

Abstract
Altered transforming growth factor-β (TGFβ) expression may contribute to inflammatory bowel disease and modulate epithelial cell restitution. Interference with TGFβ-mediated signaling inhibits excisional skin wound healing, but accelerates healing of incisional cutaneous wounds and wounds in some other tissues. Therefore, we sought to clarify the potential role of Smad3-dependent TGFβ signaling in intestinal mucosal healing in Smad3 null mice. Jejunal serosal application of filter disks saturated with 75% acetic acid yielded a circumscribed reproducible ischemic mucosal ulcer 1 day later. We compared ulcer area at 3 and 5 days to day 1 in Smad3 knockout mice and syngeneic wild-type mice, and evaluated mucosal immunoreactivity at the ulcer edge for TGFβ, phosphorylated (activated) focal adhesion kinase (pFAK), phosphorylated extracellular signal-related kinase (pERK), proliferating cell nuclear antigen and apoptosis by TUNEL. Ulcer healing in Smad3 null mice was 17% less at day 3 (n = 14, P = 0.022) and 15% less at day 5 (n = 14, P = 0.004) than in wild-type littermates. In wild-type mice, pFAK, pERK and TGFβ immunoreactivity were elevated in epithelium immediately adjacent to the ulcer compared with more distant mucosa. However, this pattern of immunoreactivity for pFAK, pERK and TGFβ was not observed in Smad3 null mice. Smad3 null mice exhibited increased epithelial proliferation and no differences in apoptotic cell death compared with wild types, suggesting that ulcer healing may reflect differences in restitutive cell migration. Thus, Smad3-dependent disruption of the TGFβ signaling pathway impairs the healing of murine intestinal mucosal ulcers and alters patterns of activated FAK and ERK immunoreactivity important for cell migration at the ulcer edge. These studies suggest a significant role for Smad3-dependent TGFβ signaling in intestinal mucosal healing.
Keywords: cell migration, inflammatory bowel disease, Smad3, TGFβ, ulcer, wound healing
Intestinal epithelial migration, proliferation and differentiation are requirements in wound healing, a process disrupted in inflammatory bowel diseases (IBDs) such as ulcerative colitis (UC) and Crohn’s disease (CD).1 Early in normal healing, epithelial cells at the edge of the injury flatten out, extend a lamellipodium and migrate across the extracellular matrix of the mucosal defect.2 This migratory process is called restitution, and independent of cell proliferation, which begins later in healing, is regulated by cytokines,3 growth factors,4-7 and modulated by integrin-dependent interactions between adhesion molecules and matrix components.8,9 One of the growth factors present at the wound site, transforming growth factor-β (TGFβ) is initially derived from accumulating platelets, but later can be overexpressed by epithelial cells involved in repairing the injury.8 Although TGFβ potently inhibits proliferation,10,11 it promotes cell migration.11-14 The TGFβ receptors exert their effects through a canonical Smad-dependent signal pathway and Smad-independent signals such as the mitogen-activated protein kinases (MAPK).9 The Smad-dependent pathway begins when activated TGFβ receptors form heterotetrameric complexes which then recruit, phosphorylate and activate the receptor-regulated SMADSs (R-Smads) Smad2/3 complexes.15 The R-Smads are composed of Smad1, Smad2, Smad3, Smad5 and Smad8, and when phosphorylated, these Smads form a complex with a common mediator, Smad4. These Smad complexes then translocate to the nucleus and act as transcription factors important in modulating proteins necessary for wound healing.10
Several signaling molecules are also critical in cellular migration and wound closure and have been implicated in the disruption of the physiologic repair system characteristic in IBD. Migration is dependent on the architecture of the cytoskeleton, including focal adhesions.11 Cellular migration of isolated human colonic lamina propria fibroblasts from CD and UC patients is significantly reduced compared to control cells, and this correlates with a reduction in phosphorylated focal adhesion kinase (FAK).12 Tumor necrosis factor (TNF)-α is a hallmark of IBD, and inhibition of TNF-induced FAK phosphorylation decreases intestinal epithelial cell migration in an in vitro wound model.13 Also important in cell migration and wound healing, the MAPK, extracellular signal-related kinase (ERK), localizes to migrating epithelium at induced ulcer margins in normal rat gastric mucosa.14 Alterations in the activation and expression of ERK have also been observed in IBD. For instance, colonic mucosal biopsies from patients with CD exhibited a threefold increase in ERK phosphorylation as detected by western blot, but these same samples exhibited a significant downregulation of expression of the total ERK2 protein.16
To investigate the role of TGFβ in intestinal mucosal healing, we induced jejunal ulcers in transgenic mice with a targeted disruption of the Smad3 gene and observed wound closure. We looked for changes in epithelial cell proliferation and apoptosis to determine their role, if any, in the differences in ulcer healing in these mice. In addition, we investigated how disruption in the TGFβ signaling pathway effected expression of the signaling molecules FAK and ERK, crucial in cell migration and physiologic repair of the intestinal mucosa.
MATERIALS AND METHODS
Transgenic Mice
All procedures involving mice were approved by our institutional Animal Investigation Committee. Smad3ex8/ex8 C57BL/6 mice were a generous gift from Dr Anita B Roberts (NCI, National Institutes of Health, Bethesda, MD, USA). These were generated by targeted disruption of the Smad3 gene by homologous recombination,17 bred in our laboratory, and verified by PCR before study (Figure 1). Male and female wild-type and Smad3 null mice (8- to 12-week-old) were used for this study after preliminary studies (not shown) found no gender differences in wound healing in this model.
Figure 1.
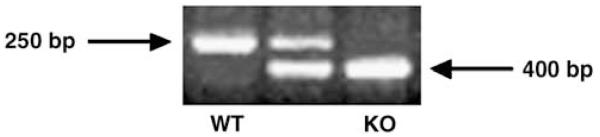
PCR products of tail lysates taken from wild-type, heterozygous and Smad3 null mice. The wild-type allele produces a 250 bp fragment, whereas the Smad3 mutant allele produces a 400-bp fragment. The presence of both fragments in the middle lane denotes a heterozygous mouse, which was not used in this study.
Genotyping Protocol
Tail samples (4 mm) were gathered from Smad3 null and wild-type littermate mice and placed in 250 μl DirectPCR lysis reagent (Viagen, CA, USA) for 24 h at 55°C in a rotating hybridization oven. The samples were then incubated in a 100°C water bath for 45 min and 2 μl of this DNA lysate material was used for each PCR reaction. The presence of the Smad3 allele was detected by primers Smad3-1 (5′-CCACTTCATTGCCATATGCCCTG-3′) and Smad3-3 (5′-CCAG ACTGCCTTGGGAAAAGC-3′), and produced a 250 bp fragment. The presence of the wild-type allele was detected using Smad3-1 (5′-CCACTTCATTGCCATATGCCCTG-3′) and Smad3-2 (5′-CCCGAACAGTTGGATTCACACA-3′), and produced a 400 bp fragment (Figure 1).
Ulcer Induction
Smad3 null and wild-type littermates were anesthetized with intraperitoneal ketamine (100 mg/kg) and xylazene (10 mg/kg), and a laparotomy was performed to expose the jejunum. Adapting a model previously used to create gastric ulcers,18 we created jejunal ulcers by placing a 75% acetic acid-saturated circular filter disk (1.77 mm2) directly on the antimesenteric serosa for 15 s, without opening the bowel. Large blood vessels were avoided in disk placement. Initial ulcer measurements were taken from killed mice 1 day after induction. To determine healing rates, we compared ulcer sizes in mice killed at days 3 or 5 after ulcer induction to their day 1 counterparts.
Ulcer Healing Measurements
After killing, jejunal samples containing the ulcers were harvested and fixed in 10% formalin for 24 h. The jejunal segments were incised along the mesenteric border (opposite the side containing the ulcer) and pinned open on dental wax in PBS to photograph the mucosal surface of the ulcer using a dissecting microscope fitted with a digital camera. The images were then analyzed with Kodak 1D Image Analysis software (Eastman Kodak Company, NY, USA) to determine ulcer area. Ulcer healing was calculated as percent closure of ulcer area at days 3 and 5 compared to the mean ulcer area of a parallel series of ulcers measured at day 1.
Histology, Immunohistochemistry, PCNA, TUNEL and Image Analysis
Jejunal samples containing the ulcers that were 5 day post-induction were fixed in 10% formalin for 24 h and embedded in paraffin. Sections (5 μm) through the area of the ulcer were placed on slides and deparaffinized, rehydrated and utilized for various experiments. Changes in TGFβ expression and cell signaling at the ulcer edge were determined by subjecting the ulcer sections to immunohistochemistry with antibodies directed against TGFβ (R&D Systems, MN, USA), pFAK397 (BD Biosciences, CA, USA), phosphorylated extracellular signal-related kinase (pERK; Cell Signaling, MA, USA) and isotype control (Sigma, MO, USA), used at a dilution of 1:50 in PBS followed by Vectastain Universal ABC kit detection (Vector Labs, CA, USA). Alterations in proliferation were detected utilizing a proliferating cell nuclear antigen (PCNA) kit (Zymed, CA, USA), and apoptosis detection was accomplished by TUNEL staining (Calbiochem, CA, USA). Sections were counterstained with hematoxylin for histological orientation in the immunohistochemical samples and PCNA, or methyl green for TUNEL, coverslipped, visualized and photographed on a Nikon Microphot-FXA.
Statistical Analysis
Immunohistochemical results were scored on a scale of 0–4 with 0 being the least amount of detectable positive staining and 4 being the highest positive staining. The score mode and range are listed in Table 1. Scores were analyzed by χ2-test for significance at P<0.001. The remaining data were expressed as mean±s.e. Statistical analysis was performed using paired or unpaired t-tests or ANOVA as appropriate. A P-value of <0.05 was considered significant.
Table 1. Immunohistochemistry scores comparing staining intensity immediately adjacent to the ulcer or distant fromthe ulcer site.
| Protein | Mode/range |
|||
|---|---|---|---|---|
| Wild type |
Smad3 null |
|||
| Immediate | Distant | Immediate | Distant | |
| TGFβ | 3/2–4 | 1/0–2 | 1/0–2 | 1/0–2 |
| pERK | 3/2–4 | 0/0–2 | 1/0–3 | 1/0–2 |
| tERK | 2/1–3 | 2/1–3 | 2/1–3 | 2/1–3 |
| pFAK | 4/2–4 | 1/0–2 | 2/1–3 | 1/0–2 |
| tFAK | 2/0–2 | 1/0–3 | 2/1–3 | 1/0–3 |
TGFβ, transforming growth factor-β; pERK, phosphorylated extracellular signal-related kinase; tERK, total extracellular signal-related kinase;pFAK, phos phorylated focal adhesion kinase; tFAK, total focal adhesion kinase.
All comparisons P< 0.001 by χ2-analysis.
RESULTS
Disruption of Smad3-Dependent TGFβ Signaling Impairs Intestinal Ulcer Healing
Mice were genotyped at postnatal day 10–14 (Figure 1) and subjected to ulcer induction between 8 and 12 weeks of age. The mice were killed at 3 or 5 days after induction of intestinal ulcers with acetic acid, and ulcer area from Smad3 null and wild-type littermates were compared to the mean ulcer area in wild-type and Smad3 null mice operated on in parallel but killed at day 1. Digital photographs of the mucosal surface of the ulcers (Figure 2) analyzed with Kodak 1D image analysis software demonstrated that ulcer healing in Smad3 null mice was 17±4.4% less than wild-type littermates at 3 days post-ulcer induction (n = 14, P = 0.022), and 15±2.3% less at 5 days post-induction (n = 14, P = 0.004; Figure 3). Thus, disruption of the TGFβ signaling pathway in mice by a targeted deletion of the Smad3 gene resulted in impaired intestinal mucosal healing.
Figure 2.
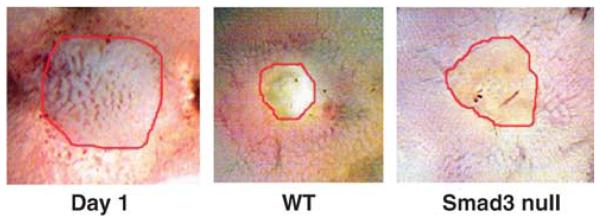
Representative digital photographs of ulcers on the mucosal surface of the jejunum at day 1 in wild-type mice and 5 days after ulcer induction by serosal acetic acid application in wild-type and Smad3 null mice. These images reflect the diminished ability of the intestinal mucosa to heal acetic acid-induced ulcers in Smad3 null mice. Original magnification, × 7.5.
Figure 3.

Ulcer healing in Smad3 null mice and wild-type littermates at 3 and 5 days post-ulcer induction. Smad3 null mice demonstrated 17% less wound healing at day 3 post-induction (n = 14, P = 0.022), and 15% less wound healing at day 5 after ulcer induction (n = 14, P = 0.004) compared to day 1.
Our model of serosal application of acetic acid to induce intestinal mucosal ulcers resulted, histologically, in a disruption of underlying matrix and smooth muscle structure at the location of the ulcer. This phenomenon did not resolve by day 5 after ulcers were induced (Figure 4).
Figure 4.

H&E-stained sections of intestinal ulcers in a wild-type and Smad3 null mice at days 1, 3 and 5 after ulcer induction. Normal matrix and smooth muscle layers are observed distant from the ulcer. Disrupted matrix and smooth muscle layers are observed at the location of the ulcer (U). Original magnification × 40.
Impaired Intestinal Epithelial Healing in Smad3 Mice Is not Due to a Decrease in Proliferation or an Increase in Apoptosis
We next evaluated the possibility that alterations in intestinal epithelial healing in Smad3 null mice might reflect changes in proliferation or apoptosis. To detect changes in proliferation between wild-type and Smad3 mice, ulcer sections from 5 day-post ulcer induction were immunostained with an antibody against PCNA, a protein produced during late G1 and S phase of the cell cycle and a common marker for proliferating cells.19 Quantification demonstrated a 9.46%±0.1 (n = 5, P<0.001) increase in PCNA-positive intestinal mucosa epithelial cells within 3 mm adjacent to the ulcer in Smad3 mice compared to wild-type mice (Figure 5). This suggested that the diminished rate of ulcer healing in the Smad3 knockout mouse was not due to a decrease in proliferating epithelial cells at the ulcer edge.
Figure 5.

Paraffin sections immunostained with proliferating cell nuclear antigen (PCNA) from jejunal ulcers 5 days after induction in wild-type (a) and Smad3 null (b) mice demonstrate more proliferating epithelial cells in intestinal mucosa epithelium in the Smad3 null mice (n = 5, P<0.001). Sections are counterstained with hematoxylin. Original magnification, ×400.
As the balance between proliferation and apoptosis is critical to maintaining the integrity of the intestinal mucosa, we investigated whether apoptotic changes could have contributed to the decrease in ulcer healing observed in the Smad3 null mice. TUNEL assay, which recognizes exposed ends of DNA fragments, revealed no detectable increase in the number of apoptotic cells in Smad3 null mice compared to wild-type (Figures 6a and b). Quantification indicated less than 1% change in apoptosis in Smad3 null mice compared to wild type (data not shown). Positive control was a mixture of HL60 cells previously incubated with actinomycin D (Figure 6c). Negative control was mouse wild-type ulcer tissue with TdT enzyme omitted from staining protocol (Figure 6d). This finding suggested that the inhibition of ulcer healing in the Smad3 null mouse was not due to an increase in apoptosis at the ulcer margin.
Figure 6.

TdT staining of 5-day ulcers (U) in wild-type (a) and Smad3 null mice (b). Smad3 null mice exhibited no detectable changes in TUNEL-positive cells in migrating epithelial cells compared with wild-type mice. Positive control slide was a mixture of HL60 cells treated with actinomycin D (c). Negative control slide was wild-type mouse ulcer tissue with TdT enzyme omitted from staining protocol (d). Sections are counterstained with methyl green. Original magnification, × 200.
TGFβ Immunoreactivity is Reduced at the Leading Edge of Migrating Epithelial Cells in Intestinal Mucosal Ulcers in Smad3 Null Mice
Previous evidence of the presence of TGFβ at wound sites and its role in cell migration led us to investigate the localization of TGFβ at the leading edge of epithelial migration in intestinal mucosal ulcers. TGFβ immunoreactivity was readily apparent in 5 day-post ulcer induction samples at the immediate edge of the ulcer border in epithelial cells and also somewhat present in the submucosa further away from the ulcer in wild-type mice (Figure 7a). Immunohistochemical assessment demonstrated that the increased TGFβ staining observed at the immediate edge of the ulcer in wild-type mice differed significantly compared to more distant mucosa (Table 1; n = 20, P<0.001). This pattern of expression was not seen in the Smad3 null mice, where there was no significant difference between TGFβ staining at the ulcer edge and more distant mucosa (Figure 7b; Table 1; n = 16, P = 0.7), raising the possibility that the reduction in ulcer healing observed in the knockout mice might reflect changes in TGFβ expression and non-Smad signaling at the edge of the ulcer.
Figure 7.
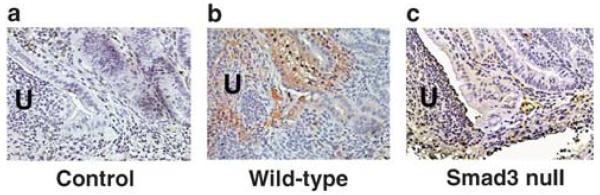
Paraffin sections of ulcers from wild-type and Smad3 null mice 5 days after induction immunostained for transforming growth factor-β (TGFβ). (a) Isotype control. TGFβ was present at the ulcer edge in wild-type mice and also in the submucosa further away (b), but this pattern of expression was not seen in Smad3 null mouse ulcers (c). Arrows point to the epithelium at the edge of the ulcer (U). Sections are counterstained with hematoxylin. Original magnification, × 400.
Signaling Molecules Crucial for Cellular Migration are Altered at the Ulcer Edge in Mice Deficient in Smad3
The diminished ability of the intestinal mucosa to heal in Smad3 null mice led us to investigate two of the intracellular signaling events that might contribute to mucosal healing. Cell migration is the first in a series of steps that lead to wound closure, and we investigated two signaling molecules important in this process. There was a significant increase in immunoreactivity for phosphorylated (activated) Focal Adhesion Kinase (pFAK) in migratory epithelial cells at the immediate ulcer edge (arrow) from 5 day-post ulcer induction samples compared to the more distant mucosa in wild-type mice (Figure 8a; Table 1; n = 12, P = 0.001). However, we were not able to detect this pattern of pFAK immunoreactivity in the epithelial cells at the ulcer edge (arrow) in Smad3 null mice and there was not a significant difference in immunoreactivity between the two areas in the Smad3 null mice (Figure 8b; Table 1; n = 12, P = 0.25). Immunohistochemical analysis of total FAK in the two areas of the mouse intestinal mucosa revealed no significant difference in either wild type or Smad3 null tissue (n = 16 in both, wild type P = 0.47, Smad3 null P = 0.34). This suggested that although FAK protein was present throughout the mucosal tissue in both wild-type and Smad3 null mice subjected to ulcers, phosphorylation of FAK was diminished at the ulcer edge only in the knockout mice.
Figure 8.

Immunohistochemical detection of phosphorylated focal adhesion kinase (pFAK) in intestinal epithelial cells (arrows) adjacent to ulcers (U) from wild-type and Smad3 null mice. pFAK was detected at the leading edge of epithelial cell migration in wild-type mice (a), but was not detectable in this area in Smad3 null mice (b). Sections are counterstained with hematoxylin. Original magnification, × 400.
Activation of ERK has previously been shown to dramatically increase up to ninefold in gastric ulcers during the initial healing period between 3 and 7 days.20 pERK immunoreactivity was detected at day 5 by immunohistochemistry in our ulcer model and was found to localize in the migrating mucosal epithelium immediately adjacent to the ulcer (arrow), but was not abundant in more distant mucosa in wild-type mice (Figure 9a; Table 1; n = 20, P<0.001). As with the phosphorylated FAK, the pERK was not significantly different between the area immediately adjacent to the ulcer (arrow) and more distant mucosa in Smad3 null mice (Figure 9b; Table 1; n = 17, P = 0.24). Immunohistochemical assessment of total ERK in the two areas of the mouse intestinal mucosa revealed no significant difference between the two areas in either wild type or Smad3 null tissue (Table 1; wild-type n = 22 P = 0.29, Smad3 null n = 19 P = 0.52). These results suggest that the phosphorylation event that occurs for ERK at the ulcer margin in wild-type mouse mucosa is impaired in Smad3 null mice.
Figure 9.

Immunohistochemical detection of phosphorylated extracellular signal-related kinase (pERK) in intestinal epithelial cells adjacent to ulcers (U) from wild-type and Smad3 null mice. (a) pERK is detected at the leading edge (arrows) of epithelial cell migration in wild-type mice, but was no detectable in this area in Smad3 null mice (b). Sections are counterstained with hematoxylin. Original magnification, × 400.
DISCUSSION
Intestinal mucosal ulcer healing was inhibited in transgenic mice with a Smad3-dependent block of the TGFβ pathway. There was no evidence of an increase in apoptotic epithelial cells adjacent to intestinal ulcers in Smad3 knockout mice. However, interestingly, we observed increased proliferation in these cells. The slower mucosal wound healing in these mice despite increased proliferation may reflect differences in epithelial cell migration. TGFβ immunoreactivity was increased in epithelial cells at the ulcer edge in wild-type mice, but not in Smad3 null mice. pFAK and ERK were also detected in migrating epithelial cells at the ulcer edge in the wild-type mice, but were below the limits of detection in epithelium surrounding ulcers. Loss of TGFβ, pFAK and pERK signaling in epithelial cells at the leading edge of intestinal ulcers could contribute to the reduction of ulcer healing in these mice.
The magnitude of ulcer healing we observed in our model is consistent with previously reported investigations. Acetic acid-induced gastric ulcers in rats treated with l-cysteine were 28% smaller than vehicle-treated ulcers, and conversely, ulcers treated with a hydrogen sulfide synthesis inhibitor demonstrated the same magnitude of delayed healing.21 Wild-type mice treated with either 1 M NaCl or capsaicin as a protective agent against ethanol-induced gastric ulcers demonstrated increased healing rates of 10 and 12%, respectively compared with saline-treated ulcers.22 These relatively small changes in healing might be biologically important in real mucosal healing because they might tip the balance between healing and nonhealing in the setting of continued reinjury by luminal noxious agents.
Although other models of mucosal injury have been utilized to demonstrate alterations in healing, including dextran sodium sulfate (DSS) model of colitis, we chose serosal application of acetic acid to induce ulcers. This model of mucosal injury was appropriate for its reproducibility and consistent initial ulcer size at day 1 that facilitated quantitation of the size of a specific isolated mucosal ulceration. This model also seemed to cause fewer side effects than have been described by others in the DSS model.23 We observed no diarrhea, no noticeable weight loss, no gross bleeding and no rectal prolapse. Such systemic aberrations could themselves lead to pathophysiologic neurohumoral or hemodynamic changes that could affect mucosal healing and conceivably might even have different consequences in the setting of a Smad3 knockout.
Epithelial proliferation begins within 3 days of ulceration.18,16 TGFβ arrests epithelial proliferation at G1.24 The loss of TGFβ at the ulcer edge in Smad3 null mice may then explain the increased proliferation near ulcers in Smad3 null mice compared to wild-type mice. Although excisional ear wounds in Smad3 null mice that heal more slowly also exhibit increased apoptosis,25 we observed very little apoptosis in the epithelial cells near the ulcer in wild type and Smad3 null mice. This apparent inconsistency may reflect differences in tissue or mechanism of injury between that previous study and this one. Although increased apoptosis could impair ear excisional wound healing in Smad3 null mice, the increased mucosal proliferation and similarly low apoptosis suggested slower enterocyte migration as an alternative mechanism for delayed ulcer healing in Smad3 nulls.
Growth factors produced at ulcers by epithelial or inflammatory cells modulate migration as well as proliferation.16,26,27 The unregulated immune response and chronic inflammation of IBD has been attributed in part to alterations in growth factors like TGFβ.28 Lamina propria mononuclear cells and T cells from patients with IBD exhibit variable levels of TGFβ1, depending on the disorder.29 Disruption of TGFβ results in either a severe inflammatory response followed by early death or 100% lethality in mice.30-34 TGFβ receptor resistance may also contribute to IBD.35-37 Although we did not investigate TGFβ receptor expression in this model of intestinal ulceration, factors that affect receptor expression could also influence mucosal healing as TGFβ expression has been implicated in immuno-regulation, matrix deposition and inhibition of proteolytic enzymes in intestinal repair, and TGFβ receptor II knockout mice demonstrate increased susceptibility to IBD.37 Our observations suggest that disruption of TGFβ signaling by Smad3 knockout impairs epithelial migration. Although TGFβ inhibits intestinal epithelial proliferation, it conversely promotes migration in vitro,38,39 and in vivo.40 TGFβ inactivation with a neutralizing antibody in mice decreases reepithelialization,40 and mice expressing a dominant-negative TGFβ RII in the intestinal epithelium are more susceptible to injury and heal more slowly in a colitis model.41 Interestingly, TGFβ levels are higher in Smad3 null mice,42,43 possibly due to mucosal T cells or platelet degranulation.43 Despite the increased TGFβ, ear punch wounds in Smad3 null mice exhibit decreased TGFβ immunoreactivity at the wound edge,25 consistent with our present observations. Together, this evidence suggests that TGFβ acts at the injury site to promote cell migration in intestinal ulcer healing.
The effects of transgenic knockouts that disrupt TGFβ signaling vary with the tissue and model studied.44-46 Smad3 null mice exhibit accelerated healing in some other tissues, including brain,47 skin25 and tail.48 Tokumasa reported that Smad3 knockout mice also heal experimental colitis more rapidly, but only heterozygous animals were utilized for the study.49 Our observations (not shown) of ulcer healing in heterozygous Smad3 mice were variable and inconsistent, leading us to study the more clearly defined homozygous knockouts. In contrast to Tokumasa’s report, but consistent with our own results, excisional ear wounds in Smad3 null mice heal more slowly.25 Differences in tissue, wound type and mechanism of injury may explain these apparent inconsistencies. Excisional wounds, such as ear punches or ulcers, exhibit absence or abnormalities of underlying matrix, support cells and vascular components. The matrix and cells across which restitutive cell motility must occur critically regulate migration.
Altered matrix proteins and structure have been described in Smad3 null mice,25,43,47 consistent with observations that TGFβ modulates matrix synthesis in vivo.50 In contrast to skin or brain, the intestine is subjected to complex repetitive forces due to peristalsis and interaction with luminal contents. Colonic contractility decreases in both animal models of colitis and in humans with IBD.44-46,51 The healing of wounds in intestinal epithelial cell monolayers in vitro is modulated by repetitive deformation in a matrix-dependent manner.52 Disruption of intestinal deformation, and alterations of muscle structure and matrix organization in an excisional type wound, could alter healing.
In addition to alterations in TGFβ expression and matrix organization, poor healing in IBD has been linked to changes in cell signaling related to cell motility. Changes in FAK have been documented in in vitro models of intestinal epithelial migration,53 including at the leading edge during cell migration in monolayer wounds.54 Inhibition with a dominant-negative FAK construct55 or siRNA56 significantly inhibits cell migration in vitro. In contrast, some studies have shown decreases in pFAK that coincide with increased wound healing and vice versa.53,55,56 Total and pFAK are decreased at the migrating front of human Caco-2 and rat IEC-6 monolayer wounds.57 This apparent discrepancy in FAK localization during cell migration and effect on wound healing may be due to several factors. Acetic acid-induced intestinal ulcers in mice are vastly different from in vitro monolayer wounds. Indeed, one study found discrepancies in cell signaling and apoptosis in both in vivo wound healing in mice with keratinocyte-specific deletion of FAK and in vitro migration of FAK null keratinocytes in culture.58 Cells in culture lose paracrine interactions with other cell types. In addition, humoral, neural and angiogenic influences are present in vivo but not in cell culture.
We also found abnormalities in pERK immunoreactivity in the mucosa of Smad3 knockout mice. ERK mediates intestinal epithelial cytokine production by IL-1 that is important in IBD.59 Although levels of phosphorylated ERK1/2 are increased in mucosal biopsies from IBD patients, the overall level of ERK expression is significantly down-regulated.60 In addition, localization of activated ERK at the wound margin is important for cell migration necessary for healing.52,61 ERK inhibition in vitro inhibits migration on collagen55 and fibronectin,52 and ERK phosphorylation levels have been shown to directly correlate with the rate of cell migration.62 We know that signals important for cell migration are altered in Smad3 null mice.25 Smad3-deficient MEF’s display reduced TGFβ-mediated activation of the ERK MAPK pathway.63 ERK and Smad3 directly effect each other’s phosphorylation after TGFβ pathway activation,64 and mediate TGFβ-induced release of connective tissue growth factor,65 and this cross talk may be disrupted in Smad3 null mice.
In summary, disruption of the Smad3-dependent branch of the TGFβ signal pathway inhibits intestinal ulcer healing in Smad3 null transgenic mice, independently of observable changes in apoptosis and despite increased proliferation. The marked loss of TGFβ, pFAK and pERK at the ulcer margin in Smad3 null mice may contribute to this effect. These results suggest that the Smad3-dependent branch of the TGFβ signal pathway is crucial for restititutive cell migration in intestinal mucosal healing.
ACKNOWLEDGEMENTS
We thank Dr Anita Roberts for the Smad3 null mice and Dr James Hatfield for technical support. This study was supported by NIH RO1 DK067257 and VA MERIT Award (MDB). This study was also supported in part by NIH RO1 DK067257 and VA MERIT Award (MDB).
References
- 1.Okamoto R, Watanabe M. Cellular and molecular mechanisms of the epithelial repair in IBD. Dig Dis Sci. 2005;50(Suppl 1):S34–S38. doi: 10.1007/s10620-005-2804-5. [DOI] [PubMed] [Google Scholar]
- 2.Basson MD, Rashid Z, Turowski GA, et al. Restitution at the cellular level: regulation of the migrating phenotype. Yale J Biol Med. 1996;69:119–129. [PMC free article] [PubMed] [Google Scholar]
- 3.Dignass AU, Podolsky DK. Interleukin 2 modulates intestinal epithelial cell function in vitro. Exp Cell Res. 1996;225:422–429. doi: 10.1006/excr.1996.0193. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki H, Hirai K, Yamamoto H, et al. HST-1/FGF-4 plays a critical role in crypt cell survival and facilitates epithelial cell restitution and proliferation. Oncogene. 2004;23:3681–3688. doi: 10.1038/sj.onc.1207348. [DOI] [PubMed] [Google Scholar]
- 5.Ramsanahie AP, Perez A, Duensing AU, et al. Glucagon-like peptide 2 enhances intestinal epithelial restitution. J Surg Res. 2002;107:44–49. doi: 10.1006/jsre.2002.6504. [DOI] [PubMed] [Google Scholar]
- 6.Nishimura S, Takahashi M, Ota S, et al. Hepatocyte growth factor accelerates restitution of intestinal epithelial cells. J Gastroenterol. 1998;33:172–178. doi: 10.1007/s005350050066. [DOI] [PubMed] [Google Scholar]
- 7.Goke MN, Schneider M, Beil W, et al. Differential glucocorticoid effects on repair mechanisms and NF-kappaB activity in the intestinal epithelium. Regul Pept. 2002;105:203–214. doi: 10.1016/s0167-0115(02)00023-x. [DOI] [PubMed] [Google Scholar]
- 8.Bascom CC, Wolfshohl JR, Coffey RJ, Jr, et al. Complex regulation of transforming growth factor beta 1, beta 2, and beta 3 mRNA expression in mouse fibroblasts and keratinocytes by transforming growth factors beta 1 and beta 2. Mol Cell Biol. 1989;9:5508–5515. doi: 10.1128/mcb.9.12.5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 11.Chai J, Baatar D, Tarnawski A. Serum response factor promotes re-epithelialization and muscular structure restoration during gastric ulcer healing. Gastroenterology. 2004;126:1809–1818. doi: 10.1053/j.gastro.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Leeb SN, Vogl D, Gunckel M, et al. Reduced migration of fibroblasts in inflammatory bowel disease: role of inflammatory mediators and focal adhesion kinase. Gastroenterology. 2003;125:1341–1354. doi: 10.1016/j.gastro.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Corredor J, Yan F, Shen CC, et al. Tumor necrosis factor regulates intestinal epithelial cell migration by receptor-dependent mechanisms. Am J Physiol Cell Physiol. 2003;284:C953–C961. doi: 10.1152/ajpcell.00309.2002. [DOI] [PubMed] [Google Scholar]
- 14.Tarnawski AS, Pai R, Wang H, et al. Translocation of MAP (Erk-1 and -2) kinases to cell nuclei and activation of c-fos gene during healing of experimental gastric ulcers. J Physiol Pharmacol. 1998;49:479–488. [PubMed] [Google Scholar]
- 15.Feng XH, Derynck R. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 16.Tarnawski A. Molecular mechanisms of ulcer healing. Drug News Perspect. 2000;13:158–168. doi: 10.1358/dnp.2000.13.3.858438. [DOI] [PubMed] [Google Scholar]
- 17.Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tarnawski A, Hollander D, Krause WJ, et al. ‘Healed’ experimental gastric ulcers remain histologically and ultrastructurally abnormal. J Clin Gastroenterol. 1990;12(Suppl 1):S139–S147. doi: 10.1097/00004836-199001001-00024. [DOI] [PubMed] [Google Scholar]
- 19.Waseem NH, Lane DP. Monoclonal antibody analysis of the proliferating cell nuclear antigen (PCNA). Structural conservation and the detection of a nucleolar form. J Cell Sci. 1990;96(Part 1):121–129. doi: 10.1242/jcs.96.1.121. [DOI] [PubMed] [Google Scholar]
- 20.Pai R, Ohta M, Itani RM, et al. Induction of mitogen-activated protein kinase signal transduction pathway during gastric ulcer healing in rats. Gastroenterology. 1998;114:706–713. doi: 10.1016/s0016-5085(98)70584-0. [DOI] [PubMed] [Google Scholar]
- 21.Wallace JL, Dicay M, McKnight W, et al. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007;21:4070–4076. doi: 10.1096/fj.07-8669com. [DOI] [PubMed] [Google Scholar]
- 22.Ohno T, Hattori Y, Komine R, et al. Roles of calcitonin gene-related peptide in maintenance of gastric mucosal integrity and in enhancement of ulcer healing and angiogenesis. Gastroenterology. 2008;134:215–225. doi: 10.1053/j.gastro.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Kong J, Zhang Z, Musch MW, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–G216. doi: 10.1152/ajpgi.00398.2007. [DOI] [PubMed] [Google Scholar]
- 24.Dennler S, Goumans MJ, ten Dijke P. Transforming growth factor beta signal transduction. J Leukoc Biol. 2002;71:731–740. [PubMed] [Google Scholar]
- 25.Arany PR, Flanders KC, Kobayashi T, et al. Smad3 deficiency alters key structural elements of the extracellular matrix and mechanotransduction of wound closure. Proc Natl Acad Sci USA. 2006;103:9250–9255. doi: 10.1073/pnas.0602473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong WM, Playford RJ, Wright NA. Peptide gene expression in gastrointestinal mucosal ulceration: ordered sequence or redundancy? Gut. 2000;46:286–292. doi: 10.1136/gut.46.2.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarnawski AS, Jones MK. The role of epidermal growth factor (EGF) and its receptor in mucosal protection, adaptation to injury, and ulcer healing: involvement of EGF-R signal transduction pathways. J Clin Gastroenterol. 1998;27(Suppl 1):S12–S20. doi: 10.1097/00004836-199800001-00004. [DOI] [PubMed] [Google Scholar]
- 28.Kader HA, Tchernev VT, Satyaraj E, et al. Protein microarray analysis of disease activity in pediatric inflammatory bowel disease demonstrates elevated serum PLGF, IL-7, TGF-beta1, and IL-12p40 levels in Crohn’s disease and ulcerative colitis patients in remission versus active disease. Am J Gastroenterol. 2005;100:414–423. doi: 10.1111/j.1572-0241.2004.40819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Del Zotto B, Mumolo G, Pronio AM, et al. TGF-beta1 production in inflammatory bowel disease: differing production patterns in Crohn’s disease and ulcerative colitis. Clin Exp Immunol. 2003;134:120–126. doi: 10.1046/j.1365-2249.2003.02250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kulkarni AB, Huh CG, Becker D, et al. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 33.Proetzel G, Pawlowski SA, Wiles MV, et al. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Souza RF, Garrigue-Antar L, Lei J, et al. Alterations of transforming growth factor-beta 1 receptor type II occur in ulcerative colitis-associated carcinomas, sporadic colorectal neoplasms, and esophageal carcinomas, but not in gastric neoplasms. Hum Cell. 1996;9:229–236. [PubMed] [Google Scholar]
- 36.Souza RF, Lei J, Yin J, et al. A transforming growth factor beta 1 receptor type II mutation in ulcerative colitis-associated neoplasms. Gastroenterology. 1997;112:40–45. doi: 10.1016/s0016-5085(97)70217-8. [DOI] [PubMed] [Google Scholar]
- 37.Hahm KB, Im YH, Parks TW, et al. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–198. doi: 10.1136/gut.49.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ciacci C, Lind SE, Podolsky DK. Transforming growth factor beta regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology. 1993;105:93–101. doi: 10.1016/0016-5085(93)90014-4. [DOI] [PubMed] [Google Scholar]
- 39.Basson MD, Modlin IM, Flynn SD, et al. Independent modulation of enterocyte migration and proliferation by growth factors, matrix proteins, and pharmacologic agents in an in vitro model of mucosal healing. Surgery. 1992;112:299–307. discussion 307-298. [PubMed] [Google Scholar]
- 40.Reynolds LE, Conti FJ, Lucas M, et al. Accelerated re-epithelialization in beta3-integrin-deficient- mice is associated with enhanced TGF-beta1 signaling. Nat Med. 2005;11:167–174. doi: 10.1038/nm1165. [DOI] [PubMed] [Google Scholar]
- 41.Beck PL, Rosenberg IM, Xavier RJ, et al. Transforming growth factor-beta mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am J Pathol. 2003;162:597–608. doi: 10.1016/s0002-9440(10)63853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang A, Ziyadeh FN, Lee EY, et al. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol. 2007;293:F1657–F1665. doi: 10.1152/ajprenal.00274.2007. [DOI] [PubMed] [Google Scholar]
- 43.Zanninelli G, Vetuschi A, Sferra R, et al. Smad3 knock-out mice as a useful model to study intestinal fibrogenesis. World J Gastroenterol. 2006;12:1211–1218. doi: 10.3748/wjg.v12.i8.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snape WJ, Jr, Williams R, Hyman PE. Defect in colonic smooth muscle contraction in patients with ulcerative colitis. Am J Physiol. 1991;261(6 Part 1):G987–G991. doi: 10.1152/ajpgi.1991.261.6.G987. [DOI] [PubMed] [Google Scholar]
- 45.Snape WJ, Jr, Kao HW. Role of inflammatory mediators in colonic smooth muscle function in ulcerative colitis. Dig Dis Sci. 1988;33(3 Suppl):65S–70S. doi: 10.1007/BF01538133. [DOI] [PubMed] [Google Scholar]
- 46.Sethi AK, Sarna SK. Colonic motor activity in acute colitis in conscious dogs. Gastroenterology. 1991;100:954–963. doi: 10.1016/0016-5085(91)90269-q. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Moges H, Bharucha Y, et al. Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex. Exp Neurol. 2007;203:168–184. doi: 10.1016/j.expneurol.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 48.Falanga V, Schrayer D, Cha J, et al. Full-thickness wounding of the mouse tail as a model for delayed wound healing: accelerated wound closure in Smad3 knock-out mice. Wound Repair Regen. 2004;12:320–326. doi: 10.1111/j.1067-1927.2004.012316.x. [DOI] [PubMed] [Google Scholar]
- 49.Tokumasa A, Katsuno T, Tanaga TS, et al. Reduction of Smad3 accelerates re-epithelialization in a murine model of colitis. Biochem Biophys Res Commun. 2004;317:377–383. doi: 10.1016/j.bbrc.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 50.Amendt C, Mann A, Schirmacher P, et al. Resistance of keratinocytes to TGFβ-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J Cell Sci. 2002;115(Part 10):2189–2198. doi: 10.1242/jcs.115.10.2189. [DOI] [PubMed] [Google Scholar]
- 51.Grossi L, McHugh K, Collins SM. On the specificity of altered muscle function in experimental colitis in rats. Gastroenterology. 1993;104:1049–1056. doi: 10.1016/0016-5085(93)90273-f. [DOI] [PubMed] [Google Scholar]
- 52.Zhang J, Owen CR, Sanders MA, et al. The motogenic effects of cyclic mechanical strain on intestinal epithelial monolayer wound closure are matrix dependent. Gastroenterology. 2006;131:1179–1189. doi: 10.1053/j.gastro.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 53.Liu YW, Sanders MA, Basson MD. Human Caco-2 intestinal epithelial motility is associated with tyrosine kinase and cytoskeletal focal adhesion kinase signals. J Surg Res. 1998;77:112–118. doi: 10.1006/jsre.1998.5369. [DOI] [PubMed] [Google Scholar]
- 54.Tilghman RW, Slack-Davis JK, Sergina N, et al. Focal adhesion kinase is required for the spatial organization of the leading edge in migrating cells. J Cell Sci. 2005;118(Part 12):2613–2623. doi: 10.1242/jcs.02380. [DOI] [PubMed] [Google Scholar]
- 55.Yu CF, Sanders MA, Basson MD. Human caco-2 motility redistributes FAK and paxillin and activates p38 MAPK in a matrix-dependent manner. Am J Physiol Gastrointest Liver Physiol. 2000;278:G952–G966. doi: 10.1152/ajpgi.2000.278.6.G952. [DOI] [PubMed] [Google Scholar]
- 56.Jiang X, Jacamo R, Zhukova E, et al. RNA interference reveals a differential role of FAK and Pyk2 in cell migration, leading edge formation and increase in focal adhesions induced by LPA in intestinal epithelial cells. J Cell Physiol. 2006;207:816–828. doi: 10.1002/jcp.20629. [DOI] [PubMed] [Google Scholar]
- 57.Basson MD, Sanders MA, Gomez R, et al. Focal adhesion kinase protein levels in gut epithelial motility. Am J Physiol Gastrointest Liver Physiol. 2006;291:G491–G499. doi: 10.1152/ajpgi.00292.2005. [DOI] [PubMed] [Google Scholar]
- 58.Essayem S, Kovacic-Milivojevic B, Baumbusch C, et al. Hair cycle and wound healing in mice with a keratinocyte-restricted deletion of FAK. Oncogene. 2006;25:1081–1089. doi: 10.1038/sj.onc.1209130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Garat C, Arend WP. Intracellular IL-1Ra type 1 inhibits IL-1-induced IL-6 and IL-8 production in Caco-2 intestinal epithelial cells through inhibition of p38 mitogen-activated protein kinase and NF-kappaB pathways. Cytokine. 2003;23:31–40. doi: 10.1016/s1043-4666(03)00182-0. [DOI] [PubMed] [Google Scholar]
- 60.Waetzig GH, Seegert D, Rosenstiel P, et al. p38 Mitogen-activated protein kinase is activated and linked to TNF-alpha signaling in inflammatory bowel disease. J Immunol. 2002;168:5342–5351. doi: 10.4049/jimmunol.168.10.5342. [DOI] [PubMed] [Google Scholar]
- 61.Matsubayashi Y, Ebisuya M, Honjoh S, et al. ERK activation propagates in epithelial cell sheets and regulates their migration during wound healing. Curr Biol. 2004;14:731–735. doi: 10.1016/j.cub.2004.03.060. [DOI] [PubMed] [Google Scholar]
- 62.Joslin EJ, Opresko LK, Wells A, et al. EGF-receptor-mediated mammary epithelial cell migration is driven by sustained ERK signaling from autocrine stimulation. J Cell Sci. 2007;120(Part 20):3688–3699. doi: 10.1242/jcs.010488. [DOI] [PubMed] [Google Scholar]
- 63.Arany PR, Rane SG, Roberts AB. Smad3 deficiency inhibits v-ras-induced transformation by suppression of JNK MAPK signaling and increased farnesyl transferase inhibition. Oncogene. 2008;27:2507–2512. doi: 10.1038/sj.onc.1210889. [DOI] [PubMed] [Google Scholar]
- 64.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 65.Leivonen SK, Hakkinen L, Liu D, et al. Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-beta-induced expression of connective tissue growth factor in human fibroblasts. J Invest Dermatol. 2005;124:1162–1169. doi: 10.1111/j.0022-202X.2005.23750.x. [DOI] [PubMed] [Google Scholar]