

Abstract
Cachexia accompanies many chronic inflammatory diseases, including cancer. Lean tissue wasting is only one component of the cancer cachexia response, which also includes anemia, anorexia, an hepatic acute phase protein response and increased susceptibility to secondary infections. The etiologies of cancer cachexia are multifactorial and include an overproduction of inflammatory mediators including cytokines produced by inappropriate activation of innate immunity. However, anti-cytokine therapies have generally not been seriously considered for cancer cachexia, in large part because of the overlapping activities of several inflammatory cytokines and the inability to prospectively identify the contributions of individual mediators. In contrast, recent evidence has focused on an immature myeloid cell population that expands dramatically in the tumors and secondary lymphoid organs of animals with some actively growing tumors. These immature GR-1+CD11b+ cells are metabolically active and secrete large quantities of inflammatory cytokines and chemokines with the potential to produce cachexia. Their expansion is temporally associated with the development of cachexia. Future studies are required to determine whether therapeutic efforts intended to block the expansion of these cells can prevent the lean tissue wasting that accompanies active tumor growth.
Keywords: wasting, sickness syndromes, cytokines, myelopoiesis
Introduction and Brief Perspective
Cachexia is a complex syndrome occurring in patients with chronic inflammatory diseases, and is represented by weight loss, muscle wasting, and derangements of intermediary metabolism, including an hepatic acute phase response. In cancer patients, the development of cachexia portends a poor outcome, and is responsible for an estimated 10–20% of cancer-related deaths (1). It is hypothesized that cachexia occurs in response to alterations in tumor-host immune interactions, yet no explanation for the cellular source of disturbance has been identified. With this uncertainty, no effective treatment modality for cancer-related cachexia has emerged other than supportive care including the use of dietary supplements, orexigenic agents, exercise, anabolic steroids and erythropoietin. Cachexia-associated morbidity and mortality continues to exact a high toll on patients with malignancy.
Cytokines in the Etiology of Cancer Cachexia
Almost twenty years ago, with the cloning of tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), we and others proposed almost simultaneously the hypothesis that an inappropriate expression of products of innate immunity and the inflammatory response could explain the development of cancer cachexia (2–4). Then and today, the concept proposed was that tumor-host interactions led to activation of innate immunity and the increased production of proinflammatory cytokines like TNF-α, IL-1 and IL-6 which in turn, contributed to the anorexia, weight loss, loss of lean tissue and body fat, and anemia (1, 5). In a variety of murine tumor models associated with increased production of these cytokines by either the tumor itself (6), or by tumor-modulated cells of the innate immune system, passive immunization against TNF-α, IL-1 or IL-6 could delay the development of cachexia (7–10). At the same time, however, it became quite clear that in several other rodent models, cachexia could develop in the absence of TNF-α, IL-1 or IL-6, while dependent on the increased production of several other inflammatory mediators or cytokines (11–13). Over the next decade, the overriding consensus was that individual cytokines produced during active tumor growth could in fact produce cachexia, but the development of cancer cachexia, especially in the clinical setting, could not be easily attributed to a single cytokine, especially in histologically diverse tumors.
The importance of this conclusion to research studying the mechanisms of cancer cachexia can not be overstated. During the 1990’s, FDA approved anti-TNF and anti-IL-1 therapeutics finally reached the clinic, not for cancer cachexia or for septic shock, as had originally been proposed (14), but for chronic autoimmune diseases including first rheumatoid arthritis, and subsequently for inflammatory bowel disease and psoriasis (15–20). Marc Feldmann and Ravinder Maini received the Lasker Award in 2003 for their pioneering studies moving anti-TNF therapies into the clinic for rheumatoid arthritis (21). There are currently three TNF inhibitors, etanercept, infliximab and adalimumab, and one IL-1 inhibitor, anakinra, approved by the FDA for the treatment of rheumatoid arthritis. Importantly, not only have these drugs revolutionized the treatment of intractable rheumatoid arthritis, but their use has provided immense insights into the role that TNF-α and IL-1 play in the sickness syndromes and cachexia associated with these diseases. Successful treatment of rheumatoid arthritis patients with these TNF inhibitors, and to a lesser extent, with the IL-1 inhibitor, attenuates the hepatic acute phase response, and improves subjective measures of quality of life (18, 22, 23). More importantly, recent studies suggest that such therapies prevent further progression of bone degradation and lean tissue wasting in rheumatoid arthritis, and can also under some conditions lead to a preferential restoration of fat free mass (23, 24). Since progression of rheumatoid arthritis is now generally accepted to be a consequence of exaggerated TNF-α and/or IL-1 production, the positive response by thousands of patients confirm that these FDA drugs are effective at blocking TNF and IL-1 activity, interrupting progression of disease, and suppressing the associated sickness syndromes.
The common feature linking diseases associated with chronic inflammation including cancer, rheumatoid arthritis, psoriasis, and inflammatory bowel disease is a chronic exaggerated production of pro-inflammatory cytokines. If this is indeed the case, the obvious question becomes: why are these cytokine inhibitors not being used in patients with cancer cachexia, or the lean tissue wasting associated with other chronic inflammatory diseases, such as COPD or cardiac cachexia? The answer is relatively simple. In the clinical setting, the causes of cancer cachexia or cachexia of other chronic inflammatory diseases do not appear to be as dependent upon a single cytokine or inflammatory mediator, as they are in rheumatoid arthritis, inflammatory bowel disease or psoriasis. Rather, the patterns of cytokine production appear to be dependent not only on the tumor type, but also upon whether the cancer is locally or widely disseminated. Increased production of a single cytokine like TNF-α may not be a common occurrence in human cancer, and it is likely that in patients with advanced cancer and active wasting disease, there is simultaneous production of multiple inflammatory mediators, with overlapping biological activities. Under some conditions, such as heart failure, blockade of TNF-α may have unexpected adverse consequences (25), and all subjects administered TNF inhibitors have to be monitored closely for secondary opportunistic infections, especially from intracellular pathogens, like mycobacterium or listeria (26, 27).
Redirecting the Research Focus Away from Individual Cytokines to Myeloid Cell Populations
With the realization that focusing on individual cytokines may not provide a viable therapeutic approach for the patient with cancer cachexia, research over the past several years has turned to an examination of the role that aberrant myelopoiesis plays in tumor progression and sickness syndromes. One of the hallmarks of tumor growth is the successful evasion of tumor cells for clearance by the innate and to a greater extent, the adaptive immune system. Paradoxically, the progression of neoplasia to malignant carcinomas appears dependent on tumor associated myeloid cells which promote angiogenesis and tissue remodeling. However, few have linked this immune suppression with the development of cancer cachexia or other components of the sickness syndrome. This has been short-sighted, and fails to consider the tight interaction between adaptive and innate immunity, and importantly, inflammation. Tumor-induced immune suppression has been well-documented in the literature and is seen as an essential component of tumor evasion of host immune defenses (28). Recent evidence suggests a role for myeloid derived suppressor cells (MDSC) in some tumor-related immune suppression (28, 29) (Figure 1).
Figure 1.

Schematic representation of how myeloid derived suppressor cells (MDSCs) impact on acquired and innate immunity. Reprinted with permission from Marx et al. (31).
MDSCs represent a heterogenous population of immature myeloid cells in varying stages of differentiation beyond common myeloid progenitors, but not fully differentiated neutrophils, monocytes/macrophages or dendritic cells. They inhabit not only the bone marrow, but also secondary lymphoid organs like the spleen and lymph nodes, and tumors themselves. Their functions are not fully known, although everyone agrees that they are metabolically active and immune suppressive (30, 31). We would argue that these MDSCs may also play a role in the inappropriate production of cytokines and inflammatory mediators that contribute to cachexia and sickness syndromes. MDSCs are heterogenous, are identified by their dual expression of GR-1 (granulocyte-differentiation antigen 1) and CD11b (a receptor for complement, fibrinogen, or Factor X), and their numbers in secondary lymphoid organs are known to increase in response to a number of diverse inflammatory stimuli, including malignancy and chronic infection (31). These cells are associated with a concurrent increase in the production of reactive oxygen species, NO, peroxynitrites, TGF-β, and IL-10, among other mediators (30). In fact, we have shown that when similar GR-1+CD11b+ cells are removed from the spleens of mice with chronic peritonitis, the cells produce increased quantities of TNF-α, IL-10 and chemokines upon stimulation with bacterial lipopolysaccharide (32). The presence of these cells, and the elaboration of cytokines that they generate have multiple effects on the immune system, including inhibition of T-cell activation, suppression of T-cell receptor mediated responses, and induction of CD8+ T-cell anergy (29, 32). These effects, which are purported to promote tumor evasion of host immunosurveillance, are also implicated in tumor bearing hosts having an increased susceptibility to bacterial infections and hyper-responsiveness to bacterial products (33, 34) (Figure 2).
Figure 2.

Proposed model by which MDSC expansion leads to increased tumor growth and the development of cachexia in a tumor bearing animal.
In addition to the direct immunosuppressive effects, altered cytokine production, and change in T-cell behavior promoted by the presence of MDSCs, it is tempting to suggest that these cells may also have the potential to alter intermediary metabolism (1). This has not been investigated to date. Specific examples of the impact that they may have in this area include increased catabolism of arginine secondary to an increase in arginase I activity and alterations in carbohydrate, lipid, and protein metabolism (35, 36) resulting in an MDSC-directed decrease in CD4+ T-cell proliferation (37). This suggests that there may be value in investigating the role of this population of cells in the development of cancer cachexia syndrome and its associated metabolic complications.
Work has been initiated in our laboratory to assess the temporal appearance of MDSCs in the cancer cachexia syndrome. Our first question was whether there was an expansion of these cell populations in murine tumor models associated with the development of cachexia and sickness syndromes. These investigations involve the implantation of a tumor known to induce the expansion of MDSC in a murine host, the 4T1 mammary adenocarcinoma (unpublished observations). As shown in Figure 3, the active growth of the tumor was associated with a progressive increase in the splenic MDSC population. By the time that the tumor had reached approximately 10% of body weight, the absolute numbers of these MDSCs had increased over 100-fold in the spleens of these animals.
Figure 3.
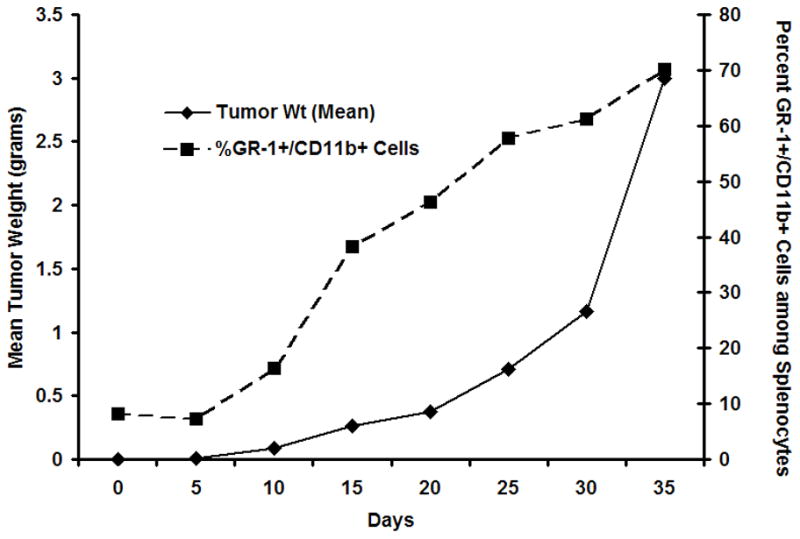
Changes in tumor weight and splenic expansion of the MDSC population. C57BL/6 mice were implanted with 4T1 mammary adenocarcinoma and followed for periods out to 35 days. At intervals, the numbers of GR-1+CD11b+ cells in the spleen were determined. With active tumor growth, there was a progressive increase in the percentage and absolute number of MDSCs in the spleen.
Our observations suggest that active tumor growth, at least of this mammary adenocarcinoma, is associated with the expansion of these immature MDSCs in the spleen. Importantly, this tumor model is also associated with several manifestations of the cachexia response. This is further supported by the observation that these CD11b+, Gr-1+ cells approach normal levels within eight days following resection of the primary tumor ((38) and unpublished data). Surprisingly, as the tumor grows, the animals demonstrate relatively few changes in gross appearance or behavior until almost three weeks after tumor implantation. After 25 and 30 days, a clear metabolic change occurs, and the animals rapidly become cachectic and debilitated. This switch in the metabolic response is characterized by a loss of non tumor body weight and lean tissue, the development of an hepatic acute phase protein response, and increased susceptibility to either a sublethal endotoxicosis or a subsequent peritonitis (data not shown).
Taken together, these physical and biochemical findings suggest a late development of the cachexia syndrome in the tumor-bearing host associated with a marked expansion of these metabolically active immature MDSCs. Our working hypothesis is that MDSCs in themselves may not be sufficient to produce cachexia and sickness syndromes, but are required in sufficient numbers before a cachexia response can develop. Expanding this hypothesis further, the increased numbers of MDSCs in the tumor bearing host predisposes the animal to the development of cachexia, by first suppressing adaptive immunity and further promoting tumor growth (and the direct release of tumor-derived cachexia factors (39)), and second by providing a cellular population responsible for the increased production of cytokines and other inflammatory mediators (Figure 2). We speculate that this increased presence of MDSC exerts effects upon reaching a threshold level with the subsequent spontaneous development of cachexia. This potentially represents a point of intersection between MDSC coordination of host immune response and the development of metabolic derangements that may result.
While further work is necessary to illustrate a definite connection between MDSC and the development of cachexia, there is reason to warrant investigation into their role in cachexia syndromes. MDSC effects on the host immune system clearly impact the host’s response to the presence of a tumor (Figure 1); however, it may be that these well-defined effects lead secondarily to the metabolic derangements of cachexia. At present, there are few approaches to preventing the expansion of the MDSC population. All trans-retinoic acid can cause the differentiation of these cells into mature macrophages through a reactive oxygen species dependent process (40), or neutralizing antibodies to GR-1 can deplete immature and mature myeloid cells (32). Whether these approaches can be used to treat cancer cachexia is presently unknown. Further research exploring the potential relationship between MDSC and cachexia syndrome is an area of great promise, may provide new answers in what has been a vexing clinical problem, and has the potential to lead to novel therapies for this devastating condition.
Acknowledgments
Supported in part by grants GM-40856 and GM-81932, awarded by the National Institute of General Medical Sciences. RDW was supported by a T32 training grant (T32 CA-106493) in surgical oncology from the National Cancer Institute, NIH. MJD was supported by a T32 training grant (T32 GM-08421) in trauma, inflammation, and burns from the National Institute of General Medical Sciences.
References
- 1.Delano MJ, Moldawer LL. The origins of cachexia in acute and chronic inflammatory diseases. Nutr Clin Pract. 2006;21:68–81. doi: 10.1177/011542650602100168. [DOI] [PubMed] [Google Scholar]
- 2.Moldawer LL, Georgieff M, Lundholm K. Interleukin 1, tumour necrosis factor-alpha (cachectin) and the pathogenesis of cancer cachexia. Clin Physiol. 1987;7:263–74. doi: 10.1111/j.1475-097x.1987.tb00169.x. [DOI] [PubMed] [Google Scholar]
- 3.Aderka D, Fisher S, Levo Y, Holtmann H, Hahn T, Wallach D. Cachectin/tumour-necrosis-factor production by cancer patients. Lancet. 1985;2:1190. doi: 10.1016/s0140-6736(85)92713-8. [DOI] [PubMed] [Google Scholar]
- 4.Oliff A, Defeo-Jones D, Boyer M, et al. Tumors secreting human TNF/cachectin induce cachexia in mice. Cell. 1987;50:555–63. doi: 10.1016/0092-8674(87)90028-6. [DOI] [PubMed] [Google Scholar]
- 5.Esper DH, Harb WA. The cancer cachexia syndrome: a review of metabolic and clinical manifestations. Nutr Clin Pract. 2005;20:369–76. doi: 10.1177/0115426505020004369. [DOI] [PubMed] [Google Scholar]
- 6.Lonnroth C, Moldawer LL, Gelin J, Kindblom L, Sherry B, Lundholm K. Tumor necrosis factor-alpha and interleukin-1 alpha production in cachectic, tumor-bearing mice. Int J Cancer. 1990;46:889–96. doi: 10.1002/ijc.2910460523. [DOI] [PubMed] [Google Scholar]
- 7.Gelin J, Moldawer LL, Lonnroth C, Sherry B, Chizzonite R, Lundholm K. Role of endogenous tumor necrosis factor alpha and interleukin 1 for experimental tumor growth and the development of cancer cachexia. Cancer Res. 1991;51:415–21. [PubMed] [Google Scholar]
- 8.Sherry BA, Gelin J, Fong Y, et al. Anticachectin/tumor necrosis factor-alpha antibodies attenuate development of cachexia in tumor models. Faseb J. 1989;3:1956–62. doi: 10.1096/fasebj.3.8.2721856. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Kishimoto H, Chua HL, et al. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am J Pathol. 2003;163:2531–41. doi: 10.1016/s0002-9440(10)63608-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strassmann G, Fong M, Kenney JS, Jacob CO. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J Clin Invest. 1992;89:1681–4. doi: 10.1172/JCI115767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander HR, Billingsley KG, Block MI, Fraker DL. D-factor/leukaemia inhibitory factor: evidence for its role as a mediator in acute and chronic inflammatory disease. Cytokine. 1994;6:589–96. doi: 10.1016/1043-4666(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 12.Billingsley KG, Fraker DL, Strassmann G, Loeser C, Fliot HM, Alexander HR. Macrophage-derived tumor necrosis factor and tumor-derived of leukemia inhibitory factor and interleukin-6: possible cellular mechanisms of cancer cachexia. Ann Surg Oncol. 1996;3:29–35. doi: 10.1007/BF02409048. [DOI] [PubMed] [Google Scholar]
- 13.Doherty GM, Alexander HR, Merino MJ, Venzon DJ, Norton JA. Role of endogenous interferon gamma in murine tumor growth and tumor necrosis factor alpha antitumor efficacy. Ann Surg Oncol. 1996;3:198–203. doi: 10.1007/BF02305801. [DOI] [PubMed] [Google Scholar]
- 14.Beutler B, Cerami A. The common mediator of shock, cachexia, and tumor necrosis. Adv Immunol. 1988;42:213–31. doi: 10.1016/s0065-2776(08)60846-9. [DOI] [PubMed] [Google Scholar]
- 15.Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N Engl J Med. 2004;350:2167–79. doi: 10.1056/NEJMra032906. [DOI] [PubMed] [Google Scholar]
- 16.Leonardi CL, Powers JL, Matheson RT, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014–22. doi: 10.1056/NEJMoa030409. [DOI] [PubMed] [Google Scholar]
- 17.Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. 1997;337:141–7. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- 18.Maini RN, Breedveld FC, Kalden JR, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum. 2004;50:1051–65. doi: 10.1002/art.20159. [DOI] [PubMed] [Google Scholar]
- 19.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–35. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 20.Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–9. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- 21.Mitka M. Gene studies, anti-TNF therapy take Lasker honors. Jama. 2003;290:1979–80. doi: 10.1001/jama.290.15.1979. [DOI] [PubMed] [Google Scholar]
- 22.Breedveld FC, Han C, Bala M, et al. Association between baseline radiographic damage and improvement in physical function after treatment of patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:52–5. doi: 10.1136/ard.2003.017160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Metsios GS, Stavropoulos-Kalinoglou A, Douglas KM, et al. Blockade of tumour necrosis factor-alpha in rheumatoid arthritis: effects on components of rheumatoid cachexia. Rheumatology (Oxford) 2007;46:1824–7. doi: 10.1093/rheumatology/kem291. [DOI] [PubMed] [Google Scholar]
- 24.Marcora SM, Chester KR, Mittal G, Lemmey AB, Maddison PJ. Randomized phase 2 trial of anti-tumor necrosis factor therapy for cachexia in patients with early rheumatoid arthritis. Am J Clin Nutr. 2006;84:1463–72. doi: 10.1093/ajcn/84.6.1463. [DOI] [PubMed] [Google Scholar]
- 25.Anker SD, Coats AJ. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int J Cardiol. 2002;86:123–30. doi: 10.1016/s0167-5273(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 26.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 27.Kesteman T, Yombi JC, Gigi J, Durez P. Listeria infections associated with infliximab: case reports. Clin Rheumatol. 2007;26:2173–5. doi: 10.1007/s10067-007-0660-8. [DOI] [PubMed] [Google Scholar]
- 28.Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–9. [PubMed] [Google Scholar]
- 29.Nagaraj S, Gupta K, Pisarev V, et al. Altered recognition of antigen is a mechanism of CD8(+) T cell tolerance in cancer. Nat Med. 2007;13:828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55:237–45. doi: 10.1007/s00262-005-0048-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marx J. Cancer immunology. Cancer’s bulwark against immune attack: MDS cells. Science. 2008;319:154–6. doi: 10.1126/science.319.5860.154. [DOI] [PubMed] [Google Scholar]
- 32.Delano MJ, Scumpia PO, Weinstein JS, et al. MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J Exp Med. 2007;204:1463–74. doi: 10.1084/jem.20062602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grossie VB., Jr Influence of the ward colon tumor on the innate and endotoxin-induced inflammatory response of the rat. Cancer Invest. 2001;19:698–705. doi: 10.1081/cnv-100106145. [DOI] [PubMed] [Google Scholar]
- 34.Grossie VB, Jr, Mailman D. Influence of the Ward colon tumor on the host response to endotoxin. J Cancer Res Clin Oncol. 1997;123:189–94. doi: 10.1007/BF01240314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popovic PJ, Zeh HJ, 3rd, Ochoa JB. Arginine and immunity. J Nutr. 2007;137:1681S–1686S. doi: 10.1093/jn/137.6.1681S. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–91. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol. 2006;176:2085–94. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 38.Sinha P, Clements VK, Ostrand-Rosenberg S. Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol. 2005;174:636–45. doi: 10.4049/jimmunol.174.2.636. [DOI] [PubMed] [Google Scholar]
- 39.Tisdale MJ. Cachexia in cancer patients. Nat Rev Cancer. 2002;2:862–71. doi: 10.1038/nrc927. [DOI] [PubMed] [Google Scholar]
- 40.Nefedova Y, Fishman M, Sherman S, Wang X, Beg AA, Gabrilovich DI. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007;67:11021–8. doi: 10.1158/0008-5472.CAN-07-2593. [DOI] [PubMed] [Google Scholar]