Zhang S. L., Crawford D. R. (2004) Multiple oxidative stress-response members of the Adapt78 family. Free Radic. Biol. Med. 37, 454–462 [DOI] [PubMed] [Google Scholar]