

Abstract
Comparative genomics provides a powerful tool to characterize the genetic differences among species that may be linked to their phenotypic variations. In the case of mosquito-associated Spiroplasma species, such approach is useful for the investigation of their differentiations in substrate utilization strategies and putative virulence factors. Among the four species that have been assessed for pathogenicity by artificial infection experiments, Spiroplasma culicicola and S. taiwanense were found to be pathogenic, whereas S. diminutum and S. sabaudiense were not. Intriguingly, based on the species phylogeny, the association with mosquito hosts and the gain or loss of pathogenicity in these species appears to have evolved independently. Through comparison of their complete genome sequences, we identified the genes and pathways that are shared by all or specific to one of these four species. Notably, we found that a glycerol-3-phosphate oxidase gene (glpO) is present in S. culicicola and S. taiwanense but not in S. diminutum or S. sabaudiense. Because this gene is involved in the production of reactive oxygen species and has been demonstrated as a major virulence factor in Mycoplasma, this distribution pattern suggests that it may be linked to the observed differences in pathogenicity among these species as well. Moreover, through comparative analysis with other Spiroplasma, Mycoplasma, and Mesoplasma species, we found that the absence of glpO in S. diminutum and S. sabaudiense is best explained by independent losses. Finally, our phylogenetic analyses revealed possible recombination of glpO between distantly related lineages and local rearrangements of adjacent genes.
Keywords: Mollicutes, Spiroplasma, mosquito, virulence factor, glycerol-3-phosphate oxidase, glpO
Introduction
Comparative analysis of gene content among related species with distinct phenotypes has provided a powerful tool to investigate the underlying genetic mechanisms. For example, examination of the presence and absence of genes between bacterial species that differ in pathogenicity can be used to identify putative virulence factors. This genome-scale screening is a high-throughput and cost-effective approach of narrowing down the list of candidate genes, which may greatly facilitate the downstream experimental verification and functional characterization. To demonstrate the utility of this comparative approach, the mosquito-associated Spiroplasma species provide a good study system.
The genus Spiroplasma contains a diverse group of wall-less bacteria that are mostly associated with various insect hosts (Whitcomb 1981; Gasparich et al. 2004; Regassa and Gasparich 2006; Gasparich 2010). To date five characterized Spiroplasma species have been found to be associated with mosquitoes, including Spiroplasma culicicola (Hung et al. 1987), S. sabaudiense (Abalain-Colloc et al. 1987), S. taiwanense (Abalain-Colloc et al. 1988), S. cantharicola (Whitcomb et al. 1993), and S. diminutum (Williamson et al. 1996). All of these five Spiroplasma species belong to the Apis clade within the genus. Interestingly, examination of their serotypes, phylogenetic placements, and the host associations of other related species suggest that the associations with mosquitoes have multiple independent origins (Gasparich et al. 2004; Lo, Ku, et al. 2013; the phylogeny and host associations are summarized in fig. 1). Because of the interests in developing these mosquito-associated bacteria for biological control of insect pests, a series of artificial infection experiments have been performed to examine the pathogenicity of these Spiroplasma species (Chastel and Humphery-Smith 1991; Humphery-Smith, Grulet, Chastel, et al. 1991; Humphery-Smith, Grulet, Le Goff, et al. 1991; Vorms-Le Morvan et al. 1991; Vazeille-Falcoz et al. 1994; Phillips and Humphery-Smith 1995). Based on these results, infection by S. taiwanense or S. culicicola increased the mortality of mosquitoes, no significant effect was found for the infection by S. diminutum or S. sabaudiense, while the effects of S. cantharicola infection remained to be tested.
Fig. 1.—
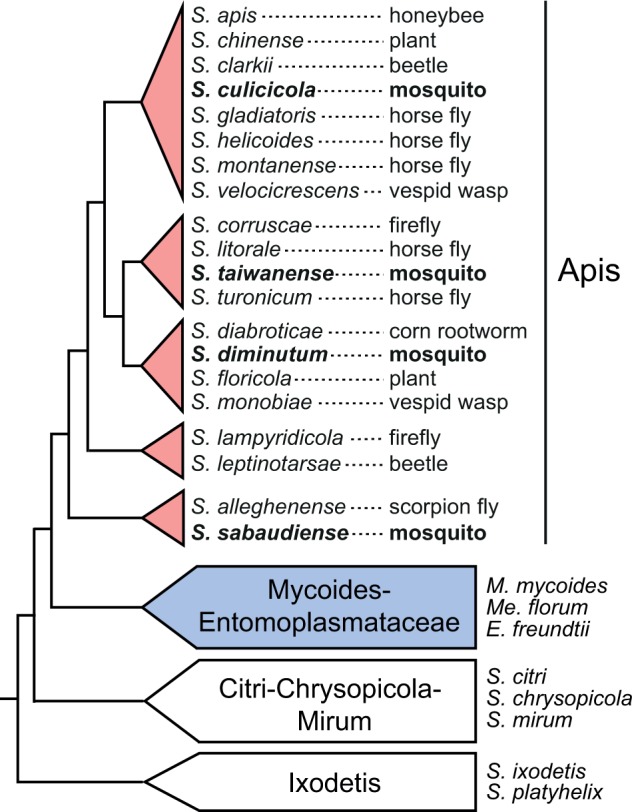
Phylogeny of representative Spiroplasma species and the Mycoides–Entomoplasmataceae clade. The isolation source of the Spiroplasma species in the Apis clade is labeled after the species name. The four mosquito-associated species analyzed in this study are highlighted in bold. The phylogeny is based on Gasparich et al. (2004) and Lo, Ku, et al. (2013).
To investigate the genetic mechanisms that may explain these observed differences in artificial infection experiments, we have determined the complete genome sequences of S. taiwanense and S. diminutum for comparative analysis (Lo, Ku, et al. 2013). One main finding from this pairwise genome comparison is that S. taiwanense has a copy of glpO encoding a glycerol-3-phosphate (G3P) oxidase, while S. diminutum does not. Because this gene is involved in reactive oxygen species (ROS) production, the presence of this gene in the S. taiwanense genome provides an explanation for the observation of tissue damage (Phillips and Humphery-Smith 1995) and increased mortality (Humphery-Smith, Grulet, Chastel, et al. 1991; Humphery-Smith, Grulet, Le Goff, et al. 1991; Vazeille-Falcoz et al. 1994) in infected hosts. Moreover, functional characterizations have provided experimental evidence that this gene is the main virulence factor in the closely related Mycoplasma mycoides (Pilo et al 2005, 2007) and the more distantly related M. pneumoniae (Hames et al. 2009).
However, several questions remained regarding the molecular evolution of glpO in mosquito-associated Spiroplasma species. For example, was the gene gained in the lineage leading to S. taiwanense or lost in S. diminutum? Do other Spiroplasma species possess this gene as well? To address these questions, we determined the complete genome sequences of the other two mosquito-associated Spiroplasma species that have been tested in artificial infection experiments, S. culicicola and S. sabaudiense, for more comprehensive comparative analyses in this study. Because the two species that have been found to be pathogenic (i.e., S. taiwanense and S. culicicola) do not form a monophyletic clade when other mosquito-associated species are considered (Lo, Ku, et al. 2013), this expansion in taxon sampling provides us with the opportunity to investigate the possibility of multiple independent gains or losses of putative virulence factors in these bacteria. Additionally, the recent increased availability of complete genome sequences from other Spiroplasma species (Ku et al. 2013, 2014) has improved our ability to establish the ancestral states of gene content and to perform molecular phylogenetic inference. Taken together, we aim to improve our understanding of the substrate utilization strategy and putative virulence factors in mosquito-associated Spiroplasma species.
Materials and Methods
Genome Sequencing
The two bacterial strains sequenced in this study, S. culicicola AES-1T and S. sabaudiense Ar-1343T, were acquired from the American Type Culture Collection (ATCC catalog numbers 35112 and 43303, respectively). For the whole genome shotgun sequencing of S. culicicola, one paired-end (∼160-bp insert; 151-bp reads; ∼0.81-Gb raw reads) and one mate-pair (∼3-kb insert; 101-bp reads; ∼1.85-Gb raw reads) library were prepared and sequenced using the Illumina HiSeq 2000 platform (Illumina, USA). For S. sabaudiense, one paired-end library (∼311-bp insert; 251-bp reads; ∼0.65-Gb raw reads) was sequenced using the Illumina MiSeq platform (Illumina).
The procedures for genome assembly and annotation were based on those described in our previous studies (Chung et al. 2013; Lo, Chen, et al. 2013). For S. culicicola, the initial de novo genome assembly was performed using ALLPATHS-LG release 42781 (Gnerre et al. 2011) because of the availability of mate-pair reads. For S. sabaudiense, the assembly was performed using VELVET v1.2.07 (Zerbino and Birney 2008). PCR primer walking and Sanger sequencing were used for gap filling and assembly verification. After the genomes were sequenced to completion, all raw reads were mapped to the final assembly by BWA v0.7.4 (Li and Durbin 2009) for variant check by SAMTOOLS v0.1.19 (Li et al. 2009) and for visual inspection by IGV v2.1.24 (Robinson et al. 2011). The programs RNAmmer (Lagesen et al. 2007), tRNAscan-SE (Lowe and Eddy 1997), and PRODIGAL (Hyatt et al. 2010) were used for gene prediction. The gene names and product descriptions were annotated based on the orthologous genes identified by OrthoMCL (Li et al. 2003) in previously published Spiroplasma genomes (Ku et al. 2013; Lo, Chen, et al. 2013; Lo, Ku, et al. 2013; Ku et al. 2014), including S. apis (GenBank accession number CP006682), S. chrysopicola (CP005077), S. diminutum (CP005076), S. melliferum (AMGI01000001-AMGI01000024), S. syrphidicola (CP005078), and S. taiwanense (CP005074-CP005075). The genes that lack identifiable orthologs were manually curated based on the BlastP (Altschul et al. 1997; Camacho et al. 2009) searches against the NCBI nonredundant (nr) protein database (Benson et al. 2012). The signal peptides of putative secreted proteins were identified using SignalP v4.0 (Petersen et al. 2011) based on the Gram-positive bacteria model. To distinguish between secreted proteins and transmembrane proteins, the predicted signal peptides were removed before the identification of transmembrane helices using TMHMM v2.0 (Krogh et al. 2001). The Conserved Domain Database (Marchler-Bauer et al. 2013) was used to identify protein domains and to provide additional annotation information. The KAAS tool (Moriya et al. 2007) provided by the KEGG database (Kanehisa and Goto 2000; Kanehisa et al. 2010) was used to classify protein-coding genes into the COG functional categories (Tatusov et al. 1997, 2003). To visualize the genomes of the four mosquito-associated Spiroplasma species, the annotated chromosomes were plotted using CIRCOS (Krzywinski et al. 2009) to show gene locations, GC-skew, and GC content.
Comparative Analysis
Two sets of comparative analyses were performed in this study. The first set includes the four mosquito-associated Spiroplasma species, all of which belong to the Apis clade within this genus. The second set expands the taxon sampling to include S. apis in the Apis clade, four Mycoplasma/Mesoplasma species in the sister Mycoides–Entomoplasmataceae clade (M. mycoides [BX293980], M. leachii [CP002108], M. putrefaciens [CP004357], and Me. florum [AE017263]) and two Spiroplasma species in the Chrysopicola clade as the outgroup (S. chrysopicola and S. syrphidicola). In these two sets of comparative analyses, the homologous gene clusters among the genomes being compared were identified by OrthoMCL (Li et al. 2003). The lists of homologous gene clusters were examined to investigate the patterns of gene presence and absence.
For the four mosquito-associated Spiroplasma species, we utilized MUMmer v3.23 (Kurtz et al. 2004) for pairwise genome alignments. We increased the minimum match length (option “−l”) to 24 from the default setting of 20 to reduce spurious hits. The chromosome of S. taiwanense was chosen as the reference to be compared with the other three species. To estimate the genome-wide sequence divergence levels, the single-copy orthologous genes shared by these four species were used for sequence alignment by MUSCLE v3.8 (Edgar 2004). The alignments of individual genes were concatenated for calculation of sequence similarities by the DNADIST and PROTDIST programs of PHYLIP v3.69 (Felsenstein 1989).
For molecular phylogenetic inference, homologous genes from selected genomes were aligned using MUSCLE v3.8 (Edgar 2004). The maximum likelihood phylogenies were inferred using PhyML v3.0 (Guindon and Gascuel 2003). The proportion of invariable sites and the gamma distribution parameter were estimated from the data set, and the number of substitute rate categories was set to four. Bootstrap supports were estimated based on 1,000 replicates generated by the SEQBOOT program of PHYLIP v3.69 (Felsenstein 1989).
Results and Discussion
Genome Sequences of Mosquito-Associated Spiroplasma Species
The genome assembly statistics and chromosomal organization of the four mosquito-associated Spiroplasma species are provided in table 1 and figure 2. Both of the two newly sequenced species contain a circular chromosome that is approximately 1.1 Mb in size (S. culicicola: 1,175,131 bp; S. sabaudiense: 1,075,953 bp). These genome sizes are slightly smaller than those reported previously based on pulsed-field gel electrophoresis (Carle et al. 1995). The values for GC content are similar to those estimates based on the buoyant density method (Abalain-Colloc et al. 1987; Hung et al. 1987).
Table 1.
Genome Assembly Statistics
| Genome | S. diminutum CUAS-1T | S. taiwanense CT-1T | S. culicicola AES-1T | S. sabaudiense Ar-1343T |
|---|---|---|---|---|
| GenBank accession | CP005076 | CP005074 | CP006681 | CP006934 |
| Chromosome size (bp) | 945,296 | 1,075,140 | 1,175,131 | 1,075,953 |
| GC content (%) | 25.5 | 23.9 | 26.4 | 30.2 |
| Coding density (%) | 92.7 | 82.5 | 92.2 | 90.0 |
| Protein-coding genes | 858 | 991 | 1,071 | 924 |
| Length distribution (Q1/Q2/Q3) (aa) | 177/283/443 | 137/247/397 | 176/283/437 | 189/296/455 |
| Hypothetical proteins | 310 | 452 | 460 | 368 |
| Annotated pseudogenes | 0 | 54 | 0 | 7 |
| rRNA genes/operons | 3/1 | 3/1 | 3/1 | 6/2 |
| tRNA genes | 29 | 29 | 29 | 30 |
| Plasmid (GenBank accession) | 0 | 1 (CP005075) | 0 | 0 |
Fig. 2.—

Chromosomal organization of the four mosquito-associated Spiroplasma species. Rings from the outside in: (1) scale marks (unit: Mb), (2 and 3), protein-coding genes on the forward and reverse strand, respectively (color coded by the functional categories), (4) GC skew (positive: green; negative: yellow), and (5) GC content (above average: orange; below average: blue; rRNA operons are labeled by black triangles).
The availability of these two additional Spiroplasma genomes, together with the previously established species phylogeny (Lo, Ku, et al. 2013), provided several insights into the genome evolution in the Apis clade. For example, the low GC content, low coding density, and high frequency of pseudogenes observed in the S. taiwanense genome (Lo, Ku, et al. 2013) appear to be derived states specific to this lineage. Additionally, S. sabaudiense belongs to the basal group and has several distinct genomic features. First, S. sabaudiense has the highest GC content (30.2%) among the Spiroplasma genomes sequenced to date. For comparison, the other three mosquito-associated Spiroplasma species have a GC content of 23.9–26.4%. Considering that the S. apis genome has a GC content of 28.3% (Ku et al. 2014) and the more distantly related Spiroplasma species in the Chrysopicola clade have a GC content of 28.8–29.2%, it is possible that a relatively high GC content of ∼28–30% represents the ancestral state of the Apis clade. Second, S. sabaudiense has one additional tRNA-Ser gene compared with other genomes in the Apis clade (Lo, Ku, et al. 2013; Ku et al. 2014), which may be the result of a lineage-specific gene duplication event. Finally, S. sabaudiense has two complete and identical rRNA operons. For comparison, all other characterized Spiroplasma genomes were found to have only one rRNA operon (Carle et al. 2010; Alexeev et al. 2012; Ku et al. 2013; Lo, Chen, et al. 2013; Lo, Ku, et al. 2013; Ku et al. 2014), while the Mycoplasma/Mesoplasma species in the sister Mycoides–Entomoplasmataceae clade have two rRNA operons. It is unclear if this pattern is due to two independent duplication events (i.e., one in the lineages leading to S. sabaudiense and one in the common ancestor of the Mycoides–Entomoplasmataceae clade) or a single duplication in the common ancestor of the Apis–Mycoides–Entomoplasmataceae clade, followed by one or more losses in the Apis clade. Future improvement in the taxon sampling of available complete genome sequences from the Apis and the Mycoides–Entomoplasmataceae clade is necessary to further investigate this issue.
When the chromosome of S. taiwanense is used as the reference for pairwise genome alignments, the patterns are consistent with the expectation based on the species phylogeny (Lo, Ku, et al. 2013) and the levels of sequence similarity (fig. 3). Compared with the most closely related S. diminutum (fig. 3A), the chromosomal organizations are largely conserved, except for the ∼0.5–0.7 Mb region that contains the putative replication terminus at ∼0.58 Mb and possibly involves an inversion. In contrast, the highly divergent S. sabaudiense exhibits low levels of sequence similarity and synteny conservation (fig. 3C).
Fig. 3.—
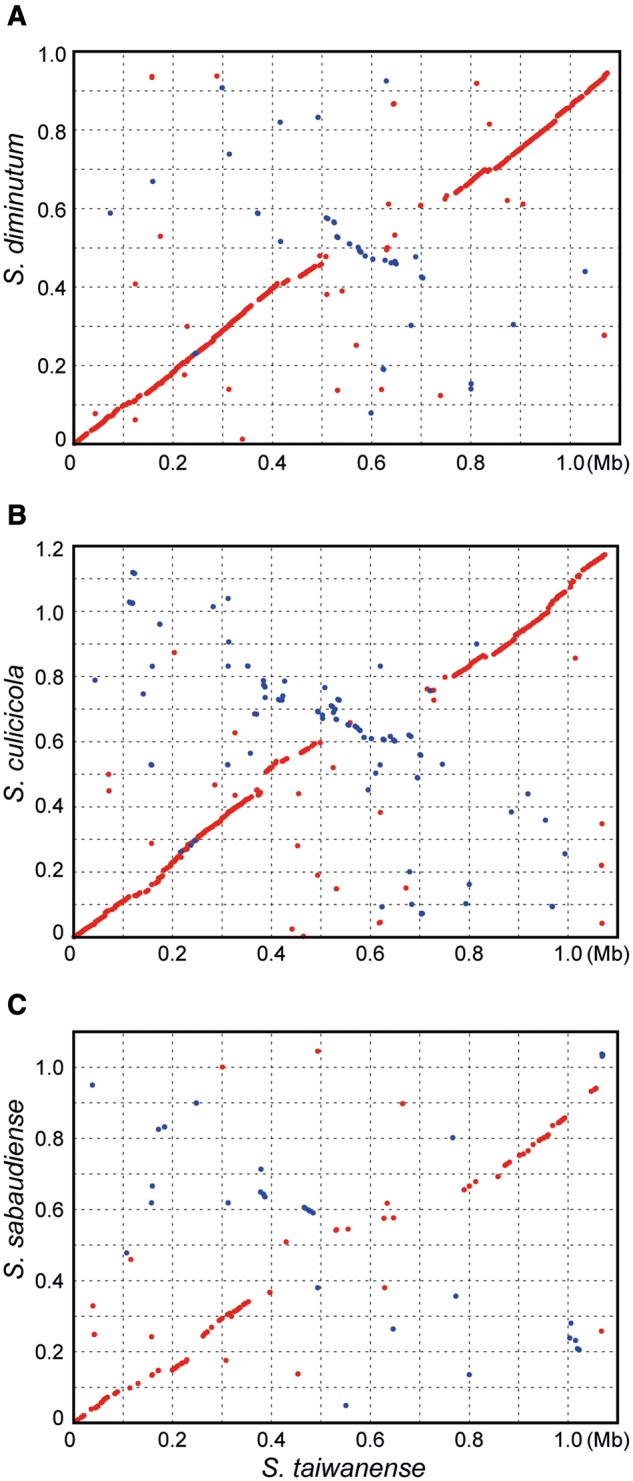
Pairwise genome alignments. The genome of S. taiwanense was used as the reference for all three alignments (red dots: matches on the same strand; blue dots: matches on the opposite strands). The sequence similarity levels were calculated based on 524 single-copy orthologous genes shared by these four species. The concatenated alignments contain 560,694 aligned nucleotide (nt) sites and 184,080 aligned amino acid (aa) sites, respectively. nt/aa similarity: (A) 76.6%/70.9%, (B) 73.3%/66.0%, and (C) 65.2%/54.2%.
Comparison of Gene Content and Substrate Utilization Strategies
A total of 1,634 homologous gene clusters were found among the four mosquito-associated Spiroplasma species (fig. 4 and supplementary table S1, Supplementary Material online). Among these, 552 are shared by all four species (>50% of all the protein-coding gene in each species). These core genes include those involved in essential cellular processes conserved among bacteria (Koonin 2003; Lapierre and Gogarten 2009; Chen et al. 2012), such as DNA replication, transcription, and translation. Furthermore, genes that have been suggested as shared between the Apis and the Citri clade within Spiroplasma (Lo, Chen, et al. 2013; Lo, Ku, et al. 2013), such as those involved in glucose uptake and utilization (ptsG and crr), fructose uptake and utilization (fruA and fruK), N-acetylglucosamine (GlcNAc) uptake and utilization (nagE, nagA, and nagB), glycolysis (pgi, pfkA, fbaA, gap, gapN, pgk, pgm, eno, and pyk; the dotted line in fig. 5), nucleotide biosynthesis (e.g., adk, apt, gmk, hprT, purA, purB, pyrG, pyrH, rdgB, tdk, thyA, tmk, upp, etc), the nonmevalonate pathway for isopentenyl pyrophosphate synthesis (dxr, dxs, ispD, ispE, ispF, ispG, and ispH), protection from oxidative stress (sufB, sufC, sufD, sufS, sufU, and tpx), and putative secreted proteins containing GH18 chitinase and SGNH hydrolase domains are found to be conserved in these four species. Intriguingly, among these conserved genes, we observed several cases of lineage-specific gene family expansions. For example, in a previous analysis of the S. taiwanense genome (Lo, Ku, et al. 2013), three oligopeptide ABC transporter genes (oppC, oppD, and oppF) were found to have three copies each. Because these genes are single copy in the other three Spiroplasma genomes compared in this study, this observation is best explained by S. taiwanense-specific tandem duplications. In addition, the 6-phospho-beta-glucosidase gene (bgl) was found to exhibit a high level of copy number variation, ranging from single copy in S. taiwanense, three copies in S. diminutum and S. sabaudiense, to eight copies in S. culicicola.
Fig. 4.—
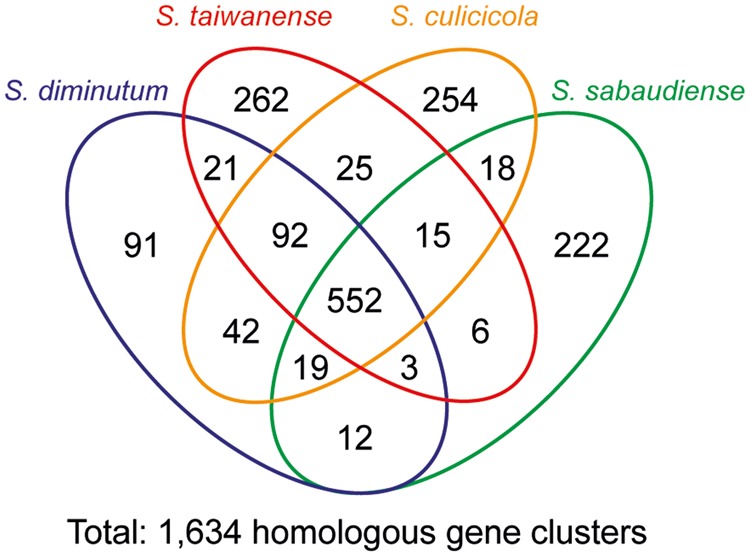
Distribution pattern of homologous gene clusters. The detailed lists of these gene clusters are provided in supplementary table S1, Supplementary Material online.
Fig. 5.—

Patterns of gene presence and absence in selected metabolic pathways and transporters.
Most of the species-specific genes are annotated as hypothetical proteins, such that we are unable to infer their functional significance. Among these four species, S. diminutum has the lowest number of species-specific gene clusters (fig. 4), possibly due to the fact that it has the smallest chromosome and the fewest protein-coding genes. Intriguingly, S. sabaudiense has a large family of species-specific hypothetical proteins with 27 copies. These genes are often found as clusters of two to four adjacent copies on the chromosome in regions with unexpected GC skew patterns (e.g., ∼0.2, ∼0.4, and ∼1.0 Mb in fig. 2; the assembly in these regions have been verified by PCR), suggesting that these DNA have been integrated recently. However, because database searches provided no identifiable homolog or conserved protein domain, the function and the origin of these hypothetical proteins remained unknown.
In the few cases that the functional roles of species-specific genes can be inferred, they reveal interesting information about the metabolism differences among these species. For example, S. sabaudiense is the only species that has the complete set of genes for arginine utilization (arcA, arcB, and arcC), which is consistent with previous biochemical tests (Abalain-Colloc et al. 1987; Hung et al. 1987; Abalain-Colloc et al. 1988; Williamson et al. 1996). Additionally, sucrose utilization (scrB and scrK) appears to be limited to S. diminutum, while glycerophosphocholine (GPC; substrate of glpU and glpQ) utilization appears to be limited to S. culicicola. Intriguingly, one putative secreted protein specific to S. culicicola (SCULI_v1c06250) was found to contain a partial Pfam03318 domain (Clostridium epsilon toxin ETX/Bacillus mosquitocidal toxin MTX2), which may contribute to its pathogenicity toward mosquitoes.
Other than the genes that are shared by all four species or specific to one of the species, genes with more variable phylogenetic distribution patterns are important in promoting our understanding of these mosquito-associated bacteria (fig. 5 and supplementary table S1, Supplementary Material online). Two sets of genes are of particular interest because of the differences in pathogenicity toward mosquitoes observed in previous artificial infection experiments (Chastel and Humphery-Smith 1991; Humphery-Smith, Grulet, Chastel, et al. 1991; Humphery-Smith, Grulet, Le Goff, et al. 1991; Vorms-Le Morvan et al. 1991; Vazeille-Falcoz et al. 1994; Phillips and Humphery-Smith 1995). For the two species without apparent pathogenicity (i.e., S. diminutum and S. sabaudiense), they were found to share murP and murQ for the uptake and utilization of N-acetylmuramic acid (MurNAc) and nrdD for the conversion of CTP to dCTP (Fontecave et al. 1989). For the two species that exhibit pathogenicity (i.e., S. culicicola and S. taiwanense), they were found to share a copy of glpO for ROS production. This finding provides further support for our previous inference that glpO is likely a virulence factor in these mosquito-associated Spiroplasma species (Lo, Ku, et al. 2013). To provide the substrate for glpO, these two species both contain the genes coding for sn-glycerol-3-phosphate ABC transporter (ugpA, ugpC, and ugpE) for direct import of G3P and glycerol kinase (glpK) for glycerol phosphorylation. Furthermore, a pseudogenized copy of glycerophosphoryl diester phosphodiesterase (glpQ) was found in the S. taiwanense genome (Lo, Ku, et al. 2013), suggesting that the metabolic capacity to utilize GPC was ancestral as well.
Molecular Evolution of the Glycerol Metabolism Genes
Based on the comparison of their substrate utilization strategies (fig. 5), glycerol metabolism and the associated production of ROS are likely to be linked to the observed pathogenicity of S. culicicola and S. taiwanense in artificial infection experiments (Chastel and Humphery-Smith 1991; Humphery-Smith, Grulet, Chastel et al. 1991; Humphery-Smith, Grulet, Le Goff, et al. 1991; Vazeille-Falcoz et al. 1994; Phillips and Humphery-Smith 1995). Our examination of the chromosomal locations of these glycerol metabolism genes revealed that the gene order of glpF-glpO-glpK is largely conserved among the Spiroplasma species with complete genome sequences available (fig. 6); S. culicicola represents the only exception due to the insertion of glpQ and glpU between glpO and glpK. For comparison, the gene order is glpO-glpK-glpF in the three Mycoplasma species belonging to the Mycoides–Entomoplasmataceae clade, which seems to be a derived state due to one or more rearrangements. Based on the phylogenetic distribution pattern of this gene cluster, its absence in S. diminutum, S. sabaudiense, and Me. florum is best explained by independent losses.
Fig. 6.—

Chromosomal locations of the glpF-glpO-glpK genes. The circular chromosomes are presented as linear for visualization; the first base of dnaA is used as the leftmost position for each chromosome. The sizes of individual genes are not drawn to scale.
For more detailed investigation of the molecular evolution of these genes, we compared the individual gene trees to the species phylogeny (fig. 7). Surprisingly, despite the conservation in gene order among the Spiroplasma species, all three gene trees support the clustering of S. taiwanense homologs with those from the Mycoides–Entomoplasmataceae clade. This unexpected conflict between gene order and gene phylogenies is difficult to explain. Future investigation that incorporates additional sequence data from more diverse lineages in the Apis and the Mycoides–Entomoplasmataceae clade is essential to confirm the gene phylogenies.
Fig. 7.—

Molecular phylogenies of conserved genes and glpF-glpO-glpK genes.The taxa are color coded according to the clade (Apis: red; Mycoides-Entomoplasmataceae: blue; Chrysopicola: black). The percentages of bootstrap support are labeled above the internal branches. (A) The species phylogeny based on the concatenated alignment of 333 single-copy genes conserved in all 11 species (122,317 aligned aa sites). (B) Molecular phylogeny of the glycerol uptake facilitator protein (glpF; 283 aligned aa sites). (C) Molecular phylogeny of the glycerol-3-phosphate oxidase (glpO; 405 aligned aa sites). (D) Molecular phylogeny of the glycerol kinase (glpK; 507 aligned aa sites). In (B–D), the genes are labeled by their locus tags; no homolog was found in the Me. florum genome.
In the examination of gene order and gene phylogenies, we found several interesting points regarding the glycerol uptake facilitator protein gene (glpF). In addition to the copy adjacent to glpO and glpK, several Spiroplasma genomes contain a second copy of glpF in other regions of the chromosomes (fig. 6). In all cases, these isolated copies exhibit high levels of sequence divergence from other homologs (fig. 7B; the locus tags are assigned sequentially starting from dnaA and reflect their relative positions on the chromosome). The second copy from S. apis (SAPIS_v1c05780; located at ∼0.69 Mb in fig. 6) was not included in the gene phylogeny because it was not grouped in the same homologous gene cluster with all other copies of glpF. This pattern of sequence divergence may be explained by the release from selective constraint for these redundant copies. For S. diminutum and perhaps also S. sabaudiense, the glpF may be in the process of nonfunctionalization because the downstream glpK has been lost (fig. 5). Such gradual degradation of gene content is common among host-associated bacteria (Ochman and Davalos 2006; McCutcheon and Moran 2012) and is likely to be driven by a combination of mutational biases toward deletions and high levels of genetic drift (Mira et al. 2001; Kuo et al. 2009; Kuo and Ochman 2009; Kuo and Ochman 2010). Eventually, these Spiroplasma lineages may lose their glpF just as what has occurred for Me. florum.
Conclusions
In summary, the gene content comparison presented in this study provides an overview on the substrate utilization strategies across diverse mosquito-associated Spiroplasma species. Moreover, our result demonstrates that glpO is conserved across diverse Spiroplasma lineage. The absence of glpO in S. diminutum and S. sabaudiense is best explained by independent losses and may be linked to the lack of pathogenicity in these two species. The clustering of the S. taiwanense glpF/glpO/glpK with those from the Mycoides–Entomoplasmataceae clade is an intriguing point that requires further investigation. Finally, future tool development for the genetic manipulation of these bacteria is necessary for the functional characterization of their putative virulence factors.
Supplementary Material
Supplementary table S1 is available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by the research grants from the Institute of Plant and Microbial Biology at Academia Sinica and the National Science Council of Taiwan (NSC 101-2621-B-001-004-MY3) to C.H.K. The Illumina sequencing services were provided by the Biofuel High Throughput Sequencing Core (Biodiversity Research Center, Academia Sinica) and Yourgene Bioscience (New Taipei City, Taiwan). The Sanger sequencing service was provided by the DNA Analysis Core Laboratory (Institute of Plant and Microbial Biology, Academia Sinica).
Literature Cited
- Abalain-Colloc ML, et al. Spiroplasma sabaudiense sp. nov. from mosquitoes collected in France. Int J Syst Microbiol. 1987;37:260–265. [Google Scholar]
- Abalain-Colloc ML, et al. Spiroplasma taiwanense sp. nov. from Culex tritaeniorhynchus mosquitoes collected in Taiwan. Int J Syst Microbiol. 1988;38:103–107. [Google Scholar]
- Alexeev D, et al. Application of Spiroplasma melliferum proteogenomic profiling for the discovery of virulence factors and pathogenicity mechanisms in host-associated spiroplasmas. J Proteome Res. 2012;11:224–236. doi: 10.1021/pr2008626. [DOI] [PubMed] [Google Scholar]
- Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, et al. GenBank. Nucleic Acids Res. 2012;40:D48–D53. doi: 10.1093/nar/gkr1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle P, et al. Partial chromosome sequence of Spiroplasma citri reveals extensive viral invasion and important gene decay. Appl Environ Microbiol. 2010;76:3420–3426. doi: 10.1128/AEM.02954-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carle P, Laigret F, Tully JG, Bove JM. Heterogeneity of genome sizes within the genus Spiroplasma. Int J Syst Bacteriol. 1995;45:178–181. doi: 10.1099/00207713-45-1-178. [DOI] [PubMed] [Google Scholar]
- Chastel C, Humphery-Smith I. Mosquito spiroplasmas. Adv Dis Vector Res. 1991;7:149–206. [Google Scholar]
- Chen L-L, Chung W-C, Lin C-P, Kuo C-H. Comparative analysis of gene content evolution in phytoplasmas and mycoplasmas. PLoS One. 2012;7:e34407. doi: 10.1371/journal.pone.0034407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W-C, Chen L-L, Lo W-S, Lin C-P, Kuo C-H. Comparative analysis of the peanut witches’-broom phytoplasma genome reveals horizontal transfer of potential mobile units and effectors. PLoS One. 2013;8:e62770. doi: 10.1371/journal.pone.0062770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP—Phylogeny Inference Package (Version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Fontecave M, Eliasson R, Reichard P. Oxygen-sensitive ribonucleoside triphosphate reductase is present in anaerobic Escherichia coli. Proc Natl Acad Sci U S A. 1989;86:2147–2151. doi: 10.1073/pnas.86.7.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparich GE. Spiroplasmas and phytoplasmas: microbes associated with plant hosts. Biologicals. 2010;38:193–203. doi: 10.1016/j.biologicals.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Gasparich GE, et al. The genus Spiroplasma and its non-helical descendants: phylogenetic classification, correlation with phenotype and roots of the Mycoplasma mycoides clade. Int J Syst Evol Microbiol. 2004;54:893–918. doi: 10.1099/ijs.0.02688-0. [DOI] [PubMed] [Google Scholar]
- Gnerre S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hames C, Halbedel S, Hoppert M, Frey J, Stülke J. Glycerol metabolism is important for cytotoxicity of Mycoplasma pneumoniae. J Bacteriol. 2009;191:747–753. doi: 10.1128/JB.01103-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphery-Smith I, Grulet O, Chastel C. Pathogenicity of Spiroplasma taiwanense for larval Aedes aegypti mosquitoes. Med Vet Entomol. 1991;5:229–232. doi: 10.1111/j.1365-2915.1991.tb00545.x. [DOI] [PubMed] [Google Scholar]
- Humphery-Smith I, Grulet O, Le Goff F, Chastel C. Spiroplasma (Mollicutes: Spiroplasmataceae) pathogenic for Aedes aegypti and Anopheles stephensi (Diptera: Culicidae) J Med Entomol. 1991;28:219–222. doi: 10.1093/jmedent/28.2.219. [DOI] [PubMed] [Google Scholar]
- Hung SHY, Chen TA, Whitcomb RF, Tully JG, Chen YX. Spiroplasma culicicola sp. nov. from the salt marsh mosquito Aedes sollicitans. Int J Syst Bacteriol. 1987;37:365–370. [Google Scholar]
- Hyatt D, et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. Comparative genomics, minimal gene-sets and the last universal common ancestor. Nat Rev Microbiol. 2003;1:127–136. doi: 10.1038/nrmicro751. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku C, Lo W-S, Chen L-L, Kuo C-H. Complete genomes of two dipteran-associated spiroplasmas provided insights into the origin, dynamics, and impacts of viral invasion in Spiroplasma. Genome Biol Evol. 2013;5:1151–1164. doi: 10.1093/gbe/evt084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku C, Lo W-S, Chen L-L, Kuo C-H. Complete genome sequence of Spiroplasma apis B31T (ATCC 33834), a bacterium associated with May disease of honeybees (Apis mellifera) Genome Announc. 2014;2:e01151–13. doi: 10.1128/genomeA.01151-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Moran NA, Ochman H. The consequences of genetic drift for bacterial genome complexity. Genome Res. 2009;19:1450–1454. doi: 10.1101/gr.091785.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Ochman H. Deletional bias across the three domains of life. Genome Biol Evol. 2009;1:145–152. doi: 10.1093/gbe/evp016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C-H, Ochman H. The extinction dynamics of bacterial pseudogenes. PLoS Genet. 2010;6:e1001050. doi: 10.1371/journal.pgen.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre P, Gogarten JP. Estimating the size of the bacterial pan-genome. Trends Genet. 2009;25:107–110. doi: 10.1016/j.tig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W-S, Chen L-L, Chung W-C, Gasparich G, Kuo C-H. Comparative genome analysis of Spiroplasma melliferum IPMB4A, a honeybee-associated bacterium. BMC Genomics. 2013;14:22. doi: 10.1186/1471-2164-14-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W-S, Ku C, Chen L-L, Chang T-H, Kuo C-H. Comparison of metabolic capacities and inference of gene content evolution in mosquito-associated Spiroplasma diminutum and S. taiwanense. Genome Biol Evol. 2013;5:1512–1523. doi: 10.1093/gbe/evt108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler-Bauer A, et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41:D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- Mira A, Ochman H, Moran NA. Deletional bias and the evolution of bacterial genomes. Trends Genet. 2001;17:589–596. doi: 10.1016/s0168-9525(01)02447-7. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Davalos LM. The nature and dynamics of bacterial genomes. Science. 2006;311:1730–1733. doi: 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Phillips RN, Humphery-Smith I. The histopathology of experimentally induced infections of Spiroplasma taiwanense (class: Mollicutes) in Anopheles stephensi mosquitoes. J Invertebr Pathol. 1995;66:185–195. [Google Scholar]
- Pilo P, et al. A metabolic enzyme as a primary virulence factor of Mycoplasma mycoides subsp. mycoides small colony. J Bacteriol. 2005;187:6824–6831. doi: 10.1128/JB.187.19.6824-6831.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo P, Frey J, Vilei EM. Molecular mechanisms of pathogenicity of Mycoplasma mycoides subsp. mycoides SC. Vet J. 2007;174:513–521. doi: 10.1016/j.tvjl.2006.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regassa L B, Gasparich GE. Spiroplasmas: evolutionary relationships and biodiversity. Front Biosci. 2006;11:2983–3002. doi: 10.2741/2027. [DOI] [PubMed] [Google Scholar]
- Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov R, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Vazeille-Falcoz M, Perchec-Merien A-M, Rodhain F. Experimental infection of Aedes aegypti mosquitoes, suckling mice, and rats with four mosquito spiroplasmas. J Invertebr Pathol. 1994;63:37–42. doi: 10.1006/jipa.1994.1007. [DOI] [PubMed] [Google Scholar]
- Vorms-Le Morvan J, Vazeille-Falcoz M-C, Rodhain F, Chastel C. Infection expérimentale de moustiques Aedes albopictus par une souche de spiroplasmes isolée de Culex annulus a Taiwan. Bull Soc Pathol Exot. 1991;84:15–24. [PubMed] [Google Scholar]
- Whitcomb RF. The biology of spiroplasmas. Ann Rev Entomol. 1981;26:397–425. [Google Scholar]
- Whitcomb RF, et al. Spiroplasma cantharicola sp. nov., from cantharid beetles (Coleoptera: Cantharidae) Int J Syst Bacteriol. 1993;43:421–424. [Google Scholar]
- Williamson DL, et al. Spiroplasma diminutum sp. nov., from Culex annulus mosquitoes collected in Taiwan. Int J Syst Bacteriol. 1996;46:229–233. doi: 10.1099/00207713-46-1-229. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.