

Abstract
Objective
To determine the long term outcomes in children with severe aplastic anemia (SAA) treated with anti-thymocyte globulin (ATG) and cyclosporine (CsA), we conducted a retrospective analysis of the pediatric patients treated at our institution in all protocols that included horse ATG + CsA.
Study design
From 1989 to 2006, 406 patients, of whom about 20% were children under the age of 18, received an initial course of immunosuppressive therapy (IST) at our institution. Here we report the outcome of 77 children who were treated with a horse ATG + CsA based regimen during this time.
Results
The overall response rate at 6 months was 74% (57/77); the cumulative incidence of relapse at 10 years was 33%, and the median time to relapse was 558 days. The cumulative incidence of evolution following IST was 8.5%; all 3 such events occurred among partial responders. Overall, there were 13 deaths (17%): four occurred within the 3 months following IST in patients who had a pre-treatment ANC of less than 100/uL, and nine deaths occurred more than 6 months after initiating IST. The median time to death was 570 days. The overall 10-year survival for the entire cohort was 80%; long term survival in children who responded to IST was 89%.
Conclusions
The long term survival in pediatrics patients who respond to IST is excellent, at about 90%. IST remains a good alternative in pediatric patients who lack an HLA-matched sibling donor and should be offered as initial therapy, prior to an alternative HSCT.
Keywords: aplastic anemia, anti-thymocyte globulin, response, immunosuppression, pediatrics, survival
From its initial description, aplastic anemia (AA) has been identified as a disease of the young. The age distribution of affected patients reveals two distinct peaks: the first between the ages of 15 and 25, and the second after the age of 60. The majority of cases occur within the younger age group (1). AA can be either inherited or acquired. Historically, patients with an inherited form of AA were identified by characteristic physical findings such as short stature, abnormal thumbs, hypopigmented and café au lait spots (Fanconi anemia); or skin pigmentation, nail dystrophy and oral leukoplakia (dyskeratosis congenita). However, an abnormal physical examination may not always be present in patients with an inherited bone marrow failure syndrome, and the identification of specific mutations in genes of the telomere complex in patients with acquired AA has blurred the distinction between the inherited and acquired forms of AA (2–4).
Although environmental exposures have been linked to the onset of severe AA (SAA), most cases are idiopathic. Regardless of etiology, patients are treated with either immunosuppressive therapy (IST) or by hematopoietic stem cell transplantation (HSCT). In general, the outcome in children following HSCT has been superior to results in adults, with a less frequent and severe graft-versus-host disease and better overall survival (5, 6). However, because the majority of children with SAA lack an HLA-histocompatible sibling, IST is often administered first. The standard IST regimen is anti-thymocyte globulin (ATG) + cyclosporine (CsA) (7).
In the past 18 years, we have treated over 400 patients with newly diagnosed SAA with IST, of which about 20% were children under the age of 18. During this period, three h-ATG-based regimens were studied: 1) standard h-ATG + CsA; 2) h-ATG + CsA+ mycophenolate mofetil (MMF); 3) h-ATG + CsA + sirolimus. The outcomes for these 3 h-ATG regimens were the same, indicating that the addition of a third immunosuppressive agent to standard h-ATG + CsA did not alter the response rate or the incidence of relapse and evolution (8, 9). In order to understand better the long term outcome of children treated with IST, we conducted a single center retrospective analysis of the incidence of response, relapse, clonal evolution and overall survival in children with acquired SAA treated with a h-ATG/CsA-based therapy.
Methods
All children less than 18 years of age who fulfilled entry criteria for SAA who, at the time of study entry, did not have an HLA-matched sibling were offered enrollment in four different immunosuppression protocols from November 1989 to December 2006 at the Warren Grant Magnuson Clinical Center and the Mark O. Hatfield Clinical Research Center at the National Institutes of Health in Bethesda, MD. Consecutive patients treated in these four sequential immunosuppression protocols were analyzed. Patients who had an HLA-matched sibling but a parent or legal guardian who opted against initial transplantation were also offered immunosuppressive therapy. All patients age seven years or above signed a minor assent and the parents (or legal guardian) signed informed consent according to protocols approved by the Institutional Review Board of the National, Heart, Lung, and Blood Institute. For protocol entry purposes, SAA was defined as bone marrow cellularity of less than 30% and severe pancytopenia with at least two of the following peripheral blood count criteria: 1) ANC < 500/uL; 2) ARC < 60 000/uL; 3) platelet count < 20 000/uL (10). Response was defined as no longer meeting criteria for SAA and was determined at 3 and 6 months following ATG (10, 11). This criteria have been previously shown to correlate strongly with both independence from transfusion and survival at 1 year (8). A complete response (CR) was defined as satisfaction of all three peripheral blood count criteria: 1) ANC > 1000/uL; 2) hemoglobin > 10 g/dL; 3) platelet count > 100 000/uL. A partial response (PR) was defined as blood counts no longer satisfying criteria for SAA but insufficient for a CR. Relapse was not defined by blood cell counts but rather as any reinstitution of immunosuppressive therapy (8).
Bone marrow biopsy and aspiration, including cytogenetics, were performed before enrollment. All patients had chromosomes assayed after in vitro exposure of lymphocytes to diepoxybutane and in some cases also to mitomycin C to exclude Fanconi anemia. Testing for paroxysmal nocturnal hemoglobinuria (PNH) was with the Ham test to 2000, when it was replaced by a flow cytometric assay (12); presence of a clone was defined as the absence of glycosylphosphatidylinositol (GPI)-anchored surface proteins greater than 1% of neutrophils or red cells; the size of the PNH clone was defined by the highest level of GPI- red blood cells or GPI- neutrophils.
Study design and treatment regimens
An ATG skin test was performed on all patients to assess for allergic hypersensitivity. H-ATG (ATGAM, Pharmacia & Upjohn Company, Kalamazoo, Mich) was administered at a dose of 40 mg/kg/d intravenously for 4 consecutive days. Serum sickness prophylaxis with oral prednisone at 1 mg/kg/d was administered prior to the first dose of ATG, continued for 10 days, and then tapered over the subsequent week. Cyclosporine was administered from the time of h-ATG at 12 mg/kg/d by mouth (15 mg/kg/day for children less than 12 years) in divided doses every 12 hours and continued for 6 months; dosing was adjusted to maintain blood levels between 200 – 400 ng/ml. Serum sickness prophylaxis, the 4-day h-ATG infusion and the 6-month CsA administration was the same for the 3 different regimens and has been described in more detail previously (10, 11). CsA was discontinued after 6 months in all but 12 patients who received standard h-ATG/CsA, which was followed by a slow CsA taper (25% dose reduction every 3 months) in the subsequent 18 months. MMF was dosed on the first day of ATG at 600 mg/m2 twice daily for children 11 years and younger and at 1 g twice daily for children 12 or over and adults, for a total of 18 months. Sirolimus was initiated on day 1 at 2 mg/d and in children less than 40 kg the dose sirolimus was 1 mg/m2/d administered for 6 months to a trough between 5 – 15 ng/ml. G-CSF was not administered routinely in conjunction with the IST. Patients underwent bone marrow biopsy at 3, 6, and 12 months, and then yearly. Evolution to myelodysplasia was defined as the appearance of a new clonal disorder on cytogenetics or characteristic dysplastic changes on bone marrow.
Statistical methods
Summary statistics, including means, proportions and their corresponding standard deviations, were used to describe patients’ age, sex, and other baseline characteristics. P-values based on multi-sample tests for proportions and the analysis of variance F-tests were used to compare patients’ baseline characteristics across the treatment groups. Sample proportions and their 95% confidence intervals were employed to describe the 6 month response rates for patients categorized by discrete risk factors. Multi-sample tests for proportions have been used to compare the 6-month response rates for patients in different risk groups. Long term survival probabilities for patients with discrete and continuous baseline risks were evaluated using the Kaplan-Meier estimates and the Cox proportional hazard models with patients who were lost to follow-up counted as censored. The numerical results were computed by S-PLUS statistical package (Insightful Inc., Seattle, WA).
Results
A total of 84 children (< 18 years of age) were treated with initial IST from 1989 to 2006 at the NIH Clinical Center. Only patients who first therapy was with a h-ATG-based immunosuppressive regimen were included in the present analysis; patients who received alternative initial investigational therapies (cyclophosphamide, rabbit ATG, or alemtuzumab) were excluded. Thus, a total of 77 children were analyzed for response, relapse, clonal evolution, and survival. The baseline characteristics of the cohort are shown in Table; their age distribution is shown in Figure 1 (available at www.jpeds.com). There were no differences in baseline characteristics among the different treatment groups.
Table 1.
Table Patient characteristics.
| All Patients | H-ATG/CsA (1989 – 2006) | H-ATG/CsA/MMF (2000 – 2003) | H-ATG/CsA/Rapa (2003 – 2005) | P-value | |||||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean% ± SD | N | Mean % ± SD | N | Mean % ± SD | N | Mean % ± SD | ||
| Total | 77 | --------- | 46 | --------- | 21 | --------- | 10 | --------- | |
|
| |||||||||
| Age (years) | 11.4 ± 0.52 | 11.6 ± 0.66 | 11 ± 1.1 | 11.8 ± 1.5 | 0.85 | ||||
|
| |||||||||
| Sex | |||||||||
| Male | 50 | 65 ± 5.5 | 29 | 63 ± 7.2 | 14 | 67 ± 10.5 | 7 | 70 ± 15.3 | 0.90 |
| Female | 27 | 35 ± 5.5 | 17 | 37 ± 7.2 | 7 | 33 ± 10.5 | 3 | 30 ± 15.3 | |
|
| |||||||||
| Etiology | |||||||||
| Idiopathic | 69 | 90 ± 3.5 | 41 | 89 ± 4.6 | 18 | 86 ± 7.8 | 10 | 100± 0.0 | 0.48 |
| Post-hepatitis | 8 | 10 ± 3.5 | 5 | 11 ± 4.6 | 3 | 14 ± 7.8 | 0 | ------ | |
|
| |||||||||
| Baseline ANC (/uL) | 289 ± 31 | 290 ± 44 | 268 ± 47 | 335 ± 81 | 0.83 | ||||
|
| |||||||||
| Baseline ALC (/uL) | 1264 ± 78 | 1295 ± 103 | 1162 ± 149 | 1335 ± 216 | 0.72 | ||||
|
| |||||||||
| Baseline ARC (/uL) | 20 540 ± 2244 | 19 232 ± 3013 | 23 021 ± 4185 | 21 344 ± 5873 | 0.76 | ||||
|
| |||||||||
| Baseline platelet (/uL) | 11 390 ± 1158 | 10 283 ± 1264 | 14 619 ± 3062 | 9700 ± 1832 | 0.23 | ||||
|
| |||||||||
| PNH | |||||||||
| < 1% | 28 | 74 ± 7.2 | 7 | 64 ± 15.2 | 13 | 72 ± 10.9 | 8 | 89 ± 11.1 | 0.46 |
| ≥ 1% | 10 | 26 ± 7.2 | 4 | 36 ± 15.2 | 5 | 28 ± 10.9 | 1 | 11 ± 11.1 | |
H-ATG/CsA, horse anti-thymocyte globulin + cyclosporine; H-ATG/CsA/MMF, horse anti-thymocyte globulin + cyclosporine + mycophenolate mofetil; H-ATG/CsA/Rapa, horse anti-thymocyte globulin + cyclosporine + sirolimus; ARC, absolute reticulocyte count; ALC, absolute lymphocyte count, ANC, absolute neutrophil count; PNH, paroxysmal nocturnal hemoglobinuria; IST, immunosuppressive therapy; SD, standard deviation. Results of Ham test prior to 2000 are not shown; only the detection of a PNH clone by flow cytometry as described in the Methods is shown.
Figure 1.

Age distribution of pediatrics patients with severe aplastic anemia treated with horse ATG-based immunosuppressive therapy.
Response to h-ATG-based IST and relapse in children < 18 years of age
The overall response rate in children at 6 months was 74% (57/77); 35% had a CR and 65% a PR, all achieved transfusion-independence. The cumulative incidence of relapse at 10 years was 33%: the median time to relapse was 558 days (Figure 2). There was no difference in the incidence of relapse between complete and partial responders. Of the 14 patients who relapsed, 1 underwent a matched sibling (alive) and 2 an alternative donor (AD) HSCT (1 died); 6 were re-treated with CsA or a repeat course of IST (all alive); 2 were lost to follow-up; and 3 patients died from infectious complications. In total, 4 patients who relapsed following initial IST died. Among the 16 patients who were non-responders at 6 months, six underwent an AD HSCT (3 died and 3 are alive); 3 underwent a matched sibling donor HSCT and are alive; 5 received a repeat course of IST (2 have responded; 1 is too early to be evaluated; 1 patient continues to receive supportive care; and 1 patient died of a fungal infection).
Figure 2.

Cumulative incidence of relapse (with 95% CI) among pediatric patients after successful initial treatment with h-ATG-based immunosuppressive therapy.
The response rate in children less than 12 (the median age of the cohort) was 80% (31/39) and for those older than 12 was 68% (26/38). In total, 37 patients had an ANC < 200/uL at baseline of which 24 (65%) responded to IST; and of the 40 patients with a baseline ANC ≥ 200/uL, 33 (83%) responded (p=0.12). When the 4 patients with an ANC < 200/uL who died prior to completing 6 months of IST are excluded, the response rate (for patients with baseline ANC < 200/uL) was 73% (24/33).
Clonal evolution and survival in children < 18 years of age
The cumulative incidence of evolution to myelodysplasia following IST was 8.5%; all 3 events occurring in partial responders (Figure 3). Cytogenetic abnormalities that were detected after IST were monosomy 7 in two patients and deletion 13q in one patient. Of these 3 patients, one is alive after a matched sibling HSCT, one patient died following an AD HSCT, and the third died of complications from marrow failure. There were no cases of evolutions to leukemia.
Figure 3.
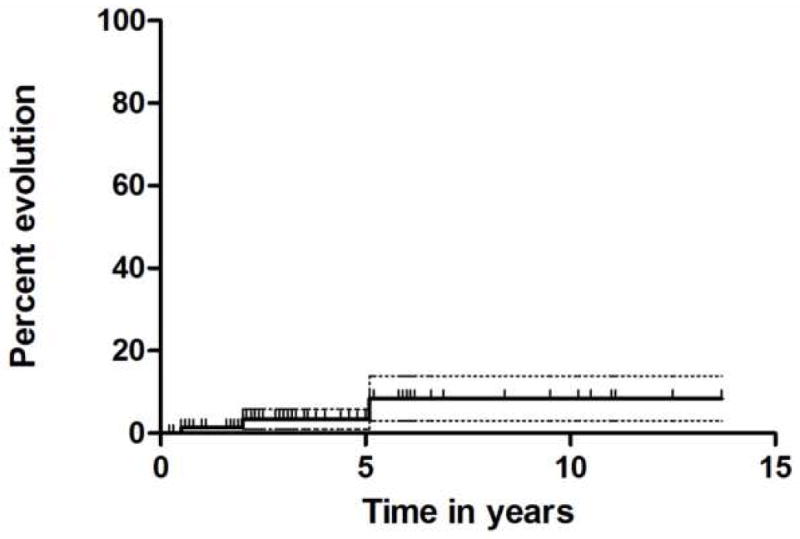
Cumulative incidence of evolution (with 95% CI) following immunosuppressive therapy with a h-ATG-based regimen.
Overall there were 13 deaths (17%): four occurred within the 3 months following IST in patients who had a pre-treatment ANC of less than 100/uL (2 fungal infections, 1 bacterial septicemia, and 1 due to gastrointestinal bleeding at a time when blood products were not available to the patient), and nine deaths occurred more than 6 months from first IST; 4 were in non-responders (3 died after an AD HSCT; 1 died of a fungal infection); 4 were relapsed patients (2 died of fungal infection; 1 died after an AD HSCT; and 1 died of sepsis); and one patient died after an AD HSCT undertaken after evolution to monosomy 7. The median time to death was 570 days. In total, 13 patients underwent HSCT; 4 from a matched sibling donor (all 4 are alive); and 9 from an AD (of whom 4 are alive). Three AD HSCTs were done in the early 1990s (all died) and one was performed in the late 1990s (alive); five AD HSCTs were performed after 2000 (2 died).
The overall 10-year survival for the entire cohort was 80% (Figure 4A); when patients who underwent HSCT were censored at time of transplantation, overall survival for the cohort was 90% (Figure 4B). Long term survival in patients who responded to IST was 89% (Figure 4C), and survival for patients who completed 6 months of IST regardless of response status was 83% (Figure 1D).
Figure 4.

Overall survival (with 95% CI) for the entire cohort including those who underwent HSCT(4A); overall survival with patients who underwent HSCT censored at the time of transplant (4B); overall survival of patients who responded to IST (4C); overall survival of patients who completed 6 months of IST regardless of response status (4D). HSCT, hematopoietic stem cell transplantation; IST, immunosuppressive therapy.
Outcome of patients ages 18–21 treated with h-ATG-based IST
Since several children’s hospitals in the US treat patients up to the age of 21 years, we conducted a separate analysis in the age group of 18–21. In total, 35 patients in this age group were treated with h-ATG-based IST from 1989 to 2006. The overall hematologic response rate was 60% (21/35). Of the 21 responders, seven (33%) relapsed. Two patients evolved to monosomy 7 and 1 patient developed a morphologic diagnosis of myelodysplasia. Among the 14 non-responders, 4 underwent HLA-matched sibling HSCT (all are alive), one underwent a haploidentical HSCT (and died); six received a second course of IST with rabbit ATG (4 responded); 2 patients died and 1 patient is alive on supportive care. Overall, there were 6 deaths in this age group: 4 in non-responders, 1 in a patient who relapsed, and 1 patient with a baseline ANC of 0/uL who died prior to completing 3 months of IST. The response rate in this age group approximates what we observe in the adult population of SAA patients treated with IST.
Discussion
In children with SAA who have an HLA-matched sibling donor, HSCT is often the initial treatment. The basis for this recommendation is from early retrospective studies that showed the superiority of transplantation over IST in children (13–15). In the 1990s, when better supportive care became available, a similar retrospective study showed comparable survival between children who underwent HSCT and IST (16). However, these reports included mainly patients treated in the 1970s and 1980s with ATG monotherapy and did encompass the current intensive regimen of an ATG + CsA, which has now been studied in children in two large prospective studies.
In a German multicenter study, 114 children were treated with the combination of ATG + CsA: response rates at 6 and 12-months was 61% and 74%, respectively; and 4-year survival was 87% (17). In a Japanese multicenter study of ATG + CsA in 119 children with SAA, the hematologic response rate was 68% and 3-year survival was 88% (18). The results from our retrospective analysis in children show an outcome very similar to those reported in the European and Japanese studies.
The response rate of 74% following IST in children is superior to our reported experience for patients of all ages (response rate of approximately 60%) (8). Children who responded to IST had long term survival of about 90%. Several of the non-responders to IST in our cohort underwent transplantation from a matched sibling or an AD with the better outcome observed among those who had a matched sibling HSCT. More recently, AD HSCT have been performed in patients who lack a genoidentical sibling with some centers reporting outcomes that rival that of a matched sibling HSCT (19). However, data from large retrospective studies in Europe and Japan suggest that the outcome for an unrelated donor HSCT remains less favorable compared with a matched-related transplant, due to a higher incidence of graft-versus-host disease, with a long term survival of about 50% (20, 21). In general, the outcome following HSCT (related and alternative donor) has been shown to be more favorable in children and in transplants performed within the past 10 years compared with earlier time periods (22–24). Despite advances in AD HSCT, prospective studies have been small, optimal conditioning is yet undefined, and follow up has been relatively short (25). Therefore, in view of the high response and survival rate among children treated with IST, it should be offered as first treatment to all children with SAA who lack a matched sibling donor.
Methods that are predictive of response to IST have been suggested but to date none have been widely accepted due to lack of practicality or standardization. A recently completed analysis of over 300 patients of all ages treated at our institution from 1989 to 2005 revealed that three factors were predictive of response at 6 months to IST: a) younger age; b) a higher pre-treatment absolute reticulocyte count (ARC); c) and a higher pre-treatment absolute lymphocyte count (ALC). In this analysis, patients with a baseline ARC ≥ 25 000/uL and ALC ≥ 1000/uL had about an 80% response rate compared with those with an ARC < 25 000/uL and ALC < 1000/uL which had a response rate of about 40%. In general, the effect of the baseline ARC was more striking than that of the baseline ALC. When a similar analysis was performed in our pediatric cohort (age < 18), the baseline ALC was not predictive of response to IST, but baseline ARC remained highly predictive. In the 77-patient pediatric cohort, those with a baseline ARC < 25 000/uL had a 65% (31/48) response rate compared with 90% (26/29) in patients with a baseline ARC ≥ 25 000/uL (p=0.02). In a large European study, a low pre-treatment ANC was predictive of response to IST in children (26). In contrast, we did not observe such association, possibly due to the smaller size of our cohort and the early deaths we observed in patients with a pre-treatment ANC of less than 200/uL.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Heart, Lung and Blood Institute.
Footnotes
95% CI is shown in all 4 figures.
The authors disclose no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alter BP, Young NS, Nathan DG, Oski FA. Hematology of Infancy and Childhood. Philadelphia: W.B. Saunders Company; 1993. The bone marrow failure syndromes; pp. 216–316. [Google Scholar]
- 2.Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand EM, Zeng W, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–30. doi: 10.1016/S0140-6736(03)14797-6. [DOI] [PubMed] [Google Scholar]
- 3.Gillio AP, Verlander PC, Batish SD, Giampietro PF, Auerbach AD. Phenotypic consequences of mutations in the Fanconi anemia FAC gene: an International Fanconi Anemia Registry study. Blood. 1997;90(1):105–10. [PubMed] [Google Scholar]
- 4.Yamaguchi H, Calado RT, Ly H, Baerlocher GM, Kajigaya S, Chanock SJ, et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N Eng J Med. 2005;352:1413–24. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 5.Horowitz MM. Current status of allogeneic bone marrow transplantation in acquired aplastic anemia. Seminars in Hematology. 2000;37:30–42. doi: 10.1016/s0037-1963(00)90028-3. [DOI] [PubMed] [Google Scholar]
- 6.Ades L, Mary JY, Robin M, Ferry C, Porcher R, Esperou H, et al. Long-term outcome after bone marrow transplantation for severe aplastic anemia. Blood. 2004;103:2490–7. doi: 10.1182/blood-2003-07-2546. [DOI] [PubMed] [Google Scholar]
- 7.Frickhofen N, Rosenfeld SJ. Immunosuppressive treatment of aplastic anemia with antithymocyte globuilin and cyclosporine. Seminars in Hematology. 2000;37:56–68. doi: 10.1016/s0037-1963(00)90030-1. [DOI] [PubMed] [Google Scholar]
- 8.Rosenfeld S, Follman D, Nu¤ez O, Young NS. Antithymocyte globulin and cyclosporine for severe aplastic anemia. Association between hematologic response and long-term outcome. Journal of the American Medical Association. 2003;289:1130–5. doi: 10.1001/jama.289.9.1130. [DOI] [PubMed] [Google Scholar]
- 9.Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anemia with combined immunosuppression: antithymocyte globulin (ATG), cyclosporin A (CSA), and mycophenolate mofetil (MMF) British Journal of Haematology. 2006;133:606–11. doi: 10.1111/j.1365-2141.2006.06085.x. [DOI] [PubMed] [Google Scholar]
- 10.Rosenfeld SJ, Kimball J, Vining D, Young NS. Intensive immunosuppression with antithymocyte globulin and cyclosporine as treatment for severe acquired aplastic anemia. Blood. 1995;85(11):3058–65. [PubMed] [Google Scholar]
- 11.Scheinberg P, Nunez O, Wu C, Young NS. Treatment of severe aplastic anaemia with combined immunosuppression: anti-thymocyte globulin, ciclosporin and mycophenolate mofetil. Br J Haematol. 2006;133(6):606–11. doi: 10.1111/j.1365-2141.2006.06085.x. [DOI] [PubMed] [Google Scholar]
- 12.Dunn DE, Tanawattanacharoen P, Boccuni P, Nagakura S, Green SW, Kirby MR, et al. Paroxysmal nocturnal hemoglobinuria cells in patients with bone marrow failure syndromes. Ann Intern Med. 1999;131(6):401–8. doi: 10.7326/0003-4819-131-6-199909210-00002. [DOI] [PubMed] [Google Scholar]
- 13.Bayever E, Champlin R, Ho W, Lenarsky C, Storch S, Ladisch S, et al. Comparison between bone marrow transplantation and antithymocyte globulin in treatment of young patients with severe aplastic anemia. Journal of Pediatrics. 1984;105:920–5. doi: 10.1016/s0022-3476(84)80078-5. [DOI] [PubMed] [Google Scholar]
- 14.Halperin DS, Grisaru D, Freedman MH, Saunders EF. Severe acquired aplastic anemia in children: 11-year experience with bone marrow transplantation and immunosuppressive therapy. American Journal of Pediatric Hematology/Oncology. 1989;11:304–9. [PubMed] [Google Scholar]
- 15.Locasciulli A, van’t Veer L, Bacigalupo A, Hows J, van Lint MT, Gluckman E, et al. Treatment with marrow transplantation or immunosuppression of childhood acquired severe aplastic anemia: a report from the EBMT SAA working party. Bone Marrow Transplantation. 1990;6:211–7. [PubMed] [Google Scholar]
- 16.Gillio AP, Boulad F, Small TN, Kernan NA, Reyes B, Childs BH, et al. Comparison of long-term outcome of children with severe aplastic anemia treated with immuno-suppression versus bone marrow transplantation. Biology of Blood and Marrow Transplantation. 1997;5:18–24. [PubMed] [Google Scholar]
- 17.Fuhrer M, Burdach S, Ebell W, Gadner H, Haas R, Harbott J, et al. Relapse and clonal disease in children with aplastic anemia (AA) after immunosuppressive therapy (IST): the SAA 94 experience. Klinische Padiatrie. 1998;210:173–9. doi: 10.1055/s-2008-1043875. [DOI] [PubMed] [Google Scholar]
- 18.Kojima S, Hibi S, Kosaka Y, Yamamoto M, Tsuchida M, Mugishima H, et al. Immunosuppressive therapy using antithymocyte globulin, cyclosporine, and danazol with or without human granulocyte colony-stimulating factor in children with acquired aplastic anemia. Blood. 2000;96:2049–54. [PubMed] [Google Scholar]
- 19.Kennedy-Nasser AA, Leung KS, Mahajan A, Weiss HL, Arce JA, Gottschalk S, et al. Comparable Outcomes of Matched-Related and Alternative Donor Stem Cell Transplantation for Pediatric Severe Aplastic Anemia. Biology of Blood and Marrow Transplantation. 2006;12(12):1277–84. doi: 10.1016/j.bbmt.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 20.Kojima S, Matsuyama T, Kato S, Kigasawa H, Kobayashi R, Kikuta A, et al. Outcome of 154 patients with severe aplastic anemia who received transplants from unrelated donors: the Japan marrow Donor Program. Blood. 2002;100:799–805. doi: 10.1182/blood.v100.3.799. [DOI] [PubMed] [Google Scholar]
- 21.Passweg JR, Perez WS, Eapen M, Camitta BM, Gluckman E, Hinterberger W, et al. Bone marrow transplants from mismatched related and unrelated donors for severe aplastic anemia. Bone Marrow Transplant. 2006 doi: 10.1038/sj.bmt.1705299. advance online publication. [DOI] [PubMed] [Google Scholar]
- 22.Locasciulli A, Oneto R, Bacigalupo A, Socie G, Korthof E, Bekassy A, et al. Outcome of patients with acquired aplastic anemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation. Haematologica. 2007;92(1):11–8. doi: 10.3324/haematol.10075. [DOI] [PubMed] [Google Scholar]
- 23.Bacigalupo A, Locatelli F, Lanino E, Marsh J, Socie G, Maury S, et al. Fludarabine, cyclophosphamide and anti-thymocyte globulin for alternative donor tranplants in acquired severe aplastic anemia: a report from the EBMT-SAA Working Party. Bone Marrow Transplantation. 2005:1–4. doi: 10.1038/sj.bmt.1705165. [DOI] [PubMed] [Google Scholar]
- 24.Deeg HJ, O’Donnell M, Tolar J, Agarwal R, Harris RE, Feig SA, et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood. 2006;108(5):1485–91. doi: 10.1182/blood-2006-03-005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108:2509–19. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuhrer M, Rampf U, Baumann I, Faldum A, Niemeyer C, Janka-Schaub G, et al. Immunosuppressive therapy for aplastic anemia in children: a more severe disease predicts better survival. Blood. 2005;106(6):2102–4. doi: 10.1182/blood-2005-03-0874. [DOI] [PubMed] [Google Scholar]