

Abstract
The histone modification H3K27me3 is involved in repression of transcription and plays a crucial role in developmental transitions in both animals and plants. It is deposited by PRC2 (Polycomb repressive complex 2), a conserved protein complex. In Arabidopsis thaliana, H3K27me3 is found at 15% of all genes. These tend to encode transcription factors and other regulators important for development. However, it is not known how PRC2 is recruited to target loci nor how this set of target genes arose during Arabidopsis evolution. To resolve the latter, we integrated A. thaliana gene families with five independent genome-wide H3K27me3 data sets. Gene families were either significantly enriched or depleted of H3K27me3, showing a strong impact of shared ancestry to H3K27me3 distribution. To quantify this, we performed ancestral state reconstruction of H3K27me3 on phylogenetic trees of gene families. The set of H3K27me3-marked genes changed less than expected by chance, suggesting that H3K27me3 was retained after gene duplication. This retention suggests that the PRC2-recruiting signal could be encoded in the DNA and also conserved among certain duplicated genes. Indeed, H3K27me3-marked genes were overrepresented among paralogs sharing conserved noncoding sequences (CNSs) that are enriched with transcription factor binding sites. The association of upstream CNSs with H3K27me3-marked genes represents the first genome-wide connection between H3K27me3 and potential regulatory elements in plants. Thus, we propose that CNSs likely function as part of the PRC2 recruitment in plants.
Keywords: ancestral state reconstruction, CNS, PRC2, PRC2 recruitment
Introduction
The histone modification H3K27me3 (trimethylation of histone H3 at lysine 27) is involved in repression of transcription in plants, animals, and fungi (Schuettengruber et al. 2007; Jamieson et al. 2013). In Arabidopsis thaliana, it plays an important role in developmental transitions (Farrona et al. 2008; Bouyer et al. 2011). Its target genes, which represent more than 15% of the Arabidopsis genome, show low and tissue-specific expression (Zhang et al. 2007). They tend to encode transcription factors or proteins with other regulatory functions.
H3K27me3 is deposited by the evolutionary conserved Polycomb repressive complex (PRC2). PRC2 was already present in the last common ancestor of plants, animals, and fungi, which was a unicellular organism. However, PRC2 target genes in both plants and animals are involved in processes related to multicellularity, implying convergent evolution (Haudry et al. 2013). In contrast to the conservation of the PRC2 protein complex, the localization of H3K27me3 differs between animals and plants. H3K27me3 in Drosophila and mammals tends to cover long genomic stretches, whereas in A. thaliana, H3K27me3 is preferentially localized at gene-coding regions. In addition, the signal that recruits PRC2 to target loci seems to vary among different groups of organisms (Zhang et al. 2007). In Drosophila, whose H3K27me3-depositing mechanism is best studied, the PRC2-recruiting signals are Polycomb response elements (PREs). PREs are several hundred base pairs long stretches of DNA without a clear consensus sequence but predicted to function as clusters of binding sites for DNA-binding proteins. Several transcription factors have been experimentally demonstrated to bind to PREs and to be required for PRC2 recruitment as well as for trimethylation at H3K27 (Simon and Kingston 2009). In mammals, the PRC2 recruiting signal is thought to consist of long noncoding RNAs (lncRNAs), CpG-rich regions, and other unknown factors (Margueron and Reinberg 2011). In Arabidopsis, the PRC2-recruiting signal is unknown. However, recent studies on two loci suggest two potentially compatible options for PRC2 recruitment. One study reported that regulation of FLOWERING LOCUS C (FLC) expression involves lncRNAs in H3K27me3 establishment (Heo and Sung 2011). The second study proposed the upstream region of LEAFY COTYLEDON 2 (LEC2) to facilitate PRC2 recruitment (Berger et al. 2011). Hence, PRC2 might utilize these mechanisms independently in a gene-specific context. Alternatively, recruitment to its targets could depend on a so far unexplored combination of factors as proposed for animals.
Studying how the set of H3K27me3-marked genes evolved could further our understanding of this epigenetic system. The finding that H3K27me3 is highly conserved between rice and maize orthologs (Makarevitch et al. 2013) suggests conservation after species divergence. Furthermore, an analysis of a small sample of A. thaliana gene families shows that some families contain higher than expected proportion of H3K27me3-marked genes (Lafos et al. 2011). Because gene families evolved mostly by gene duplication, it is tempting to speculate that H3K27me3 is retained after duplication. If indeed H3K27me3 is retained after gene duplication and speciation, it suggests the existence of a conserved signal in DNA that is directly or indirectly involved in PRC2-mediated deposition of the modification.
Our previous work (Berke et al. 2012) is consistent with this hypothesis: A. thaliana paralogs from the latest (α) whole-genome duplication (WGD) that are both marked with H3K27me3 show more similar expression patterns and more conserved upstream regions. Such conserved upstream regions could represent conserved noncoding sequences (CNSs) (Thomas et al. 2007; Baxter et al. 2012; Haudry et al. 2013 and others). CNSs in plants are more often located upstream of genes, generally associated with lowly expressed genes, and enriched with transcription factor binding motifs such as the G-box (Freeling et al. 2007; Baxter et al. 2012; Spangler et al. 2012; Haudry et al. 2013). They are also relatively short, rarely surpassing 100 bp in length, which hinders their detection. The function of CNSs is mostly unknown.
In this study, we reconstructed the ancestral states of H3K27me3 in A. thaliana gene families to show that H3K27me3 tends to be retained after gene duplication. Retention of H3K27me3 after duplication helps to explain the set of H3K27me3-marked genes and is consistent with the PRC2 recruitment signal being encoded in conserved DNA elements. We sought for the cause of retention by comparing H3K27me3-marked genes with CNS-rich genes. The two sets overlap significantly; moreover, we observed an even more significant overlap between H3K27me3-marked genes and genes with CNSs in their upstream regions. This evolutionarily conserved association holds for CNSs defined at the level of paralogs as well as orthologs and suggests a role for CNSs in PRC2 recruitment.
Material and Methods
Data Sets
Arabidopsis thaliana chromosome, intergenic and protein sequences (representative model), as well as annotation files v. TAIR10 were obtained from TAIR (http://www.arabidopsis.org, last accessed March 4, 2014). Binary data on H3K27me3-marked genes were obtained from ChIP-chip and ChIP-seq experiments (Zhang et al. 2007; Bouyer et al. 2011; Farrona et al. 2011; Lafos et al. 2011; Lu et al. 2011). We extracted a high-confidence list of genes with H3K27me3 by collecting only genes that appear in at least three data sets.
We used four CNS data sets (Freeling et al. 2007; Baxter et al. 2012; Haudry et al. 2013), with minor modifications. From the Freeling data set, we removed pairs designated as “our additional.” From the Baxter paralogous data set, we excluded 12 pairs of paralogs that do not exist in the Bowers data set of α WGD paralogs (Bowers et al. 2003) in order to perform hypergeometric test. From the Haudry data set, we used only the CNSs that did not overlap more than 2 bp with any previously described element in the genome (listed in ftp://anonymous@ftp.arabidopsis.org/home/tair/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes_transposons.gff, last accessed March 4, 2014). The remaining Haudry CNSs were then assigned to the closest gene or transposable element gene, because transposable elements are also marked by H3K27me3 (Lafos et al. 2011). The CNSs were then classified as upstream or downstream. Overrepresentation (Venn diagrams) was calculated using hypergeometric test. Motif search was performed on concatenated CNSs of genes with more than five CNSs with MEME v. 4.9 (Bailey and Elkan 1994) (motif width 6–8 nt, as in Haudry et al. [2013]; oops search mode, included reverse complement) using second-order Markov model of A. thaliana intergenic CNSs as background.
We used two rice H3K27me3 data sets (He et al. 2010; Hu et al. 2012). Protein coding sequences were obtained from Phytozome v. 9.1. (Goodstein et al. 2012) (rice genome v. 7.0; Ouyang et al. 2007).
Clusters, Trees, and Ancestral State Reconstruction
We used BlastP (filtered query sequence, e-value cutoff 1e−3) version 2.2.18 to find similar sequences and MCL (Enright et al. 2002) (v.12-068; inflation parameter 2) to create families. Families containing between 4 and 200 proteins were aligned using MAFFT v. 7.027b (Katoh and Standley 2013) (parameter genafpair). Phylogenetic trees were based on protein sequences and calculated using RAxML (Stamatakis 2006) version 7.2.8 alpha (substitution model PROTGAMMAWAG, 100 runs). Blast, MAFFT, and RAxML used protein (and not DNA) sequences as input; throughout the manuscript we refer to the resulting families as “gene families” for simplicity.
As ancestral state reconstruction requires rooted trees, we used midpoint rooting for all inferred trees (there is no outgroup which would allow for different type of rooting). Ancestral state reconstruction for H3K27me3 was performed using original Sankoff parsimony as described (Clemente et al. 2009). Ties were resolved according to ACCTRAN algorithm (Farris 1970; Swofford and Maddison 1987; Wiley and Lieberman 2011); ties at the level of root were resolved by setting the root value to “1.” Parameter sweep was performed with gain and loss costs varying in steps of 0.1 while the sum of both amounted to 2 (i.e., gain 0.1, loss 1.9; gain 0.2, loss 1.8; etc.). To obtain the randomized data set, we permuted the H3K27me3 marks within each family and performed ancestral state reconstruction. This was repeated 100 times.
Results
H3K27me3 Tends to be Retained after Gene Duplications
To uncover the fate of H3K27me3 after gene duplications, we systematically analyzed the distribution of H3K27me3 in A. thaliana gene families. As a first step, we obtained H3K27me3-marked genes from already published genome-wide data sets (Zhang et al. 2007; Bouyer et al. 2011; Farrona et al. 2011; Lafos et al. 2011; Lu et al. 2011). These H3K27me3 data sets differ in their experimental approach (ChIP-seq and ChIP-chip) and analyzed tissues (supplementary fig. S1A and data set S1, Supplementary Material online). They were combined into a high-confidence set of H3K27me3-marked genes by selecting only the 5,118 genes that appeared in at least three out of five genome-wide analyses. Next, we inferred A. thaliana gene families. Single-gene families (4,216) and those with more than 200 genes (4) were discarded. In total, 3,661 gene families were used for further analysis. Most of them contained little or no H3K27me3-marked genes (fig. 1A and supplementary fig. S1B, Supplementary Material online). Although a substantial number of nonmarked families is expected by chance given their paucity in the genome (18% of all genes), mostly nonmarked (0–20%) as well as mostly marked (80–100% marked genes) gene families were significantly overrepresented (P < 0.001, permutation test; expected values are averages of 1,000 permutations of H3K27me3 among all families) (fig. 1B). Moreover, we observed a depletion of approximately 18% marked gene families, on the contrary to what one would expect if comparing with genome average (supplementary fig. S1C, Supplementary Material online). Large families had a higher proportion of marked genes (supplementary fig. S2A, Supplementary Material online). Yet, this correlation did not confound our results as both small (2–6 genes) and large (7–200 genes) gene families showed a bimodal distribution, with enrichment in mostly marked and mostly nonmarked gene families (supplementary fig. S2B–E, Supplementary Material online). Thus, bimodality in distribution of gene families showed strong influence of shared ancestry on H3K27me3 distribution and suggests H3K27me3 is inherited after gene duplication.
Fig. 1.—

Gene families are enriched in either marked or nonmarked genes. (A) Distribution of gene families according to the proportion of H3K27me3-marked genes. (B) Bar heights show an average log2 ratio of observed/expected number of gene families in each bin for 1,000 permutations. Stars indicate bars where observed values were more extreme than the expected values for all 1,000 permutations.
Measuring the rate of change from H3K27me3-marked to nonmarked genes and vice versa requires knowing the evolutionary relationships among genes. Phylogenetic trees were thus inferred from families consisting of 4 to 200 genes and rooted at midpoints. One of the 1,579 inferred trees contained a group of genes encoding AP2-domain proteins, transcription factors with diverse roles in development (fig. 2A). With 9 of its 19 genes marked, this family at first seemed to deviate from the enrichment pattern seen earlier. However, its phylogenetic tree revealed two partitions in the H3K27me3 distribution. One of the clades contained eight H3K27me3-marked genes. They were AINTEGUMENTA (ANT) and ANT-LIKE (AIL) genes with diverse regulatory functions ranging from root stem cell maintenance to flower development (Elliott et al. 1996; Aida et al. 2004). In contrast, the rest of the family except for a single gene (1/11) was not H3K27me3-marked.
Fig. 2.—
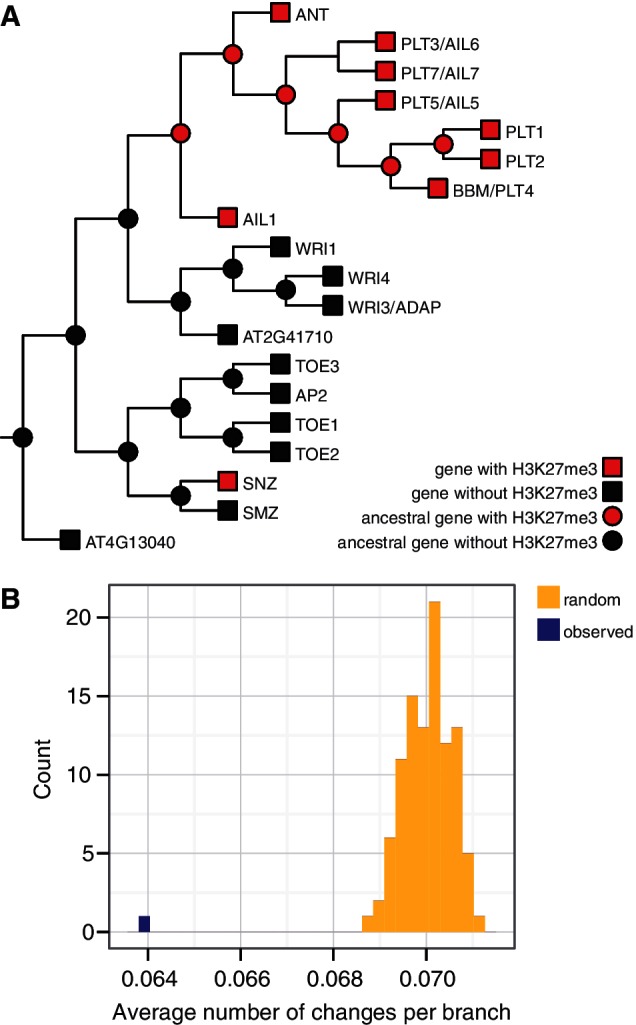
H3K27me3 distribution in gene families reflects their phylogeny. (A) An example phylogenetic tree with ancestral state reconstruction shows AP2-domain transcription factors. H3K27me3 data are represented as squares at the tips. Reconstructed ancestral states are shown as circles. (B) Histogram of average number of H3K27me3 changes per branch when gain penalty is 1.1. Observed number of changes is lower than number of changes in any of the randomized cases.
To obtain a more detailed picture of the rate of change of PRC2 target genes, we reconstructed the ancestral states of H3K27me3 on phylogenetic trees. We used Sankoff parsimony with two states: H3K27me3 is either present or absent. This method uses different costs for gains and losses of H3K27me3. To see their effect on reconstruction, we used a range of cost combinations (supplementary fig. S3, Supplementary Material online). Sankoff parsimony does not use information on the rate of protein evolution, represented by the length of branches in the tree. This is appropriate for our reconstructions, as rate of protein evolution is not necessarily correlated to rate of H3K27me3 change. Ancestral state reconstruction is shown for the example case of AP2-domain transcription factors (fig. 2A) where, for a wide range of cost parameters, the most parsimonious explanation is that H3K27me3 was gained twice: once at the base of the ANT/AIL clade and once on a branch leading to a single protein (SCHNARCHZAPFEN [SNZ]).
The rate of change of H3K27me3 (sum of gains and losses) on trees was defined as the fraction of the number of branches in the phylogeny where a change occurred (number of gains and losses) in relation to total number of branches in the tree. Next, we randomly reassigned H3K27me3 within each gene family (while keeping the topology of the phylogenetic tree) and reconstructed ancestral states for the entire set of families 100 times. The average observed rate of change per branch was always lower than the rate for randomized data (P < 0.01) (fig. 2B and supplementary fig. S4, Supplementary Material online), revealing that H3K27me3 tends to be retained after gene duplications.
CNSs Are Associated with H3K27me3-Marked Genes in Arabidopsis Paralogs
The observed tendency of H3K27me3 to be retained after gene duplication suggests that the signal recruiting PRC2 could be in the DNA sequence and that it is the conservation of this signal that directly or indirectly causes the retention of H3K27me3 after duplication. As H3K27me3 is important for correct gene expression, PRC2-recruiting signals are likely to be conserved. Indeed, paralogs that arose in the α WGD and retained their mark are enriched for long (>18 nt) stretches of high sequence similarity (Berke et al. 2012). To directly test whether CNSs tend to occur more often between gene pairs that also have H3K27me3, we first focused on published CNSs between α paralogs, as their sequences are likely most conserved. A manually curated subset of these paralogs with longer upstream regions and more CNSs, named bigfoot genes (Freeling et al. 2007) (table 1), showed an enrichment of H3K27me3-marked gene pairs from the α WGD (hypergeometric test, P = 4.0e − 11) (fig. 3A). This enrichment was not observed in gene pairs without or with only one marked gene. Moreover, an independent set of paralogs with CNSs (Baxter et al. 2012) showed the same association with marked paralogs (P = 6.8e − 12) (fig. 3B) and hence confirmed the association between H3K27me3-marked genes and CNSs in α WGD paralogs.
Table 1.
Overview of CNS Data Sets
| Name of Data Set | Type of Comparison | Breadth of Species Sampling | Reference |
|---|---|---|---|
| Freeling bigfoot | Paralogs | Arabidopsis thaliana | Freeling et al. (2007) |
| Baxter paralogous | Paralogs | A. thaliana | Baxter et al. (2012) |
| Haudry | Orthologs | Brassicaceae | Haudry et al. (2013) |
| Baxter orthologous | Orthologs | Dicots | Baxter et al. (2012) |
Fig. 3.—

Paralogous CNSs are associated with H3K27me3. (A) Overlap between bigfoot gene pairs and α WGD gene pairs with H3K27me3 where both genes (two red squares), one gene (a red and a black square), or none (two black squares) are marked. (B) Overlap between paralogous pairs with CNSs (as determined by Baxter et al. 2012) with H3K27me3-marked pairs. Background gene set consists of all α paralogous pairs (3,817 pairs).
H3K27me3-Marked Genes Are Enriched in Genes with Orthologous CNSs
To see whether the association between CNSs and H3K27me3 could be generalized beyond paralogs to any H3K27me3-marked gene, we made use of a recently published analysis that reported more than 90,000 CNSs (Haudry et al. 2013). These CNSs (the Haudry data set) were based on the conservation among nine genomes from the Brassicaceae family, including A. thaliana. We used 45,947 CNSs that did not overlap with any annotated genomic element and assigned them to the closest gene. Even with this crude approach of assigning CNSs to genes, genes with >10 CNSs were significantly overrepresented among marked genes (P = 8.9e − 21) (fig. 4A). To see whether width of phylogenetic sampling for CNS discovery (Freeling and Subramaniam 2009; Baxter et al. 2012) influences the comparison with H3K27me3-marked genes, we also used a dicot CNS data set that was derived by comparing the genomes of A. thaliana, papaya (Carica papaya), poplar (Populus trichocarpa), and grapevine (Vitis vinifera) (Baxter et al. 2012). Genes with CNSs showed significant overlap with H3K27me3-marked genes (P = 4.4e − 38) (fig. 4B), independently corroborating the association of CNSs with H3K27me3.
Fig. 4.—
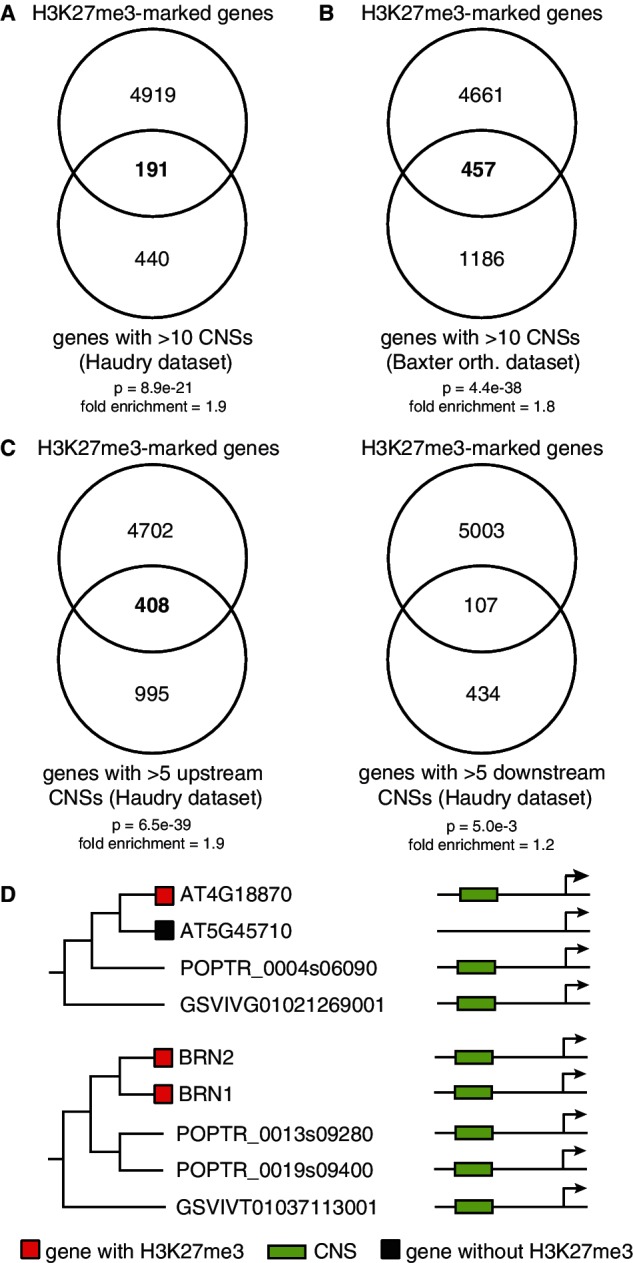
Orthologous CNSs are associated with H3K27me3. (A) Overlap between genes with H3K27me3 and genes from the Haudry CNS data set with >10 CNS. (B) Overlap between marked genes and genes with Baxter data set CNSs. (C) Overlap between genes with >5 CNSs and H3K27me3-marked genes for upstream and downstream CNSs. The background data set for all Venn diagrams contains 32,678 genes. (D) Two examples of CNS inheritance and H3K27me3. Left panel: only one of Arabidopsis homologs is H3K27me3-marked and has a CNS conserved with poplar and grapevine orthologs. Right panel: both Arabidopsis paralogs have H3K27me3 and a shared CNS. There is no H3K27me3 data for grapevine and poplar.
Previous analyses showed that upstream and downstream CNSs differ in their properties (Spangler et al. 2012). To see whether they also differ their association with H3K27me3, we divided the Haudry CNSs into upstream and downstream CNSs. Genes with >5 upstream CNSs showed an even more significant overlap between H3K27me3 and CNSs (P = 6.5e − 39) (fig. 4C), whereas genes with >5 downstream CNSs were only weakly associated with H3K27me3-marked genes (P = 0.005). Therefore, upstream CNSs showed a much stronger link with H3K27me3 than the downstream CNSs.
As CNSs have been shown to be enriched in transcription factor binding sites (Haudry et al. 2013), we wanted to see whether specific motifs are more common in upstream CNSs that are assigned to H3K27me3-marked genes. Seven motifs occured in at least one of the CNSs of each H3K27me3-marked gene (supplementary fig. S5, Supplementary Material online). They exhibited either only partial or no similarity to known transcription factor binding sites. Thus, a single motif in the upstream regions involved in direct or indirect PRC2 recruitment is unlikely to exist.
Discussion
In this article, we examine the fate of histone modification H3K27me3 after gene duplications at two levels. We first found that the distribution of H3K27me3 in families is bimodal: gene families were mostly marked or mostly nonmarked. We also observed this biomodality in rice (supplementary fig. S6, Supplementary Material online) but could not perform subsequent analyses as the two rice H3K27me3 data sets are much less congruent and consequently much less suitable for our analysis than the data of A. thaliana (supplementary fig. S6A, Supplementary Material online). The ancestral reconstruction of H3K27me3 in A. thaliana, incorporating phylogenetic information, showed that H3K27me3 is retained after gene duplication.
The retention of H3K27me3 between paralogs is consistent with the possibility that the signal directing PRC2 to target genes is encoded in the DNA. We suggest that the conservation of this signal directly or indirectly causes the retention. Indeed, genes with upstream CNSs overlap significantly with H3K27me3-marked genes. Why is the overlap not larger? First, there are both biological and technical reasons for nonmarked genes with CNSs. Nonmarked genes with CNSs confirm that the functions of CNSs are diverse (Thomas et al. 2007; Spangler et al. 2012; Hupalo and Kern 2013) and only some will be connected to H3K27me3. The observation that genes with more CNSs are more likely to be H3K27me3-marked (see >10 CNS cutoff for the Venn diagram in fig. 4A–C) supports this: the more CNSs a gene has, the more substantial the overlap with H3K27me3-marked genes. Apart from biological reasons, technical reasons also account for part of the discrepancy. False negatives are likely present in our H3K27me3 data set, either due to the stringent cutoff to obtain the dataset or due to the limited data for different tissues. As H3K27me3 is a tissue-specific mark, genes specifically marked in flower tissues, for example, are missing from our data set.
Second, H3K27me3-marked genes without CNSs also contribute to smaller overlap. CNS discovery in plants, where CNSs are short, is difficult and thwarted by transcription factor binding site turnover (Moses et al. 2006; Freeling and Subramaniam 2009; Habib et al. 2012). The overlap between paralogous and orthologous CNSs is surprisingly small even when using the same CNS detection method (Baxter et al. 2012). In other words, if upstream regions of two paralogs are compared with respective orthologs, the resulting two CNSs are not necessarily alignable or homologous. An important factor in CNS discovery is also the width of phylogenetic sampling (Freeling and Subramaniam 2009); distant clades will have less CNSs in common. For example, a comparison of monocots and dicots yielded only 18 syntenic CNSs (D'Hont et al. 2012). A more biological suggestion is that CNSs are only one of the ways to recruit PRC2 to target loci: H3K27me3 might be tightly regulated for some genes and a passive consequence of lack of transcriptional activity for others (Klose et al. 2013). This duality in the regulation of H3K27me3 would help to explain why not all marked genes have a CNS.
Epigenetic modifications have indeed been suggested to not be equally important for correct regulation of all genes (Meagher 2010). The “different targets-different mode of repression” paradigm has been suggested before, with different effect on expression for different genes after PRC2 loss (Farrona et al. 2011), different modes of repression for different subsets of genes (Yang et al. 2013), or H3K27me3 reprogramming at different time points (He et al. 2012). Accordingly, the redistribution of H3K27me3 to new loci that follows from loss of DNA methylase MET1 suggests varying “importance” of targets (Deleris et al. 2012). For DNA methylation, this has been already established: there are less DNA methylation polymorphisms at loci with transposable elements than at other loci, presumably because repression of transposable elements is crucial (Vaughn et al. 2007). In A. thaliana and maize, intraspecies variation in H3K27me3-marked genes is small and affects a seemingly random set of genes (Moghaddam et al. 2011; Dong et al. 2012; Makarevitch et al. 2013), confirming that H3K27me3 may not be equally important for correct expression of all genes.
The significant overlap between H3K27me3-marked and CNS-containing genes strongly suggests coupling of transcription factors, or expression levels, and PRC2 recruitment. We suggest a model where some CNSs act as repressive elements that restrict expression to smaller domains via (possibly indirect) recruitment of PRC2. CNSs have already been shown to be negatively correlated with expression level (Spangler et al. 2012), and the model suggests that this negative correlation is at least partly mediated by the deposition of H3K27me3. Previous observation that CNSs are rich in transcription factor binding sites (Baxter et al. 2012) and the lack of a single specific motif in the CNSs of H3K27me3-marked genes suggest that such highly conserved transcription factor binding islands could be involved in recruitment of PRC2. Two examples (fig. 4D) demonstrate the model. BEARSKIN1 (BRN1) and BRN2 (Bennett et al. 2010) are H3K27me3-marked α WGD paralogs with a CNS that is conserved between them as well as among their more distant coorthologs in other dicots. An interesting case is the paralogs AT4G18870 and AT5G45710: AT4G18870 shares a CNS with its orthologs in other dicots and is marked by H3K27me3 while AT5G45710, which arose after a recent duplication event, lost the CNS and H3K27me3. This is consistent with detailed investigations of the regulation of gene expression of a number of developmentally important Arabidopsis loci. For example, the expression domain of SHOOT MERISTEMLESS (STM), an H3K27me3-target gene in all five data sets, is restricted by an upstream sequence that is conserved between monocots and dicots (Uchida et al. 2007). The same region was shown to be bound by CURLY LEAF (CLF), a PRC2 subunit (Schubert et al. 2006). In light of our model and previous knowledge, this conserved sequence appears likely to be involved in PRC2 recruitment and H3K27me3 deposition.
Three additional loci, all with CNSs that can be found in the Haudry data set, are also consistent with our model. A CNS upstream of LEC2 influences H3K27me3 deposition (Berger et al. 2011). Additionally, one of three CNSs upstream of FLOWERING LOCUS T (FT) (Adrian et al. 2010), an H3K27me3-marked gene, was shown to repress expression of FT and influence the binding of an H3K27me3-interacting protein LHP1, agreeing with our proposed model. Finally, SHORT VEGETATIVE PHASE (SVP) was found to bind a CArG box upstream of SUPPRESOR OF OVEREXPRESSION OF CO 1 (SOC1) (Li et al. 2008). This CArG box is embedded in a longer CNS and acts in restricting expression of SOC1 to a narrower domain.
Based on in silico analysis and published experimental examples, we propose that CNSs are likely to function as part of PRC2 recruitment in plants. However, further experimental work is needed to fully establish whether the correlation between H3K27me3 marking and CNS depends on direct or indirect mechanisms of PRC2 recruitment.
Note added in proof
A recent publication experimentally characterized a PRC2-recruiting sequence upstream of the developmentally essential Arabidopsis gene KNUCKLES (KNU) (Sun et al. 2014). Although not reported in their work, this sequence also overlaps with a previously found CNS (Haudry et al. 2013) and shows that PRC2-recruiting mechanism is DNA sequence-dependent.
Supplementary Material
Supplementary figures S1–S6 and data sets S1–S3 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Michael F. Seidl for the discussions on the manuscript and providing the parsimony script, as well as Gabino F. Sanchez-Perez and Bas Rutjens for helpful comments. They are also grateful to Alessia Peviani, Eelco Tromer, Leny M. van Wijk, and Ethel Gilliquet for reading and discussing the manuscript and Linda McPhee for language editing. They would also like to thank two anonymous referees for their comments.
Literature Cited
- Adrian JJ, et al. cis-Regulatory elements and chromatin state coordinately control temporal and spatial expression of FLOWERING LOCUS T in Arabidopsis. Plant Cell. 2010;22:1425–1440. doi: 10.1105/tpc.110.074682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aida M, et al. The PLETHORA genes mediate patterning of the Arabidopsis root stem cell niche. Cell. 2004;119:109–120. doi: 10.1016/j.cell.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Baxter L, et al. Conserved noncoding sequences highlight shared components of regulatory networks in dicotyledonous plants. Plant Cell. 2012;24:3949–3965. doi: 10.1105/tpc.112.103010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett T, et al. SOMBRERO, BEARSKIN1, and BEARSKIN2 regulate root cap maturation in Arabidopsis. Plant Cell. 2010;22:640–654. doi: 10.1105/tpc.109.072272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger NN, Dubreucq BB, Roudier FF, Dubos CC, Lepiniec LL. Transcriptional regulation of Arabidopsis LEAFY COTYLEDON2 involves RLE, a cis-element that regulates trimethylation of histone H3 at lysine-27. Plant Cell. 2011;23:4065–4078. doi: 10.1105/tpc.111.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke L, Sanchez-Perez GF, Snel B. Contribution of the epigenetic mark H3K27me3 to functional divergence after whole genome duplication in Arabidopsis. Genome Biol. 2012;13:R94. doi: 10.1186/gb-2012-13-10-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer D, et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 2011;7:e1002014. doi: 10.1371/journal.pgen.1002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers JE, Chapman BA, Rong J, Paterson AH. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature. 2003;422:433–438. doi: 10.1038/nature01521. [DOI] [PubMed] [Google Scholar]
- Clemente JC, Ikeo K, Valiente G, Gojobori T. Optimized ancestral state reconstruction using Sankoff parsimony. BMC Bioinformatics. 2009;10:51. doi: 10.1186/1471-2105-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hont A, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488:213–217. doi: 10.1038/nature11241. [DOI] [PubMed] [Google Scholar]
- Deleris A, et al. Loss of the DNA methyltransferase MET1 induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet. 2012;8:e1003062. doi: 10.1371/journal.pgen.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, et al. Natural variation of H3K27me3 distribution between two Arabidopsis accessions and its association with flanking transposable elements. Genome Biol. 2012;13:R117. doi: 10.1186/gb-2012-13-12-r117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott RC, et al. AINTEGUMENTA, an APETALA2-like gene of Arabidopsis with pleiotropic roles in ovule development and floral organ growth. Plant Cell. 1996;8:155–168. doi: 10.1105/tpc.8.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris JS. Methods for computing Wagner trees. Syst Zool. 1970;19:83–92. [Google Scholar]
- Farrona S, Coupland G, Turck F. The impact of chromatin regulation on the floral transition. Semin Cell Dev Biol. 2008;19:560–573. doi: 10.1016/j.semcdb.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Farrona S, et al. Tissue-specific expression of FLOWERING LOCUS T in Arabidopsis is maintained independently of polycomb group protein repression. Plant Cell. 2011;23:3204–3214. doi: 10.1105/tpc.111.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Rapaka L, Lyons E, Pedersen B, Thomas BC. G-boxes, bigfoot genes, and environmental response: characterization of intragenomic conserved noncoding sequences in Arabidopsis. Plant Cell. 2007;19:1441–1457. doi: 10.1105/tpc.107.050419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M, Subramaniam S. Conserved noncoding sequences (CNSs) in higher plants. Curr Opin Plant Biol. 2009;12:126–132. doi: 10.1016/j.pbi.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Goodstein DM, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Wapinski I, Margalit H, Regev A, Friedman N. A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol. 2012;8:619. doi: 10.1038/msb.2012.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudry A, et al. An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet. 2013;45:891–898. doi: 10.1038/ng.2684. [DOI] [PubMed] [Google Scholar]
- He CC, Chen XX, Huang HH, Xu LL. Reprogramming of H3K27me3 is critical for acquisition of pluripotency from cultured Arabidopsis tissues. PLoS Genet. 2012;8:e1002911–e1002911. doi: 10.1371/journal.pgen.1002911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JB, Sung S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011;331:76–79. doi: 10.1126/science.1197349. [DOI] [PubMed] [Google Scholar]
- Hu Y, et al. CHD3 protein recognizes and regulates methylated histone H3 lysines 4 and 27 over a subset of targets in the rice genome. Proc Natl Acad Sci U S A. 2012;109:5773–5778. doi: 10.1073/pnas.1203148109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hupalo D, Kern AD. Conservation and functional element discovery in 20 angiosperm plant genomes. Mol Biol Evol. 2013;30:1729–1744. doi: 10.1093/molbev/mst082. [DOI] [PubMed] [Google Scholar]
- Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU. Regional control of histone H3 lysine 27 methylation in Neurospora. Proc Natl Acad Sci U S A. 2013;110:6027–6032. doi: 10.1073/pnas.1303750110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose RJ, Cooper S, Farcas AM, Blackledge NP, Brockdorff N. Chromatin sampling—an emerging perspective on targeting polycomb repressor proteins. PLoS Genet. 2013;9:e1003717. doi: 10.1371/journal.pgen.1003717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafos M, et al. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011;7:e1002040. doi: 10.1371/journal.pgen.1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, et al. A repressor complex governs the integration of flowering signals in Arabidopsis. Dev Cell. 2008;15:110–120. doi: 10.1016/j.devcel.2008.05.002. [DOI] [PubMed] [Google Scholar]
- Lu F, Cui X, Zhang S, Jenuwein T, Cao X. Arabidopsis REF6 is a histone H3 lysine 27 demethylase. Nat Genet. 2011;43:715–719. doi: 10.1038/ng.854. [DOI] [PubMed] [Google Scholar]
- Makarevitch I, et al. Genomic distribution of maize facultative heterochromatin marked by trimethylation of H3K27. Plant Cell. 2013;25:780–793. doi: 10.1105/tpc.112.106427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher RB. The evolution of epitype. Plant Cell. 2010;22:1658–1666. doi: 10.1105/tpc.110.075481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghaddam AMB, et al. Additive inheritance of histone modifications in Arabidopsis thaliana intra-specific hybrids. Plant J. 2011;67:691–700. doi: 10.1111/j.1365-313X.2011.04628.x. [DOI] [PubMed] [Google Scholar]
- Moses AM, et al. Large-scale turnover of functional transcription factor binding sites in Drosophila. PLoS Comput Biol. 2006;2:e130. doi: 10.1371/journal.pcbi.0020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang S, et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007;35:D883–D887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, et al. Silencing by plant Polycomb-group genes requires dispersed trimethylation of histone H3 at lysine 27. EMBO J. 2006;25:4638–4649. doi: 10.1038/sj.emboj.7601311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128:735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol. 2009;10:697–708. doi: 10.1038/nrm2763. [DOI] [PubMed] [Google Scholar]
- Spangler JB, Subramaniam S, Freeling M, Feltus FA. Evidence of function for conserved noncoding sequences in Arabidopsis thaliana. New Phytol. 2012;193:241–252. doi: 10.1111/j.1469-8137.2011.03916.x. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Sun B, et al. Timing mechanism dependent on cell division is invoked by Polycomb eviction in plant stem cells. Science. 2014;343:1248559. doi: 10.1126/science.1248559. [DOI] [PubMed] [Google Scholar]
- Swofford DL, Maddison WP. Reconstructing ancestral character states under Wagner parsimony. Math Biosci. 1987;87:199–229. [Google Scholar]
- Thomas BC, Rapaka L, Lyons E, Pedersen B, Freeling M. Arabidopsis intragenomic conserved noncoding sequence. Proc Natl Acad Sci U S A. 2007;104:3348–3353. doi: 10.1073/pnas.0611574104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Townsley B, Chung KH, Sinha N. Regulation of SHOOT MERISTEMLESS genes via an upstream-conserved noncoding sequence coordinates leaf development. Proc Natl Acad Sci U S A. 2007;104:15953–15958. doi: 10.1073/pnas.0707577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn MW, et al. Epigenetic natural variation in Arabidopsis thaliana. PLoS Biol. 2007;5:e174. doi: 10.1371/journal.pbio.0050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley EO, Lieberman BS. Phylogenetics: theory and practice of phylogenetic systematics. 2nd ed. Wiley-Blackwell; 2011. [Google Scholar]
- Yang C, et al. VAL- and AtBMI1-mediated H2Aub initiate the switch from embryonic to postgerminative growth in Arabidopsis. Curr Biol. 2013;23:1324–1329. doi: 10.1016/j.cub.2013.05.050. [DOI] [PubMed] [Google Scholar]
- Zhang X, et al. Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 2007;5:e129. doi: 10.1371/journal.pbio.0050129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.