

Abstract
ATP-binding cassette (ABC) transporters form a large superfamily of ATP-dependent protein complexes that mediate transport of a vast array of substrates across membranes. The 14 currently available structures of ABC transporters have greatly advanced insight into the transport mechanism and revealed a tremendous structural diversity. Whereas the domains that hydrolyze ATP are structurally related in all ABC transporters, the membrane-embedded domains, where the substrates are translocated, adopt four different unrelated folds. Here, we review the structural characteristics of ABC transporters and discuss the implications of this structural diversity for mechanistic diversity.
ATP-binding cassette (ABC) transporters are a large group of membrane protein complexes that couple transport of a substrate across the membrane to the hydrolysis of the phosphate bond between the γ- and the β-phosphate of ATP (Ames et al., 1990; Higgins, 1992; Davidson et al., 2008; Rees et al., 2009). The free energy released when ATP is converted into ADP and orthophosphate (Pi; approximately −50 kJ mol−1 in many cells) can be used to accumulate the transported substrates in, or to remove them from, cellular compartments.
In prokaryotes, ABC transporters are localized to the plasma membrane, and ATP is hydrolyzed on the cytoplasmic side. In eukaryotes, ABC transporters are also found in organellar membranes. ATP hydrolysis by organellar ABC transporters takes place on the cytosolic side of the membrane, except for transporters from mitochondria and chloroplasts where the ATP-binding domains of the transporters are located on the matrix or stroma side. The side of the membrane where ATP is bound and hydrolyzed is termed the cis-side, and the opposite side is called the trans-side.
ABC transporters can be classified as exporters or importers. Exporters move substrates from the cis-side to the trans-side of the membrane, from the hydrophobic core of the lipid bilayer to the trans-side, or transfer substrates between the inner and outer leaflets of the bilayer. In contrast, importers move substrates from the trans-side to the cis-side. There are a few ABC transporters that do not have a bona fide transport function. Notable examples include the CFTR, which is a gated chloride channel (Aleksandrov et al., 2007), and the sulfonylurea receptor SUR1, which is a regulatory complex associated with a potassium channel (Bryan et al., 2007). There has been tremendous interest in ABC transporters not only from a mechanistic point of view but also because malfunctioning of human ABC transporters leads to a plethora of diseases (see for instance, Silverton et al., 2011), and some ABC exporters are involved in the drug resistance of bacteria and cancer cells (Ambudkar et al., 2003; Davidson et al., 2008).
Many excellent reviews on ABC transporters have been published over the past few years and cover the history, structure, mechanism, physiology, and pharmacology of these proteins (Davidson et al., 2008; Rees et al., 2009; Parcej and Tampé, 2010; Eitinger et al., 2011; George and Jones, 2012; Lewis et al., 2012). Because a wealth of new crystal structures has been determined lately, here we provide an update on the structural diversity of ABC transporters.
Overview
Similar to other membrane transport proteins, ABC transporters adopt at least two conformations in which the substrate-binding site is accessible from either the cis-side or the trans-side. Alternation between the two conformations allows substrate translocation across the membrane (“alternating access” model; Jardetzky, 1966; Tanford, 1982). The binding of substrate on one side of the membrane and release on the opposite side are coordinated by ATP binding and hydrolysis, and ADP and Pi release. Several ABC transporters have been crystallized in different conformations (see Table 1 for details). These crystal structures provide profound insight in the transport mechanism, but a comprehensive understanding of the transport cycle is still lacking.
Table 1.
Crystal structures of ABC transporters
| Name (organism) | Remarks | Substrate bound | Resolution (Å) | PDB accession no. | Reference |
| Type I importers | |||||
| Molybdate transporter ModB2C2-A complex (Archaeoglobus fulgidus) | Inward-facing conformation, with SBP bound | Mg2+, PO43-, WoO42− | 3.10 | 2ONK | Hollenstein et al., 2007 |
| ModBC (Methanosarcina acetivorans) | Inward-facing conformation, without SBP bound, in a trans-inhibited state | Mg2+, WoO42− | 3.00 | 3D31 | Gerber et al., 2008 |
| MalFGK2-MBP maltose uptake transporter (E. coli) | Outward-facing conformation stabilized by a mutation in the NBDs (MalK E159Q), with MBP | Maltose, ATP | 2.80 | 2R6G | Oldham et al., 2007 |
| MalFGK2 maltose uptake transporter (E. coli) | TM helix 1 deleted, in inward-facing conformation, in resting state, without MBP | — | 4.50 | 3FH6 | Khare et al., 2009 |
| MalFGK2-MBP maltose uptake transporter (E. coli) | Pre-translocation intermediate state, with mutations in MBP (G69C/S337C) that stabilize the substrate-bound closed conformation by a cross-link | AMP-PNP, Mg2+, maltose | 2.90 | 3PUZ | Oldham and Chen, 2011a |
| Outward-facing conformation state, with MBP | AMP-PNP, Mg2+, maltose | 3.10 | 3PUY | ||
| Pre-translocation intermediate state, with mutations in MBP (G69C/S337C) that stabilize the substrate-bound closed conformation by a cross-link | Maltose | 3.10 | 3PV0 | ||
| MalFGK2-MBP maltose uptake transporter (E. coli) | Outward-facing conformation, with MBP | AMP-PNP, Mg2+, maltose | 2.20 | 3RLF | Oldham and Chen, 2011b |
| Outward-facing conformation, with MBP | ADP · VO43−, Mg2+, maltose | 2.40 | 3PUV | ||
| Outward-facing conformation, with MBP | ADP · AlF4−, Mg2+, maltose | 2.30 | 3PUW | ||
| Outward-facing conformation, with MBP | ADP · BeF3−, Mg2+, maltose | 2.30 | 3PUX | ||
| MalFGK2-MBP maltose uptake transporter (E. coli) | Complex with its regulatory protein EIIAglc, inward-facing conformation | — | 3.91 | 4JBW | Chen et al., 2013 |
| MalFGK2-MBP maltose uptake transporter (E. coli) | Outward-facing conformation, with MBP | Maltoheptaose | 2.90 | 4KHZ | Oldham et al., 2013 |
| Outward-facing conformation, with MBP | ANP, α-d-glycose | 2.38 | 4KI0 | ||
| MetNI methionine uptake transporter (E. coli) | Inward-facing conformation | — | 3.70 | 3DHW | Kadaba et al., 2008 |
| Inward-facing conformation, at higher resolution (detergent Cymal5) | ADP | 2.90 | 3TUI | Johnson et al., 2012 | |
| Inward-facing conformation, C2 domains repositioned (detergent decylmaltoside) | — | 4.00 | 3TUJ | ||
| Type II importers | |||||
| BtuC2D2 vitamin B12 transporter (E. coli) | Outward-facing conformation, no SBP bound | V4O124− | 3.20 | 1L7V | Locher et al., 2002 |
| BtuC2D2-F complex (E. coli) | BtuC in asymmetric conformation. The translocation pore is closed from both sites. BtuF is in an open state. | — | 2.60 | 2QI9 | Hvorup et al., 2007 |
| BtuC2D2-F complex (E. coli) | Intermediate occluded state, nucleotide bound | AMP-PNP | 3.47 | 4FI3 | Korkhov et al., 2012b |
| BtuC2D2-F complex (E. coli) | E159Q mutation in NBD abolished ATP hydrolysis activity, BtuC in asymmetric conformation. The translocation pore is closed from both sites. | — | 3.49 | 4DBL | Korkhov et al., 2012a |
| HI1470/1471 putative metal chelate–type ABC transporter (H. influenzae) | Inward-facing conformation, without SBP, renamed MolB2C2 (as later was shown to bind WoO42−/MoO42−) | — | 2.40 | 2NQ2 | Pinkett et al., 2007 |
| HmuU2V2 heme transporter (Yersinia pestis) | Outward-facing conformation | — | 3.00 | 4G1U | Woo et al., 2012 |
| ECF-type importers | |||||
| RibU S-component for riboflavin (S. aureus) | Substrate bound | Riboflavin | 3.60 | 3P5N | Zhang et al., 2010 |
| ThiT S-component for thiamin (L. lactis) | Substrate bound | Thiamin | 2.00 | 3RLB | Erkens et al., 2011 |
| BioY S-component for biotin (L. lactis) | Substrate bound | Biotin | 2.10 | 4DVE | Berntsson et al., 2012 |
| NikM S-component for Ni2+ (Thermoanaerobacter tengcongensis) | Substrate bound | Ni2+/Co2+ | 1.83–2.5 | — | Yu et al., 2013 |
| ECF-FolT transporter (L. brevis) | Substrate free, inward-facing conformation | — | 3.00 | 4HUQ | Xu et al., 2013 |
| ECF-HmpT transporter (L. brevis) | Substrate free, inward-facing conformation | — | 3.53 | 4HZU | Wang et al., 2013 |
| Exporters | |||||
| Sav18662 multidrug transporter (S. aureus) | Outward-facing conformation | ADP | 3.00 | 2HYD | Dawson and Locher, 2006 |
| Outward-facing conformation | AMP-PNP | 3.40 | 2ONJ | Dawson and Locher, 2007 | |
| Heterodimeric ABC transporter TM287-TM288 (T. maritima) | Inward-facing conformation | AMP-PNP | 2.90 | 3QF4 | Hohl et al., 2012 |
| MsbA2 lipid “flippase” (Salmonella typhimurium) | Outward-facing conformation, with nucleotide bound | ANP-PNP | 4.50 | 3B5Y | Ward et al., 2007 |
| Outward-facing conformation, with nucleotide bound | ANP-PNP | 3.70 | 3B60 | ||
| Outward-facing conformation, with nucleotide bound | ADP, VO43− | 4.20 | 3B5Z | ||
| MsbA2 lipid “flippase” E. coli | Inward-facing conformation, open apo structure | — | 5.30 | 3B5W | |
| MsbA2 lipid “flippase” Vibrio cholera | Inward-facing conformation, closed apo structure | — | 5.50 | 3B5X | |
| Multidrug transporter P-glycoprotein (Mus musculus) | Inward-facing conformation | — | 3.80 | 3G5U | Aller et al., 2009 |
| Inward-facing conformation | Cyclic-tris-(R)-valineselenazole | 4.40 | 3G60 | ||
| Inward-facing conformation | Cyclic-tris-(S)-valineselenazole | 4.35 | 3G61 | ||
| Re-refined, inward-facing conformation | — | 3.80 | 4M1M | Li et al., 2014 | |
| Multidrug transporter P-glycoprotein (Caenorhabditis elegans) | Inward-facing conformation | — | 3.40 | 4F4C | Jin et al., 2012 |
| ABCB10 mitochondrial ABC transporter (Homo sapiens) | Inward-facing conformation | AMPPCP | 2.85 | 4AYT | Shintre et al., 2013 |
| Inward-facing conformation; different crystal form (rod/plate) | AMP-PCP/AMP-PNP | 2.90/3.30 | 4AYX/4AYW | ||
| Inward-facing conformation | — | 2.85 | 3ZDQ | ||
SBP, substrate-binding protein; MBP, maltose-binding protein; outward and inward facing, the translocation pathway in the TMDs exposed to the trans-side or the cis-side of the membrane, respectively.
All ABC transporters have a core with the same modular architecture: two transmembrane (TM) domains (TMDs) or subunits and two nucleotide-binding domains (NBDs) or subunits. The NBDs, which are highly conserved in structure and sequence among all ABC transporters, are the hallmark of the family. NBDs do not always associate with TMDs but may also be involved in various functions that do not occur at the membrane (see for instance, Boël et al., 2014, and references therein). However, the name “ABC transporter” is only used when the NBDs form a complex with TMDs, and NBDs that are not associated with TMDs will not be discussed here.
In contrast to the conserved NBDs, several unrelated folds of the TMDs have been found. These different folds, which are defined by the connectivity and three-dimensional arrangement of the secondary structure elements, do not share significant sequence similarity. Because TMDs with the same fold may also lack sequence similarity, structure determination is necessary for fold assignment. So far, four types of ABC transporters have been identified based on the TMDs folds as determined by the crystal structures (Fig. 1).
Figure 1.
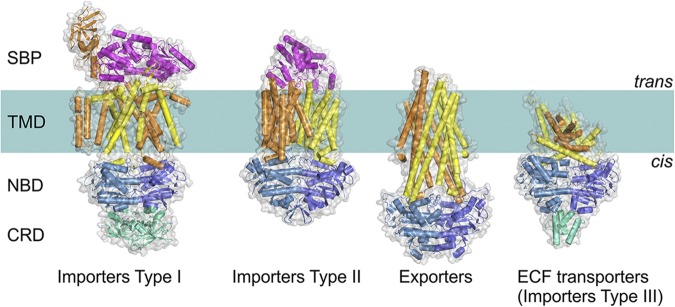
Four distinct folds of ABC transporters. All share a similar general architecture: two NBDs (blue and sky blue) are attached to two TMDs (orange and yellow). In some transporters, additional domains are present (green), which often have a regulatory function (C-terminal regulatory domain [CRD]). In Type I and II importers, the transported compounds are delivered to TMDs by SBPs (or SBDs; magenta) located in periplasm (Gram-negative bacteria) or external space (Gram-positive bacteria and Archaea). ECF, energy coupling factor.
Three ABC transporter types appear to be associated exclusively with import functions (transport of substrates from the trans-side to the cis-side of the membrane): Type I and Type II importers, and energy coupling factor (ECF) transporters (also named Type III importers). All three types of ABC importers are found only in prokaryotes. The fourth fold is found in all structurally characterized exporters. ABC transporters with the exporter fold are present in both prokaryotes and eukaryotes. ABC transporters with different TMD folds probably also differ in the mechanistic details of transport (see below).
Prokaryotic ABC transporters are often assembled from separate protein subunits (two TMDs and two NBDs; Biemans-Oldehinkel et al., 2006). The two NBDs and TMDs can be identical (homodimeric) or different proteins (heterodimeric). In the latter case, the two NBD subunits are invariably structurally similar. The TMDs in single transporters are also usually similar in structure, with the notable exception of the two TMDs in ECF-type importers, which are completely unrelated (Wang et al., 2013; Xu et al., 2013; Slotboom, 2014). Sometimes two, and occasionally three, subunits are fused into a multi-domain protein in the prokaryotic transporters. In particular, the two NBDs are occasionally fused, and in many prokaryotic exporters, the TMDs are fused with the NBDs (for instance, in bacterial exporters MsbA2 [Ward et al., 2007], Sav18662 [Dawson and Locher, 2006], and TM287/TM288 [Hohl et al., 2012], for which crystal structures have been solved). Eukaryotic exporters are generally composed either of one polypeptide chain containing all the domains (e.g., P-glycoprotein) or of a dimer of two polypeptides, each of which contains an NBD and a TMD (as in the bacterial exporters).
The Type I and II ABC importers depend on additional soluble substrate-binding domains (SBDs) or substrate-binding proteins (SBPs) (Fig. 1), which capture the transported substrate on the trans-side and deliver it to the TMDs (Quiocho and Ledvina, 1996; Berntsson et al., 2010). In some cases, the SBD is fused with a TMD into a multi-domain subunit (Biemans-Oldehinkel et al., 2006). ECF transporters and exporters do not require SBPs (Rodionov et al., 2009). Many prokaryotic and eukaryotic ABC transporters contain additional domains or subunits, such as regulatory domains (Fig. 1) or extra TMDs of unknown function. These additional domains are very diverse and will not be discussed here (Biemans-Oldehinkel et al., 2006; Parcej and Tampé, 2010).
It is unknown whether the different ABC transporter folds have evolved to address specific mechanistic challenges. It is possible that the structural differences between ABC exporters and importers are related to the opposing directions in which the substrate is pumped, which may lead to different mechanistic requirements. Alternatively, the differences between the exporter and the importer folds may be related to the range of transported substrates. ABC exporters are involved in the transport of hydrophobic compounds such as lipids, fatty acids, cholesterol, and drugs, as well as larger molecules such as proteins (toxins, hydrolytic enzymes, S-layer proteins, lantibiotics, bacteriocins, and competence factors). In addition, most drug exporters can transport a large variety of drugs (of different sizes) out of the cell and are therefore called multidrug-resistant transporters. In contrast, importers generally are selective for a single or a few related water-soluble substrates.
The three different types of ABC importers have overlapping substrate specificities, and it is therefore not clear why three importer folds have evolved. In general (but not exclusively), the substrates of Type I importers are compounds required in bulk (such as sugars and amino acids), whereas Type II importers and ECF transporters are more often specific for compounds needed in small quantities (metal chelates, vitamins; Davidson et al., 2008; Eitinger et al., 2011). It is possible that Type I ABC importers are more suitable for high capacity, low affinity transport, whereas Type II and ECF importers may better serve high affinity, low capacity transport. However, this distinction is blurred, and it is possible that the variation in kinetics within an importer type may be as large as the variation between the different types.
NBD
All ABC transporters contain two NBDs, also called ATPases or ABCs, which bind and hydrolyze ATP. The NBDs from ABC transporters are a subgroup of the diverse superfamily of P-loop NTPases (Vetter and Wittinghofer, 1999) and depend on magnesium ions for catalysis. Each NBD has a core of ∼200 amino acids and consists of two subdomains: the larger RecA-like domain, which is also found in other P-loop ATPases, and the structurally more diverse α-helical domain, which is unique to ABC transporters (Fig. 2).
Figure 2.
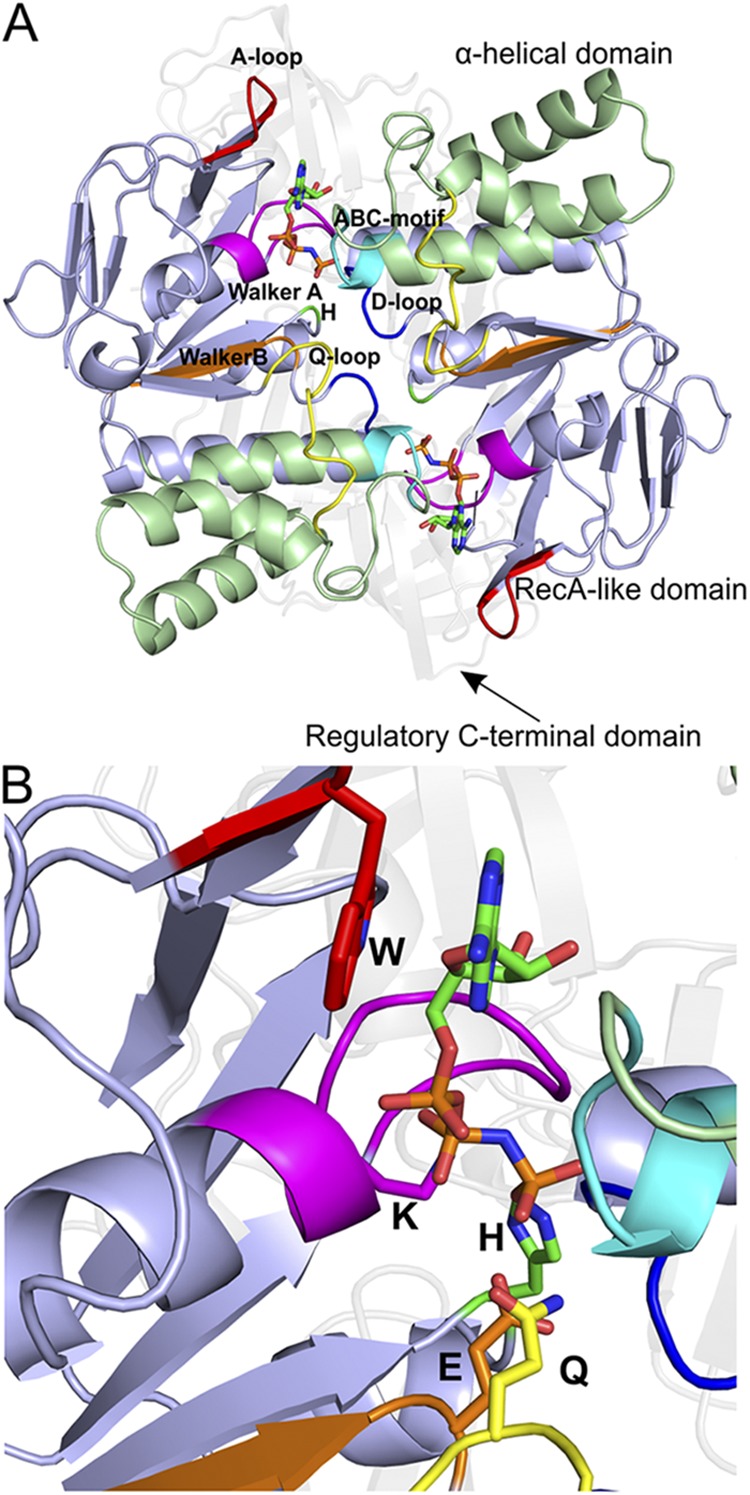
The structure of the NBDs, as exemplified by the MalK dimer of the maltose transporter MalEFGK2 (Protein Data Bank accession no. 3RLF). (A) View along an axis perpendicular to the membrane plane from the trans-side onto the NBDs (The TMDs and SBP have been removed for clarity). Domains and highly conserved sequence motifs are color-coded: green, α-helical domain; light blue, RecA-like domain; faded gray, regulatory C-terminal domain; red, A-loop; magenta, Walker A; orange, Walker B; blue, D-loop; green, H-loop; cyan, ABC motif; yellow, Q-loop. The ATP analogue AMP-PNP is shown in sticks. (B) A closer look onto the nucleotide-binding site. The key amino acids are indicated (see NBD for details).
Structures of NBDs in the absence of their corresponding TMDs were determined before the first full ABC transporter structures were solved (Oswald et al., 2006), and have provided crucial insight into the architecture of the catalytic site and mechanism of ATP hydrolysis (Smith et al., 2002; Verdon et al., 2003; Zaitseva et al., 2005). However, to fully understand the catalytic mechanism, high resolution structures of the full complexes are indispensable.
NBDs can be identified at the sequence level by a specific set of seven highly conserved motifs (Figs. 2 A and 3 C):
Figure 3.
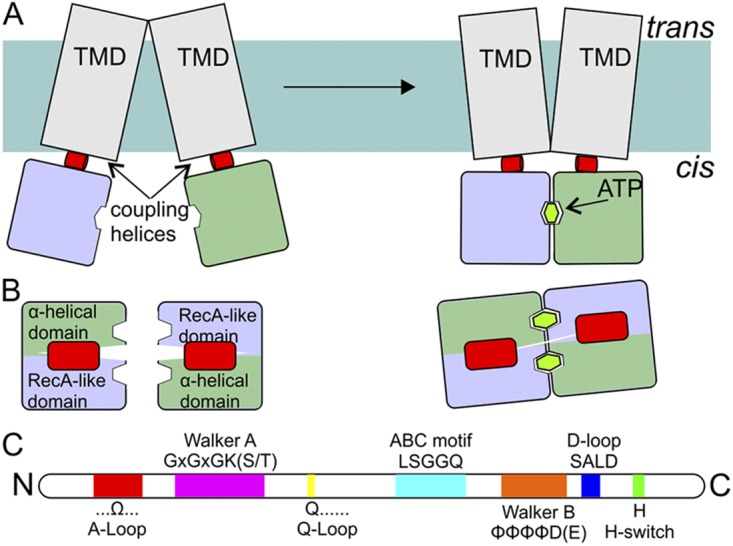
Schematic representation of NBDs and coupling helices. (A) Side view (from the membrane plane) of an ABC transporter. NBDs (blue and green; colors of the domains are as in Fig. 2) are attached to the TMDs (gray) via so-called coupling helices (red) present in loops of the TMDs. ATP binding and hydrolysis cause rearrangements in the NBDs, which are propagated to the TMDs via the coupling helices. (B) Top view (along an axis perpendicular to the membrane) of the NBDs and the coupling helices from the TMDs. (C) The relative positions of sequence motifs in NBDs (see also Fig. 2).
(1) The A-loop contains a conserved aromatic residue (usually a tyrosine) that helps to position the ATP via stacking with the adenine ring (Fig. 2 B).
(2) The P-loop or Walker A motif (GXXGXGK(S/T)) is a phosphate-binding loop that contains the highly conserved lysine residue. Backbone amide nitrogens and the ε-amino group of this lysine residue form a network of interactions with β- and γ-phosphate of ATP.
(3) The Walker B motif (ϕϕϕϕDE, where ϕ is a hydrophobic amino acid) helps to coordinate the magnesium ion via the conserved aspartate residue. The second acidic residue at the end of the Walker B motif (often a glutamate residue) very likely is the general base that polarizes the attacking water molecule. This role of the glutamate residue has long been under debate, but recent crystal structures of the maltose transporter MalEFGK2 from Escherichia coli strongly favor its function as the general base (Oldham and Chen, 2011b).
(4) The D-loop (motif: SALD) directly follows the Walker B motif. The D-loops from the two monomers in the dimeric ensemble run alongside each other. Changes in the conformation of the D-loop affect the geometry of the catalytic site and help to form the ATP hydrolysis site.
(5) The H-loop (or switch region) contains a highly conserved histidine residue that forms a hinge between a β strand and an α helix near the C terminus of the NBD. The histidine residue interacts with the conserved aspartate from the D-loop, the proposed general base (glutamate residue of the Walker B motif) and with the γ-phosphate of the ATP. It assists with the positioning of the attacking water, the general base, and the magnesium ion.
(6) The Q-loop is a stretch of approximately eight residues with a conserved glutamine residue at its N terminus. It is located at the interface between the RecA-like subdomain and the α-helical subdomain. Conformational changes in the Q-loop allow the conserved glutamine residue to move in and out of the active site during the hydrolysis cycle, forming the active site when Mg-ATP is bound and disrupting it once ATP is hydrolyzed. The Q-loop is also a major site of interaction with the TMDs (see below).
(7) The ABC signature motif (or C motif, LSGGQ) is found in the α-helical subdomain and is a characteristic feature of the ABC superfamily, not present in other P-loop NTPases such as the F1-ATPase. This LSGGQ motif is located at the N-terminal end of a long helix that directs the positive charge of the helical dipole toward the γ-phosphate of ATP.
The two NBDs in ABC transporters can adopt different orientations relative to each other (Fig. 3): they can tightly pack against each other (closed conformation) or partially dissociate (open conformation). There are two ATP-binding sites at the interface between the two monomers, which are related by twofold (pseudo)symmetry. ATP binding promotes the formation of the closed conformation because each ATP molecule interacts with motifs from both NBDs: the ABC signature motif of one monomer is located close to the Walker A and B motifs and the A, H, and Q loop of the other domain (Fig. 2). Only when the monomers are packed against each other can ATP hydrolysis take place. The release of Pi and ADP after ATP hydrolysis destabilizes the dimer and allows the NBDs to move apart. Moreover, during this catalytic cycle, the RecA-like subdomain and the α-helical subdomain within each NBD rotate toward each other when ATP is bound and away from each other after hydrolysis and ADP and Pi release. In this way, the chemical energy of ATP hydrolysis is transformed into conformational energy that can be transmitted to the TMDs to promote alternating access of the substrate–translocation pathway to the two sides of the membrane.
Because NBD dimers have two ATP hydrolysis sites, it is tempting to assume that ABC transporters use the hydrolysis of two ATP molecules for a complete transport cycle. Although a 2:1 stoichiometry (ATP to transported substrate) has indeed been determined experimentally for the glycine-betaine importer OpuA from the bacterium Lactococcus lactis (Patzlaff et al., 2003), it cannot be concluded that this stoichiometry is conserved among all ABC transporters, for two reasons.
First, it has been very difficult to accurately measure the stoichiometry of transport, because many purified ABC transporters have basal ATPase activity in the absence of the transported substrate (Lewinson et al., 2010). Although the basal activity may be an artifact of purified ABC transporters, it is also possible that some degree of futile ATP hydrolysis takes place in vivo. The degree of futile cycling may differ for different members of the family.
Second, some ABC transporters have heterodimeric NBDs that contain only a single complete ATPase site. The second site is degenerate and cannot hydrolyze ATP because of mutation(s) in the conserved motifs (Procko et al., 2009; Jones and George, 2013). Combinations of canonical and degenerate sites are found frequently in ABC exporters (both prokaryotic and eukaryotic) and possibly indicate that the hydrolysis of a single ATP molecule takes place per transport cycle. Additionally, mutagenesis experiments have shown that a single active ATP hydrolysis site may also be sufficient for transporters with homodimeric NBDs and two canonical ATPase sites (in the histidine transporter, HisP2MQJ from E. coli; Nikaido and Ames, 1999). However, this is not a universal property; for instance, in the maltose transporter MalEFGK2, two functional ATPase sites are required (Davidson and Sharma, 1997). These observations suggest that different transport stoichiometries can be found in ABC transporters, indicating the use of multiple mechanisms of transport, even though all NBDs are structurally related. The functional consequences of differences in stoichiometry between different members of the ABC transport superfamily are not clear. Thermodynamically, the coupled hydrolysis of two ATP molecules rather than one per substrate could lead to greater membrane gradients of the transported substrate.
A combination of a consensus and a degenerate ATP-binding site in the NBD dimer introduces obvious asymmetry. But even in the presence of two consensus sites, ATP hydrolysis is not likely to be simultaneous at both sites, and thus asymmetry may be a generic feature of ABC transporters (Mittal et al., 2012; Jones and George, 2013).
TMD
In all four ABC transporter types, the TMDs constitute a translocation pathway, which is alternately accessible from the cis-side and trans-side of the membrane to enable the transport of substrate (Fig. 1).
The two TMDs of Type I importers are either identical (homodimers) or structurally similar (e.g., the two TMDs of the maltose transporter MalEFGK2 share only 13% sequence identity but are structurally related), with a core membrane topology of five TM helices per TMD (Fig. 4 A). In many cases, an additional N-terminal helix is present that wraps around the helices of the other TMDs and intertwines the TMDs, making a total of 12 TM helices (Fig. 4 A). However, some TMDs contain up to eight TM helices (Fig. 4 A). The translocation pathway is located at the interface between the two TMDs.
Figure 4.
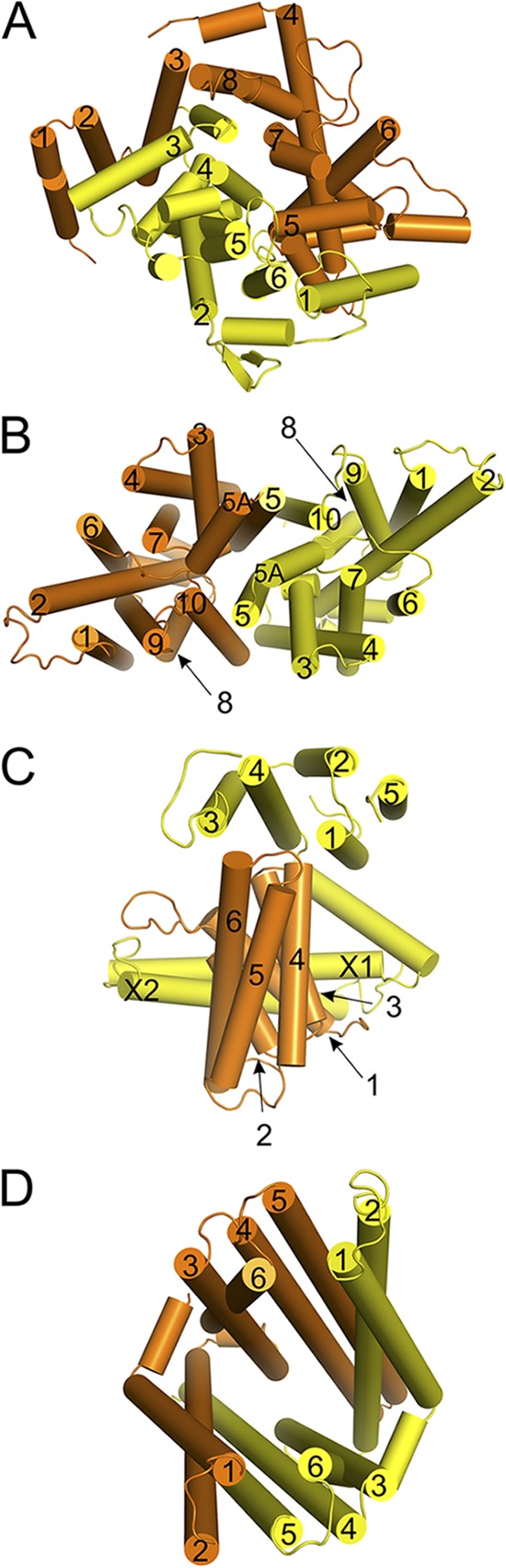
Arrangement of the membrane helices in ABC transporters. Viewpoints are from the outside (trans-side) along an axis perpendicular to the membrane plane. (A) The MalF and MalG subunits of the maltose transporter MalEFGK2, (B) the BtuC dimer of the vitamin B12 transporter BtuC2D2F, (C) the EcfT subunit (yellow) and the S-component from the ECF-HmpT transporter, and (D) the membrane domains of the TM287 and TM288 subunits of the exporter from T. maritima. TM helices are numbered according to their occurrence in the sequence, with the one located most closely to the N terminus numbered as 1. The two coupling helices in the EcfT subunit are labeled X1 and X2.
Type II ABC importers have two identical TMDs, each comprised of 10 TM helices (Fig. 4 B). In the Type II fold, the TMDs are lined up next to each other (Fig. 4 B); they do not have helices that cross over to the other TMD. In each TMD, there is a pseudo twofold symmetry between the segments containing TM helices 2–5 and TM helices 7–10. These two subdomains have a similar helical packing but with opposite orientation with respect to the membrane. The helices of a single TMD are tightly packed together, and the two TMDs line a translocation pore at the interface.
In ECF-type ABC importers, the two TMDs are structurally and functionally unrelated. One TMD is termed the EcfT subunit (or T-component). In the available crystal structures of ECF transporters, this subunit has five TM helices (Fig. 4 C). However, in other ECF transporters, EcfT subunits are predicted to have four to eight TM helices (Eitinger et al., 2011). The second TMD is termed the S-component and binds the transported substrate with high affinity (Duurkens et al., 2007; Eudes et al., 2008; Erkens and Slotboom, 2010; Berntsson et al., 2012). In contrast to Type I and II importers, which require water-soluble SBPs for high affinity substrate recognition, ECF transporters only need the hydrophobic integral membrane S-component. S-components have a core of six TM helices, but a few S-components have an additional N-terminal helix (Yu et al., 2013). In ECF transporters, the translocation pathway is probably not located at the interface between the TMDs but confined to the S-component, which uses a unique alternating access mechanism (see below).
Crystal structures of ABC exporters show a common structural fold consisting of a core six TM helices per TMD (Fig. 4 D). The two TMDs in the dimer may be identical or structurally similar. The 12 TM helices extend a considerable distance into the cytoplasm, with the NBDs located ∼25 Å away from the membrane surface (Fig. 1). This is very different from the importers, where the NBDs are located very close to the membrane. In exporters, the translocation path most likely is located at the interface of the dimeric assembly. In all crystallized ABC exporters, the NBDs are fused to the TMDs, and two helices of each TMD cross over to the other TMD.
Coupling helix
A crucial mechanistic question is how alternating access in the TMDs is coupled to conformational changes in the NBDs when binding and hydrolysis of ATP and release of Pi and ADP take place. So-called coupling helices have been identified in the TMDs of ABC exporters and Type I and II importers (Dawson et al., 2007). A coupling helix is a short α helix in one of the cytoplasmic protrusions of the TMD that fits into a groove of an NBD monomer. In this way, each NBD is connected to a TMD, and the conformational changes in the NBDs can be transduced to conformational changes in the TMDs, leading to alternating access. Some coupling helices contain a conserved sequence (EAA motif) (Mourez et al., 1997), but in most cases, sequence similarity is lacking in the coupling helices. Coupling helices are found between TM helix 3 and 4 in the core of Type I importers. These helices correspond to helices 4 and 5 in MalG (which has an additional N-terminal helix) and helices 6 and 7 in MalF (three additional helices) in Fig. 4 A. In Type II importers, they are located in TM helices 6 and 7, and in ABC exporters, the coupling helix region is found in the intracellular loop (ICL)2 between TM helices 4 and 5. In exporters where the TMDs are fused to the NBDs, the coupling helix of one TMD interacts with the NBD that is linked to the other subunit. Although the arrangements are different in the different types of transporters, all coupling helices interact in a similar way with the NBDs. The region of the NBDs that interacts with the coupling helix of the TMDs contains the Q-loop. The cleft for the coupling helices in the NBDs is located exactly at the interface between the α-helical subdomain and the RecA-like subdomain, which rotate toward each other in response to ATP binding for ATP hydrolysis.
In the Type I maltose importer (MalEFGK2), the coupling helices are not the only site of interaction between the TMDs and the NBDs, because the C-terminal segment of one of the TMDs (MalG) is partially inserted between the two NBDs and seems to further order the Q-loop region. Additional interactions are also seen in Type II importers, where BtuC2D2 has a helical segment next to the coupling helix that also interacts with the NBDs. The crystallized exporters contain the most extensive additional interaction area, with a second cytoplasmic coupling helix. This helix is located between TM helices 2 and 3 (ICL1) and interacts directly with the NBD regions that bind the nucleotide adenine ring. Therefore, they shield the nucleotide and the active site of the NBDs from the bulk solvent in the ATP-bound closed state. The NBDs of exporters contain an additional motif (the X-loop: TEVGERG) that interacts with both coupling helices (ICL1 and ICL2) of the TMDs. This motif is located just before the ABC signature motif. Based on this X-loop motif, it was suggested that the coupling mechanism of ATP hydrolysis and transport would occur through a distinct mechanism in exporters (Dawson and Locher, 2006).
In the case of ECF transporters, only one of the two TMDs (the EcfT subunit or T-component) interacts with the NBDs. The EcfT subunit contains two long helices in a single cytoplasmic region that interact with the two NBDs at the similar location as the coupling helices in the Type I and II importers and the exporters. The other TMD (the S-component) interacts extensively with EcfT but barely with the NBDs. The asymmetry in the TMD–NBD interaction in ECF transporters most likely leads to a distinct mechanism of transport.
Substrate binding
Type I and II ABC importers.
The SBD (or SBP) is a soluble constituent of ABC transporters that is located on the trans-side of the membrane. SBPs for different substrates display widely varying binding affinities (Quiocho and Ledvina, 1996; Berntsson et al., 2010). Dissociation constants are often in the range of 0.01 to 1 µM (Davidson et al., 2008) but occasionally are much lower or higher (e.g., OpuBC from Bacillus subtilis has an affinity of 30 µM for choline [Pittelkow et al., 2011], MolA from Haemophilus influenzae has an affinity of ∼100 µM for molybdate and tungstate [Tirado-Lee et al., 2011], and TbpA from E. coli has an affinity of 2.3 nM for thiamin [Soriano et al., 2008]). SBPs are either linked in a single polypeptide with the TMD of the transporter, connected to the membrane via a lipid anchor or separate TM helix, or freely diffusible in the periplasm (the latter only in Gram-negative bacteria; Biemans-Oldehinkel et al., 2006). Even though SBPs can vary considerably in sequence and size, and have very different substrate specificities, they share highly conserved general architecture with two domains or lobes that are connected via a hinge region (Fig. 5). Based on their structure, SBPs can be categorized into six different clusters (Table 2) (Berntsson et al., 2010). It is notable that all Type II importers for which crystal structures have been determined make exclusive use of SBPs from cluster A, whereas all Type I importers use SBPs from group D. It is possible that the use of SBPs from different clusters correlates with the use of different TMD folds, and that more TMD folds remain to be discovered.
Figure 5.
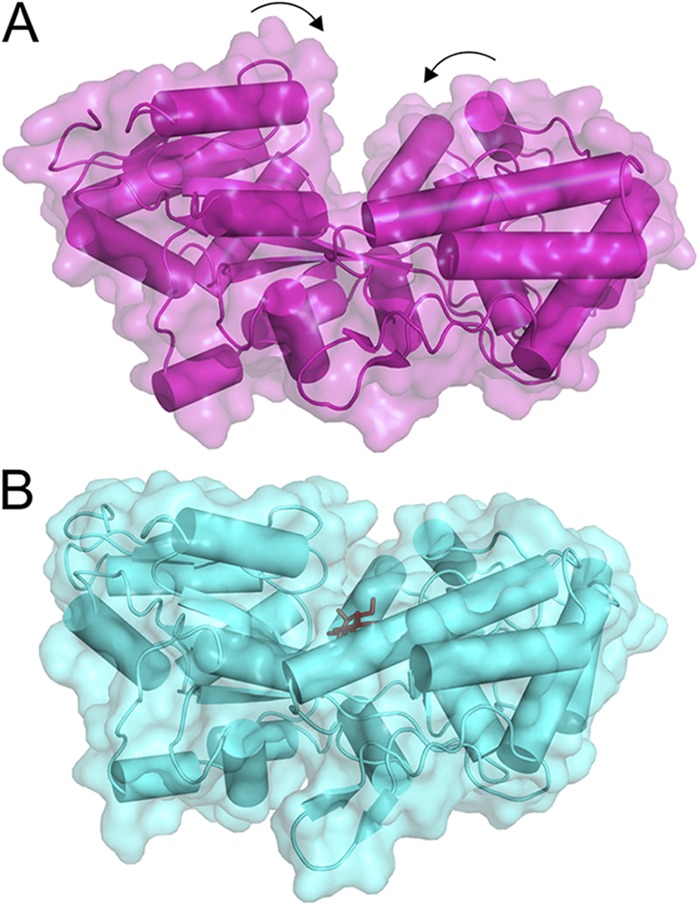
Rearrangements in MBP upon the substrate binding. (A) In the substrate-free form (Protein Data Bank accession no. 1ANF), the cavity between two protein lobes connected by the hinge is accessible. (B) Upon the binding of substrate (dark sticks; Protein Data Bank accession no. 1EZ9), the cavity becomes occluded.
Table 2.
Clusters of soluble SBPs
| Cluster | Subcluster | Types of ligands | Main feature |
| A | I | Metal ions | Single rigid α helix connects the two domains |
| II | Siderophores | ||
| B | Carbohydrates, Leu, Ile, Val, autoinducer-2 | Three interconnecting segments between the two domains | |
| C | Di- and oligopeptides, Arg, cellubiose, nickel | An additional domain. These SBPs are significantly larger. | |
| D | I | Carbohydrates | Two relative short hinges between the two domains |
| II | Putrescine, thiamine | ||
| III | Tetrahedral oxyanions | ||
| E | Sialic acid, 2-keto acids, ectoine, pyroglutamic acid | Large flexible helix in between the two domains. Only associated with TRAP transporters. | |
| F | I | Trigonal planar anions, unknown ligands | Two hinges in between the two domains as in cluster D, but these hinges are almost twice as long, giving the SBP more flexibility |
| II | Methionine | ||
| III | Compatible solutes | ||
| IV | Amino acids |
Data taken from Berntsson et al. (2010).
The vast amount of structural information on SBPs has provided profound insight in the mechanism of substrate binding. In the absence of a ligand, the two lobes exist predominantly in an open conformation, but upon substrate binding, they close to trap the ligand (the “Venus fly trap” model [Quiocho and Ledvina, 1996]; Fig. 5) and eventually deliver it to the TMDs. SBPs interact with both TMDs, with each lobe interacting with one of the TMDs.
Some of the Type I and II ABC importers can transport more than one substrate, which is possible either because their SBPs can recognize various substrates (e.g., the SBP of the multi-sugar transporter Msm of Streptococcus mutans can recognize melibiose, sucrose, raffinose, isomaltotriose, and isomaltotetraose [Russell et al., 1992]), or because the transporter can interact with various SBPs. Examples of the latter are the His/Lys/Arg transport system in Enterobacteriaceae (Higgins and Ames, 1981), the peptide transporter OppBCDF from Enterococcus faecalis (Leonard et al., 1996), and the oligopeptide/muramyl peptide transport system of E. coli (Park et al., 1998). However, the interaction of different SBPs with the same translocator is relatively rare.
In the Type I maltose transporter MalEFGK2, one of the TMDs (MalF) contains a second binding site for maltose (in addition to the binding site in the SBP MalE), located along the translocation path at the center of the bilayer (Oldham et al., 2007). It is likely that substrate moves from the SBP to the central membrane-embedded site during the translocation cycle. To date, this is the only crystal structure where an additional binding site has been identified, but this may be a feature of more Type I importers. In the histidine transporter (HisMQP2-HisJ/LAO), mutations in the NBDs (HisP2) lead to transport of histidine in the absence of the SBP (HisJ/LAO), indicating the presence of a second binding site (Speiser and Ames, 1991). No crystal structure of the His transporter is available, but its TMDs (HisM and HisQ) are predicted to have five TM helices. Therefore it is probably a Type I transporter.
No specific binding sites have been found in the translocation pathway between the TMDs of the Type II importers. It has been hypothesized that the translocation pathways of the Type II ABC importers are inert, “Teflon”-like, with little or no affinity for substrate (Korkhov et al., 2012b). This would mean that the specificity of these transporters depends entirely on the SBP.
In some Type I transporters, binding sites for the transported substrates are present in additional domains connected to the NBDs. These binding sites are not required for transport but have regulatory functions (Gerber et al., 2008; Kadaba et al., 2008). When cytosolic concentrations of the transported substrate are high, and no further transport is needed, substrate binding to the regulatory sites keeps the NBDs dissociated from each other, in an inhibited state. This type of regulation has been named “trans-inhibition,” a name that may appear confusing because the regulatory site is located on the cis-side of the membrane.
ECF-type ABC importers.
In ECF-type ABC importers, one of the TMDs (the S-component) binds the substrate without the need of an SBP. S-components have very high affinities for their substrates, with dissociation constants in the low or subnanomolar range (Duurkens et al., 2007; Eudes et al., 2008; Erkens and Slotboom, 2010; Berntsson et al., 2012). To date, four crystal structures of individual S-components with bound substrates (RibU, ThiT, BioY, and NikM for uptake of riboflavin, thiamine, biotin, and Ni2+, respectively; Table 1), as well as two complete substrate-free ECF transporters for folate and pyridoxin uptake, have been elucidated. The crystal structures of the solitary S-components (in the absence of the other subunits) indicate that the substrates are bound to a site located near the extracellular surface of the protein (Zhang et al., 2010; Erkens et al., 2011; Berntsson et al., 2012; Yu et al., 2013). Access to the binding site from the external solution requires movement of the flexible loop between TM helices 1 and 2, which serves as a lid for the binding site (Fig. 6) (Majsnerowska et al., 2013).
Figure 6.
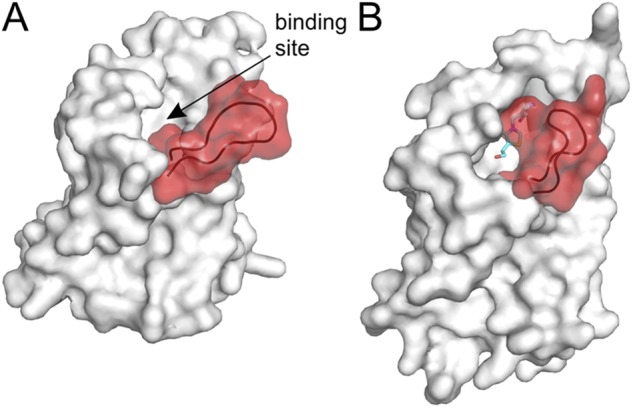
Structural changes in the S-component of ECF transporters (substrate-binding TM protein of ECF transporters). The loop between TM helices 1 and 2 is highlighted in red. (A) S-component from the ECF–HmpT complex (Protein Data Bank accession no. 4HZU) in the substrate-free form. The shape of the binding pocket is visible, indicated by the arrow. (B) Thiamin-specific ThiT (Protein Data Bank accession no. 3RLB) in the substrate-bound state. The loop between TM helices 1 and 2 closes the binding pocket. The substrate thiamin is shown as sticks.
It must be noted though that it is difficult to unambiguously determine the membrane orientation of the S-components in the absence of the NBDs and the EcfT subunit based solely on the crystal structures of these proteins in detergent solution. However, the predicted orientation is supported by molecular dynamics simulations of the solitary S-components, which are consistent with the interpretation that the substrate-binding site is located close to the extracellular side of the membrane (Majsnerowska et al., 2013; Song et al., 2013).
No substrates were found bound to the S-components in the structures of the complete (four-subunit) ECF transporters (Wang et al., 2013; Xu et al., 2013). Surprisingly, in the context of the complete complexes, the empty binding sites of the S-components are located close to the cytoplasmic side of the membrane, because the S-components have toppled over compared with the (predicted) membrane orientation of the solitary S-components. The toppling seems to be essential for the alternating access mechanism in ECF transporters.
ABC exporters.
Based on the crystal structures, the substrate-binding sites in ABC exporters are poorly defined compared with those of ABC importers. A possible cause is that some exporters are multidrug transporters, which bind many structurally different substrates, and are unlikely to have a single well-defined binding site (Ambudkar et al., 2003). In addition, the location of the binding site may not be conserved among the different members of the exporter family, because some exporters bind the substrate from the cis-side and others take up the substrate from the lipid bilayer.
There are no crystal structures available of exporters in complex with transported substrates, but in P-glycoprotein of mouse, the binding sites for two inhibitors have been located (Aller et al., 2009; Li et al., 2014). These binding sites probably overlap partially with substrate-binding sites. The inhibitor-binding sites are located in the membrane-spanning part of the TMDs and are lined with hydrophobic and aromatic residues (in accordance with the fact that P-glycoprotein substrates are hydrophobic). It must be noted that the original crystal structures of mouse P-glycoprotein contained errors, which had to be corrected (Li et al., 2014).
Mechanistic diversity
The structural diversity in the ABC transporter superfamily suggests differences in transport mechanisms. Below, we will briefly discuss some mechanistic features of a Type I importer (the maltose importer MalEFGK2), a Type II importer (the cobalamin importer BtuC2D2F), ECF transporters (represented by ECF-FolT and ECF-HmpT, and the S-components ThiT, RibU, BioY, and NikM), and exporters (represented by two bacterial proteins, Sav18662 and TM288/287). This selection of ABC transporters is based on the availability of high quality crystal structures and biochemical data, but it is unlikely to cover the entire mechanistic diversity of the ABC transporter superfamily. Notably, it is possible that there are also mechanistic differences between members of the ABC transporter family that share the same fold. Because many separate reviews and overviews have been published over the past few years on the mechanistic details of each of the selected transporters (for instance, Davidson et al., 2008; Rees et al., 2009; Chen, 2013; Zhang, 2013; Slotboom, 2014), we will only discuss the gross differences between the four types.
Type I ABC importers.
The maltose transporter MalEFGK2 from E. coli is one of the best-characterized Type I importers. MalF and MalG are the TMDs, MalK2 is the homodimer of NBDs, and MalE is the periplasmic maltose-binding protein (MBP). The protein has been captured in crystals in several states (Table 1 and Fig. 7), allowing deduction of a tentative mechanism of transport (Chen, 2013).
Figure 7.

The transport mechanism of Type I importers (exemplified by MalEFGK2) based on the available structures (A) and in schematic representation (B). Coloring is as in Fig. 1. Structures have been determined for the inward-facing, pre-translocation, and outward-facing conformations (Protein Data Bank accession nos.: 4JBW, 4KHZ, and 4KI0; see Table 1 for details of all available structures of ABC transporters). Substrate-loaded MalE docks onto the resting MalFGK2 transporter. This brings the NBDs closer to each other, allows ATP binding, and consequently causes MalK closure and TMD reorientation (outward-facing), leading to MBP opening. The released substrate diffuses toward the binding site in TMD, followed by ATP hydrolysis and resetting of the transporter into the inward-facing state.
In the absence of nucleotides, the two NBDs are separated and the two TMDs line a cavity that is accessible from the cytoplasmic side of the membrane and sealed by a hydrophobic gate on the outside (periplasmic side). The binding of a substrate-loaded, MBP (MalE) causes a conformational change in the TMDs and the NBDs. The NBDs come into closer proximity (the pre-translocation state), which allows binding of ATP. When ATP binds, the NBD dimer closes, with the two ATP molecules located at the interface. In this state (catalytic intermediate state), the cavity between the two TMDs is sealed on the cytoplasmic side by a gate formed by the TMDs themselves. The cavity is open toward the outside, but the MBP is still attached and prevents access between the cavity and the periplasm. The MBP is in an open conformation and has released the substrate maltose into the cavity between the TMDs, where it binds to a specific site on MalF. ATP hydrolysis does not appear to affect this state, but subsequent release of Pi and ADP is expected to allow the TMDs to return into a conformation with the cavity exposed to the cytoplasm, which will result in release of the maltose and completion of the transport cycle.
The sequence of events suggested by the crystal structures is in good agreement with a large number of biochemical experiments as well as structural studies using spectroscopic techniques (such as electron paramagnetic resonance) (Davidson et al., 1992; Chen et al., 2001; Lu et al., 2005; Grote et al., 2008, 2009; Orelle et al., 2008, 2010; Bordignon et al., 2010; Jacso et al., 2012; Böhm et al., 2013; Chen, 2013), but further refinement of the model may be needed to explain details. It must be noted that Bao and Duong (2013, 2012) has proposed a radically different mechanistic model, which cannot be reconciled with the mechanism described above.
Type II ABC importers.
The vitamin B12 transporter BtuC2D2F from E. coli is the best-characterized Type II importer, with crystal structures available in three states (for a recent overview of the mechanism see Korkhov et al., 2012b). The NBDs (BtuC subunits), TMDs (BtuD subunits), and the periplasmic SBP (BtuF) adopt different conformations depending on whether the transported substrate and nucleotides are present (Fig. 8).
Figure 8.

The transport mechanism of Type II importers (exemplified by the BtuC2D2F transporter) based on the available structures (A) and in schematic representation (B). Coloring is as in Fig. 1. Structures have been determined for an outward-open, occluded nucleotide-bound, and closed ATP-free asymmetric transporter (Protein Data Bank accession nos.: 1L7V, 4DBL, and 2QI9; see Table 1 for details of all available structures of ABC transporters). Substrate-loaded BtuF docks onto the resting BtuC2D2 transporter in the outward conformation. ATP binding and concomitant rearrangements in the TMDs lead to the trapping of the substrate between TMDs (substrate was not visible in the crystal structure). The hydrolysis of ATP causes opening of the cytoplasmic gate and allows substrate release, after which the gate is asymmetrically closed, possibly to prevent the leakage of small molecules.
In the absence of BtuF, the transporter BtuC2D2 adopts a conformation in which the cavity between the TMDs is open toward the outside (periplasmic side of the membrane). This cavity is sufficiently large to fit almost the entire molecule of vitamin B12 (cobalamin) and is lined by hydrophobic residues. The cavity is closed at the cytoplasmic side. The outward-facing conformation is adopted in the absence of nucleotides, which is different from the maltose transporter (Type I importer), where the inward-facing conformation is favored in the absence of nucleotides.
The binding of the ATP analogue AMP-PNP together with the SBP BtuF results in a transporter complex in which the translocation pathway is sealed from both ends by the TMDs, and a central occluded cavity, large enough to harbor the entire molecule of cobalamin, is present. This cavity does not contain a specific binding site for cobalamin (compared with the Type I maltose transporter where a maltose-binding site is present in MalF), indicating that the affinity of BtuC2D2 for its substrate may be low.
To release the substrate from the cavity, the transporter needs to open the cytoplasmic gate. Opening probably takes place upon ATP hydrolysis and release of Pi and ADP, which allows dissociation of the NBD dimer and propagation of the conformational changes to the TMDs via the coupling helices. In this way, the substrate is squeezed out of the cavity and the complex converts into an asymmetric state in which the translocation path is collapsed. The latter may be required to prevent the leakage of small molecules. In detergent solution, this collapsed transporter has an exceptionally high affinity for BtuF (KD of 10−13 M) (Lewinson et al., 2010). It is likely that ATP binding is required to release the BtuF subunit.
The tight binding of the SBP is characteristic for Type II transporters. For the maltose (MalFGK2) and other ABC Type I importers, the SBPs have a low affinity for the TMDs of the transporter and only interact with the transporter transiently (Lewinson et al., 2010; Vigonsky et al., 2013).
Type III or ECF-type importers.
These have recently been reviewed in Eitinger et al. (2011), Erkens et al. (2012), Zhang (2013), and Slotboom (2014). Two crystal structures are available for complete ECF transporter complexes: ECF–FolT (specific for folate transport) and ECF–HmpT (predicted to transport pyridoxin) from the bacterium Lactobacillus brevis. One of the TMDs (the EcfT subunit) and the two NBDs (the EcfA and EcfA’ subunits) form a module that is identical in the two complexes and can associate with different S-components (FolT and HmpT, and five more S-components present in the bacterium; Rodionov et al., 2009; Slotboom, 2014). The modular nature (different S-components interacting with the same EcfAA’T complex) is a characteristic feature of many ECF transporters. Both crystallized complexes are in the same conformational state, with neither nucleotides nor transported substrates bound.
The orientation of the S-components FolT and HmpT in the complexes is highly unusual. Crystal structures of the S-components in the absence of the EcfAA’T module (Zhang et al., 2010; Erkens et al., 2011; Berntsson et al., 2012; Yu et al., 2013) indicated that the N and C termini of the proteins are exposed to the cytosol, and that the substrate-binding site is located close to the extracellular (or cytoplasmic) space (see above and Fig. 6). Surprisingly, the FolT and HmpT subunits in the complexes have “toppled over” by almost 90°, and helices 1–4 lie parallel to the membrane plane, an unprecedented orientation for membrane proteins. In the crystallized ECF–FolT and ECF–HmpT complexes, the binding sites in the S-components are empty and accessible from the cytoplasm. Thus, it appears that the toppling of the S-components is mechanistically important, as it physically moves the bound substrate from the outside to the cytoplasm (Fig. 9).
Figure 9.

Possible transport mechanism of ECF transporters (Type III importers) based on the available structures (A) and in schematic representation (B). Structures have been determined of the nucleotide- and substrate-free transporter (ECF-HmpT, Protein Data Bank accession no. 4HZU, is shown here; see Table 1 for details of all available structures of ABC transporters). The S-component (orange) is predicted to rotate in the membrane (“topple over”) to bring the substrate from the trans-side to the cis-side.
The coupling mechanism between nucleotide binding, hydrolysis and release in the NBDs, and the hypothesized toppling of the S-components is not clear, because the crystal structure of only a single conformational state of the complexes has been captured: the nucleotide-free state. Nonetheless, this crystal structure provides some clues about the mechanism of transport, as it shows that the NBDs interact almost exclusively with the EcfT subunit, and not directly with the S-component. The S-components interact extensively with the EcfT subunit only. Therefore, the coupling between events in the NBDs and the proposed toppling of the S-components must be mediated by EcfT. EcfT has two long X-shaped α helices that interact with the NBDs on one side and the S-component on the other. A scissor-like movement of these helices may affect the orientation of the S-components. It is also possible that the S-components dissociate from the complex during the catalytic cycle (Henderson et al., 1979; Rodionov et al., 2009; Slotboom, 2014). The solitary S-components may then spontaneously assume an orientation with the substrate-binding site located on the trans-side of the membrane.
ABC exporters
In all crystallized ABC exporters, the NBDs are linked to the TMDs in a single protein. Exporters have been captured in two different states: the outward-facing conformation, represented by Sav18662 from Staphylococcus aureus, which is a homodimer (Dawson and Locher, 2006), and the inward-facing conformation, represented by TM287/288 from Thermotoga maritima, which is a heterodimer (Fig. 10) (Hohl et al., 2012). It must be noted that a few other crystal structure of ABC exporters (MsbA and mouse P-glycoprotein) initially contained large errors and had to be corrected (Ward et al., 2007; Li et al., 2014).
Figure 10.

The transport mechanism of exporters (exemplified by the structures of Sav18662 and TM287/288) based on the available structures (A) and in schematic representation (B). Coloring is as in Fig. 1. Structures of outward-open and nucleotide-bound inward-facing transporter are shown (Protein Data Bank accession nos. 2HYD and 3QF4; see Table 1 for details of all available structures of ABC transporters). The hydrolysis of ATP in the outward-open conformation results in the reorientation of TMDs, leading to the inward-open conformation where substrate and ATP binding take place. This in turn orients the transporter back into the outward-open conformation, and the substrate is expelled.
In the inward-facing state, the transported substrates can be bound in a cavity between the two subunits, although the exact location of the binding sites has not been resolved with x-ray crystallography. In the outward-facing state, the NBDs are in close contact and have nucleotides bound at their interface. The cavity between the TMDs is now exposed to the outside, and substrate can be expelled. Compared with Type I and II importers, the crystal structures of exporters have shed less light on the interplay between the binding of transported substrates, the conformational changes in the TMDs, the binding and hydrolysis of ATP, and the release of Pi and ADP.
A distinct feature of some exporters with nonidentical NBDs is the presence of a degenerate nucleotide-binding site (Procko et al., 2009; Hohl et al., 2012). In such exporters, one site is referred to as the consensus site (and contains all the conserved motifs usually found in NBDs; see above), and the second is termed degenerate (because of the deviations from the consensus sequence). The heterodimeric ABC exporter TM287/288 from T. maritima (crystallized in the inward conformation) is one of the transporters with a degenerate and a consensus site. In the reported structure, the two NBDs are not fully disengaged but still interact with each other. A bound nucleotide (AMP-PNP) was found only in the degenerate site, and its presence may prevent further dissociation of the NBDs. Based on analysis of interactions, Hohl et al. (2012) suggest that ATP hydrolysis is blocked at the degenerate site as a result of the increased distance to γ-phosphate caused by replacement of the glutamate from Walker B motif to aspartate. Whether the ABC exporters that have two consensus sites are mechanistically different from the ones with a degenerate site remains to be elucidated.
Concluding remarks
The recent surge in crystal structures of ABC transporters has revealed a remarkable structural diversity and suggests unanticipated mechanistic diversity in the superfamily. Crystal structures alone obviously are not sufficient to elucidate transport mechanisms. They provide snapshots of “states.” The number of states that can be crystallized may not be enough to describe the complete transport cycle. Moreover, what a state represents, and how such a state relates to physiological conditions, is often loosely defined. For example, the name “resting state” has been used to describe the nucleotide-free transporters, but it is unlikely that nucleotide-free conditions are physiologically relevant. In addition, the names “high energy state” and “intermediate state” are sometimes used without solid definition.
Because crystal structures are determined in detergent solution, an environment very different than that of the lipid bilayer, it is essential that models based on crystal structures be tested in membrane environments. Spectroscopic techniques such as electron paramagnetic resonance and (single-molecule) FRET are suitable to obtain structural and dynamic information in lipid bilayers (Erkens et al., 2013; Hänelt et al., 2013; Majsnerowska et al., 2013). Such techniques may reveal states that cannot be captured in crystals (Böhm et al., 2013). In addition, classical biochemical experiments in liposomes can be used to test mechanistic models.
Finally, a few high profile examples of crystal structures with huge errors have made many non-crystallographers skeptical about the validity and relevance of any crystal structure (Chang et al., 2006). Although such errors have damaged the reputation of crystallography, it is also important to note that other crystallographers have detected these errors, leading to corrections or retractions. Arguably, crystal structures are some of the most scrutinized biochemical data, which—eventually—warrants high standards. The currently available high quality structures of ABC transporters underscore the powerful role of protein crystallography to provide mechanistic insight. Therefore, crystal structures of new states of the four known ABC transporter types, as well as structures of novel ABC transporter folds, are very welcome, and expected to further advance our understanding of this large, important, and fascinating superfamily of membrane transporters.
Acknowledgments
This work was funded by The Netherlands Organisation for Scientific Research (TopTalent grant to J. ter Beek, and vici and ECHO grants to D.J. Slotboom), and the European Research Council (starting grant to D.J. Slotboom).
The authors declare no competing financial interests.
Elizabeth M. Adler served as editor.
Footnotes
Abbreviations used in this paper:
- ABC
- ATP-binding cassette
- ECF
- energy coupling factor
- ICL
- intracellular loop
- MBP
- maltose-binding protein
- NBD
- nucleotide-binding domain
- Pi
- orthophosphate
- SBD
- substrate-binding domain
- SBP
- substrate-binding protein
- TM
- transmembrane
- TMD
- TM domain
References
- Aleksandrov A.A., Aleksandrov L.A., Riordan J.R. 2007. CFTR (ABCC7) is a hydrolyzable-ligand-gated channel. Pflugers Arch. 453:693–702 10.1007/s00424-006-0140-z [DOI] [PubMed] [Google Scholar]
- Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R., Harrell P.M., Trinh Y.T., Zhang Q., Urbatsch I.L., Chang G. 2009. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 323:1718–1722 10.1126/science.1168750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar S.V., Kimchi-Sarfaty C., Sauna Z.E., Gottesman M.M. 2003. P-glycoprotein: from genomics to mechanism. Oncogene. 22:7468–7485 10.1038/sj.onc.1206948 [DOI] [PubMed] [Google Scholar]
- Ames G.F., Mimura C.S., Shyamala V. 1990. Bacterial periplasmic permeases belong to a family of transport proteins operating from Escherichia coli to human: Traffic ATPases. FEMS Microbiol. Rev. 6:429–446 10.1016/S0168-6445(05)80008-7 [DOI] [PubMed] [Google Scholar]
- Bao H., Duong F. 2012. Discovery of an auto-regulation mechanism for the maltose ABC transporter MalFGK2. PLoS ONE. 7:e34836 10.1371/journal.pone.0034836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H., Duong F. 2013. ATP alone triggers the outward facing conformation of the maltose ATP-binding cassette transporter. J. Biol. Chem. 288:3439–3448 10.1074/jbc.M112.431932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berntsson R.P.-A., Smits S.H.J., Schmitt L., Slotboom D.J., Poolman B. 2010. A structural classification of substrate-binding proteins. FEBS Lett. 584:2606–2617 10.1016/j.febslet.2010.04.043 [DOI] [PubMed] [Google Scholar]
- Berntsson R.P.-A., ter Beek J., Majsnerowska M., Duurkens R.H., Puri P., Poolman B., Slotboom D.J. 2012. Structural divergence of paralogous S components from ECF-type ABC transporters. Proc. Natl. Acad. Sci. USA. 109:13990–13995 10.1073/pnas.1203219109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biemans-Oldehinkel E., Doeven M.K., Poolman B. 2006. ABC transporter architecture and regulatory roles of accessory domains. FEBS Lett. 580:1023–1035 10.1016/j.febslet.2005.11.079 [DOI] [PubMed] [Google Scholar]
- Boël G., Smith P.C., Ning W., Englander M.T., Chen B., Hashem Y., Testa A.J., Fischer J.J., Wieden H.-J., Frank J., et al. 2014. The ABC-F protein EttA gates ribosome entry into the translation elongation cycle. Nat. Struct. Mol. Biol. 21:143–151 10.1038/nsmb.2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böhm S., Licht A., Wuttge S., Schneider E., Bordignon E. 2013. Conformational plasticity of the type I maltose ABC importer. Proc. Natl. Acad. Sci. USA. 110:5492–5497 10.1073/pnas.1217745110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon E., Grote M., Schneider E. 2010. The maltose ATP-binding cassette transporter in the 21st century—towards a structural dynamic perspective on its mode of action. Mol. Microbiol. 77:1354–1366 10.1111/j.1365-2958.2010.07319.x [DOI] [PubMed] [Google Scholar]
- Bryan J., Muñoz A., Zhang X., Düfer M., Drews G., Krippeit-Drews P., Aguilar-Bryan L. 2007. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 453:703–718 10.1007/s00424-006-0116-z [DOI] [PubMed] [Google Scholar]
- Chang G., Roth C.B., Reyes C.L., Pornillos O., Chen Y.-J., Chen A.P. 2006. Retraction. Science. 314:1875 10.1126/science.314.5807.1875b [DOI] [PubMed] [Google Scholar]
- Chen J. 2013. Molecular mechanism of the Escherichia coli maltose transporter. Curr. Opin. Struct. Biol. 23:492–498 10.1016/j.sbi.2013.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Sharma S., Quiocho F.A., Davidson A.L. 2001. Trapping the transition state of an ATP-binding cassette transporter: Evidence for a concerted mechanism of maltose transport. Proc. Natl. Acad. Sci. USA. 98:1525–1530 10.1073/pnas.98.4.1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Oldham M.L., Davidson A.L., Chen J. 2013. Carbon catabolite repression of the maltose transporter revealed by X-ray crystallography. Nature. 499:364–368 10.1038/nature12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.L., Sharma S. 1997. Mutation of a single MalK subunit severely impairs maltose transport activity in Escherichia coli. J. Bacteriol. 179:5458–5464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.L., Shuman H.A., Nikaido H. 1992. Mechanism of maltose transport in Escherichia coli: transmembrane signaling by periplasmic binding proteins. Proc. Natl. Acad. Sci. USA. 89:2360–2364 10.1073/pnas.89.6.2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson A.L., Dassa E., Orelle C., Chen J. 2008. Structure, function, and evolution of bacterial ATP-binding cassette systems. Microbiol. Mol. Biol. Rev. 72:317–364 10.1128/MMBR.00031-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson R.J.P., Locher K.P. 2006. Structure of a bacterial multidrug ABC transporter. Nature. 443:180–185 10.1038/nature05155 [DOI] [PubMed] [Google Scholar]
- Dawson R.J.P., Locher K.P. 2007. Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581:935–938 10.1016/j.febslet.2007.01.073 [DOI] [PubMed] [Google Scholar]
- Dawson R.J.P., Hollenstein K., Locher K.P. 2007. Uptake or extrusion: crystal structures of full ABC transporters suggest a common mechanism. Mol. Microbiol. 65:250–257 10.1111/j.1365-2958.2007.05792.x [DOI] [PubMed] [Google Scholar]
- Duurkens R.H., Tol M.B., Geertsma E.R., Permentier H.P., Slotboom D.J. 2007. Flavin binding to the high affinity riboflavin transporter RibU. J. Biol. Chem. 282:10380–10386 10.1074/jbc.M608583200 [DOI] [PubMed] [Google Scholar]
- Eitinger T., Rodionov D.A., Grote M., Schneider E. 2011. Canonical and ECF-type ATP-binding cassette importers in prokaryotes: diversity in modular organization and cellular functions. FEMS Microbiol. Rev. 35:3–67 10.1111/j.1574-6976.2010.00230.x [DOI] [PubMed] [Google Scholar]
- Erkens G.B., Slotboom D.J. 2010. Biochemical characterization of ThiT from Lactococcus lactis: A thiamin transporter with picomolar substrate binding affinity. Biochemistry. 49:3203–3212 10.1021/bi100154r [DOI] [PubMed] [Google Scholar]
- Erkens G.B., Berntsson R.P.-A., Fulyani F., Majsnerowska M., Vujičić-Žagar A., Ter Beek J., Poolman B., Slotboom D.J. 2011. The structural basis of modularity in ECF-type ABC transporters. Nat. Struct. Mol. Biol. 18:755–760 10.1038/nsmb.2073 [DOI] [PubMed] [Google Scholar]
- Erkens G.B., Majsnerowska M., ter Beek J., Slotboom D.J. 2012. Energy coupling factor-type ABC transporters for vitamin uptake in prokaryotes. Biochemistry. 51:4390–4396 10.1021/bi300504v [DOI] [PubMed] [Google Scholar]
- Erkens G.B., Hänelt I., Goudsmits J.M.H., Slotboom D.J., van Oijen A.M. 2013. Unsynchronised subunit motion in single trimeric sodium-coupled aspartate transporters. Nature. 502:119–123 10.1038/nature12538 [DOI] [PubMed] [Google Scholar]
- Eudes A., Erkens G.B., Slotboom D.J., Rodionov D.A., Naponelli V., Hanson A.D. 2008. Identification of genes encoding the folate- and thiamine-binding membrane proteins in Firmicutes. J. Bacteriol. 190:7591–7594 10.1128/JB.01070-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George A.M., Jones P.M. 2012. Perspectives on the structure-function of ABC transporters: The Switch and Constant Contact models. Prog. Biophys. Mol. Biol. 109:95–107 10.1016/j.pbiomolbio.2012.06.003 [DOI] [PubMed] [Google Scholar]
- Gerber S., Comellas-Bigler M., Goetz B.A., Locher K.P. 2008. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 321:246–250 10.1126/science.1156213 [DOI] [PubMed] [Google Scholar]
- Grote M., Bordignon E., Polyhach Y., Jeschke G., Steinhoff H.-J., Schneider E. 2008. A comparative electron paramagnetic resonance study of the nucleotide-binding domains’ catalytic cycle in the assembled maltose ATP-binding cassette importer. Biophys. J. 95:2924–2938 10.1529/biophysj.108.132456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote M., Polyhach Y., Jeschke G., Steinhoff H.-J., Schneider E., Bordignon E. 2009. Transmembrane signaling in the maltose ABC transporter MalFGK2-E: Periplasmic MalF-P2 loop communicates substrate availability to the ATP-bound MalK dimer. J. Biol. Chem. 284:17521–17526 10.1074/jbc.M109.006270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänelt I., Wunnicke D., Bordignon E., Steinhoff H.-J., Slotboom D.J. 2013. Conformational heterogeneity of the aspartate transporter Glt(Ph). Nat. Struct. Mol. Biol. 20:210–214 10.1038/nsmb.2471 [DOI] [PubMed] [Google Scholar]
- Henderson G.B., Zevely E.M., Huennekens F.M. 1979. Mechanism of folate transport in Lactobacillus casei: Evidence for a component shared with the thiamine and biotin transport systems. J. Bacteriol. 137:1308–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins C.F. 1992. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 8:67–113 10.1146/annurev.cb.08.110192.000435 [DOI] [PubMed] [Google Scholar]
- Higgins C.F., Ames G.F. 1981. Two periplasmic transport proteins which interact with a common membrane receptor show extensive homology: complete nucleotide sequences. Proc. Natl. Acad. Sci. USA. 78:6038–6042 10.1073/pnas.78.10.6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl M., Briand C., Grütter M.G., Seeger M.A. 2012. Crystal structure of a heterodimeric ABC transporter in its inward-facing conformation. Nat. Struct. Mol. Biol. 19:395–402 10.1038/nsmb.2267 [DOI] [PubMed] [Google Scholar]
- Hollenstein K., Frei D.C., Locher K.P. 2007. Structure of an ABC transporter in complex with its binding protein. Nature. 446:213–216 10.1038/nature05626 [DOI] [PubMed] [Google Scholar]
- Hvorup R.N., Goetz B.A., Niederer M., Hollenstein K., Perozo E., Locher K.P. 2007. Asymmetry in the structure of the ABC transporter-binding protein complex BtuCD-BtuF. Science. 317:1387–1390 10.1126/science.1145950 [DOI] [PubMed] [Google Scholar]
- Jacso T., Schneider E., Rupp B., Reif B. 2012. Substrate transport activation is mediated through second periplasmic loop of transmembrane protein MalF in maltose transport complex of Escherichia coli. J. Biol. Chem. 287:17040–17049 10.1074/jbc.M112.340679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky O. 1966. Simple allosteric model for membrane pumps. Nature. 211:969–970 10.1038/211969a0 [DOI] [PubMed] [Google Scholar]
- Jin M.S., Oldham M.L., Zhang Q., Chen J. 2012. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 490:566–569 10.1038/nature11448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson E., Nguyen P.T., Yeates T.O., Rees D.C. 2012. Inward facing conformations of the MetNI methionine ABC transporter: Implications for the mechanism of transinhibition. Protein Sci. 21:84–96 10.1002/pro.765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.M., George A.M. 2013. Mechanism of the ABC transporter ATPase domains: catalytic models and the biochemical and biophysical record. Crit. Rev. Biochem. Mol. Biol. 48:39–50 10.3109/10409238.2012.735644 [DOI] [PubMed] [Google Scholar]
- Kadaba N.S., Kaiser J.T., Johnson E., Lee A., Rees D.C. 2008. The high-affinity E. coli methionine ABC transporter: Structure and allosteric regulation. Science. 321:250–253 10.1126/science.1157987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare D., Oldham M.L., Orelle C., Davidson A.L., Chen J. 2009. Alternating access in maltose transporter mediated by rigid-body rotations. Mol. Cell. 33:528–536 10.1016/j.molcel.2009.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkhov V.M., Mireku S.A., Hvorup R.N., Locher K.P. 2012a. Asymmetric states of vitamin B12 transporter BtuCD are not discriminated by its cognate substrate binding protein BtuF. FEBS Lett. 586:972–976 10.1016/j.febslet.2012.02.042 [DOI] [PubMed] [Google Scholar]
- Korkhov V.M., Mireku S.A., Locher K.P. 2012b. Structure of AMP-PNP-bound vitamin B12 transporter BtuCD-F. Nature. 490:367–372 10.1038/nature11442 [DOI] [PubMed] [Google Scholar]
- Leonard B.A., Podbielski A., Hedberg P.J., Dunny G.M. 1996. Enterococcus faecalis pheromone binding protein, PrgZ, recruits a chromosomal oligopeptide permease system to import sex pheromone cCF10 for induction of conjugation. Proc. Natl. Acad. Sci. USA. 93:260–264 10.1073/pnas.93.1.260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewinson O., Lee A.T., Locher K.P., Rees D.C. 2010. A distinct mechanism for the ABC transporter BtuCD-BtuF revealed by the dynamics of complex formation. Nat. Struct. Mol. Biol. 17:332–338 10.1038/nsmb.1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis V.G., Ween M.P., McDevitt C.A. 2012. The role of ATP-binding cassette transporters in bacterial pathogenicity. Protoplasma. 249:919–942 10.1007/s00709-011-0360-8 [DOI] [PubMed] [Google Scholar]
- Li J., Jaimes K.F., Aller S.G. 2014. Refined structures of mouse P-glycoprotein. Protein Sci. 23:34–46 10.1002/pro.2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher K.P., Lee A.T., Rees D.C. 2002. The E. coli BtuCD structure: A framework for ABC transporter architecture and mechanism. Science. 296:1091–1098 10.1126/science.1071142 [DOI] [PubMed] [Google Scholar]
- Lu G., Westbrooks J.M., Davidson A.L., Chen J. 2005. ATP hydrolysis is required to reset the ATP-binding cassette dimer into the resting-state conformation. Proc. Natl. Acad. Sci. USA. 102:17969–17974 10.1073/pnas.0506039102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majsnerowska M., Hänelt I., Wunnicke D., Schäfer L.V., Steinhoff H.-J., Slotboom D.J. 2013. Substrate-induced conformational changes in the S-component ThiT from an energy coupling factor transporter. Structure. 21:861–867 10.1016/j.str.2013.03.007 [DOI] [PubMed] [Google Scholar]
- Mittal A., Böhm S., Grütter M.G., Bordignon E., Seeger M.A. 2012. Asymmetry in the homodimeric ABC transporter MsbA recognized by a DARPin. J. Biol. Chem. 287:20395–20406 10.1074/jbc.M112.359794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourez M., Hofnung M., Dassa E. 1997. Subunit interactions in ABC transporters: a conserved sequence in hydrophobic membrane proteins of periplasmic permeases defines an important site of interaction with the ATPase subunits. EMBO J. 16:3066–3077 10.1093/emboj/16.11.3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido K., Ames G.F. 1999. One intact ATP-binding subunit is sufficient to support ATP hydrolysis and translocation in an ABC transporter, the histidine permease. J. Biol. Chem. 274:26727–26735 10.1074/jbc.274.38.26727 [DOI] [PubMed] [Google Scholar]
- Oldham M.L., Chen J. 2011a. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science. 332:1202–1205 10.1126/science.1200767 [DOI] [PubMed] [Google Scholar]
- Oldham M.L., Chen J. 2011b. Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 108:15152–15156 10.1073/pnas.1108858108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M.L., Khare D., Quiocho F.A., Davidson A.L., Chen J. 2007. Crystal structure of a catalytic intermediate of the maltose transporter. Nature. 450:515–521 10.1038/nature06264 [DOI] [PubMed] [Google Scholar]
- Oldham M.L., Chen S., Chen J. 2013. Structural basis for substrate specificity in the Escherichia coli maltose transport system. Proc. Natl. Acad. Sci. USA. 110:18132–18137 10.1073/pnas.1311407110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C., Ayvaz T., Everly R.M., Klug C.S., Davidson A.L. 2008. Both maltose-binding protein and ATP are required for nucleotide-binding domain closure in the intact maltose ABC transporter. Proc. Natl. Acad. Sci. USA. 105:12837–12842 10.1073/pnas.0803799105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C., Alvarez F.J.D., Oldham M.L., Orelle A., Wiley T.E., Chen J., Davidson A.L. 2010. Dynamics of α-helical subdomain rotation in the intact maltose ATP-binding cassette transporter. Proc. Natl. Acad. Sci. USA. 107:20293–20298 10.1073/pnas.1006544107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C., Holland I.B., Schmitt L. 2006. The motor domains of ABC-transporters. Naunyn Schmiedebergs Arch. Pharmacol. 372:385–399 10.1007/s00210-005-0031-4 [DOI] [PubMed] [Google Scholar]
- Parcej D., Tampé R. 2010. ABC proteins in antigen translocation and viral inhibition. Nat. Chem. Biol. 6:572–580 10.1038/nchembio.410 [DOI] [PubMed] [Google Scholar]
- Park J.T., Raychaudhuri D., Li H., Normark S., Mengin-Lecreulx D. 1998. MppA, a periplasmic binding protein essential for import of the bacterial cell wall peptide l-alanyl-γ-d-glutamyl-meso-diaminopimelate. J. Bacteriol. 180:1215–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzlaff J.S., van der Heide T., Poolman B. 2003. The ATP/substrate stoichiometry of the ATP-binding cassette (ABC) transporter OpuA. J. Biol. Chem. 278:29546–29551 10.1074/jbc.M304796200 [DOI] [PubMed] [Google Scholar]
- Pinkett H.W., Lee A.T., Lum P., Locher K.P., Rees D.C. 2007. An inward-facing conformation of a putative metal-chelate-type ABC transporter. Science. 315:373–377 10.1126/science.1133488 [DOI] [PubMed] [Google Scholar]
- Pittelkow M., Tschapek B., Smits S.H.J., Schmitt L., Bremer E. 2011. The crystal structure of the substrate-binding protein OpuBC from Bacillus subtilis in complex with choline. J. Mol. Biol. 411:53–67 10.1016/j.jmb.2011.05.037 [DOI] [PubMed] [Google Scholar]
- Procko E., O’Mara M.L., Bennett W.F.D., Tieleman D.P., Gaudet R. 2009. The mechanism of ABC transporters: general lessons from structural and functional studies of an antigenic peptide transporter. FASEB J. 23:1287–1302 10.1096/fj.08-121855 [DOI] [PubMed] [Google Scholar]
- Quiocho F.A., Ledvina P.S. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol. Microbiol. 20:17–25 10.1111/j.1365-2958.1996.tb02484.x [DOI] [PubMed] [Google Scholar]
- Rees D.C., Johnson E., Lewinson O. 2009. ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10:218–227 10.1038/nrm2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov D.A., Hebbeln P., Eudes A., ter Beek J., Rodionova I.A., Erkens G.B., Slotboom D.J., Gelfand M.S., Osterman A.L., Hanson A.D., Eitinger T. 2009. A novel class of modular transporters for vitamins in prokaryotes. J. Bacteriol. 191:42–51 10.1128/JB.01208-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R.R., Aduse-Opoku J., Sutcliffe I.C., Tao L., Ferretti J.J. 1992. A binding protein-dependent transport system in Streptococcus mutans responsible for multiple sugar metabolism. J. Biol. Chem. 267:4631–4637 [PubMed] [Google Scholar]
- Shintre C.A., Pike A.C.W., Li Q., Kim J.-I., Barr A.J., Goubin S., Shrestha L., Yang J., Berridge G., Ross J., et al. 2013. Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. USA. 110:9710–9715 10.1073/pnas.1217042110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverton L., Dean M., Moitra K. 2011. Variation and evolution of the ABC transporter genes ABCB1, ABCC1, ABCG2, ABCG5 and ABCG8: implication for pharmacogenetics and disease. Drug Metabol. Drug Interact. 26:169–179 10.1515/DMDI.2011.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotboom D.J. 2014. Structural and mechanistic insights into prokaryotic energy-coupling factor transporters. Nat. Rev. Microbiol. 12:79–87 10.1038/nrmicro3175 [DOI] [PubMed] [Google Scholar]
- Smith P.C., Karpowich N., Millen L., Moody J.E., Rosen J., Thomas P.J., Hunt J.F. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell. 10:139–149 10.1016/S1097-2765(02)00576-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Ji C., Zhang J.Z.H. 2013. Unveiling the gating mechanism of ECF Transporter RibU. Sci Rep. 3:3566 10.1038/srep03566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano E.V., Rajashankar K.R., Hanes J.W., Bale S., Begley T.P., Ealick S.E. 2008. Structural similarities between thiamin-binding protein and thiaminase-I suggest a common ancestor. Biochemistry. 47:1346–1357 10.1021/bi7018282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser D.M., Ames G.F. 1991. Salmonella typhimurium histidine periplasmic permease mutations that allow transport in the absence of histidine-binding proteins. J. Bacteriol. 173:1444–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanford C. 1982. Simple model for the chemical potential change of a transported ion in active transport. Proc. Natl. Acad. Sci. USA. 79:2882–2884 10.1073/pnas.79.9.2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Lee L., Lee A., Rees D.C., Pinkett H.W. 2011. Classification of a Haemophilus influenzae ABC transporter HI1470/71 through its cognate molybdate periplasmic binding protein, MolA. Structure. 19:1701–1710 10.1016/j.str.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdon G., Albers S.V., Dijkstra B.W., Driessen A.J.M., Thunnissen A.-M.W.H. 2003. Crystal structures of the ATPase subunit of the glucose ABC transporter from Sulfolobus solfataricus: Nucleotide-free and nucleotide-bound conformations. J. Mol. Biol. 330:343–358 10.1016/S0022-2836(03)00575-8 [DOI] [PubMed] [Google Scholar]
- Vetter I.R., Wittinghofer A. 1999. Nucleoside triphosphate-binding proteins: different scaffolds to achieve phosphoryl transfer. Q. Rev. Biophys. 32:1–56 10.1017/S0033583599003480 [DOI] [PubMed] [Google Scholar]
- Vigonsky E., Ovcharenko E., Lewinson O. 2013. Two molybdate/tungstate ABC transporters that interact very differently with their substrate binding proteins. Proc. Natl. Acad. Sci. USA. 110:5440–5445 10.1073/pnas.1213598110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Fu G., Pan X., Wu J., Gong X., Wang J., Shi Y. 2013. Structure of a bacterial energy-coupling factor transporter. Nature. 497:272–276 10.1038/nature12045 [DOI] [PubMed] [Google Scholar]
- Ward A., Reyes C.L., Yu J., Roth C.B., Chang G. 2007. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA. 104:19005–19010 10.1073/pnas.0709388104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo J.-S., Zeltina A., Goetz B.A., Locher K.P. 2012. X-ray structure of the Yersinia pestis heme transporter HmuUV. Nat. Struct. Mol. Biol. 19:1310–1315 10.1038/nsmb.2417 [DOI] [PubMed] [Google Scholar]
- Xu K., Zhang M., Zhao Q., Yu F., Guo H., Wang C., He F., Ding J., Zhang P. 2013. Crystal structure of a folate energy-coupling factor transporter from Lactobacillus brevis. Nature. 497:268–271 10.1038/nature12046 [DOI] [PubMed] [Google Scholar]
- Yu Y., Zhou M., Kirsch F., Xu C., Zhang L., Wang Y., Jiang Z., Wang N., Li J., Eitinger T., Yang M. 2013. Planar substrate-binding site dictates the specificity of ECF-type nickel/cobalt transporters. Cell Res. In press 10.1038/cr.2013.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J., Jenewein S., Jumpertz T., Holland I.B., Schmitt L. 2005. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 24:1901–1910 10.1038/sj.emboj.7600657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. 2013. Structure and mechanism of energy-coupling factor transporters. Trends Microbiol. 21:652–659 10.1016/j.tim.2013.09.009 [DOI] [PubMed] [Google Scholar]
- Zhang P., Wang J., Shi Y. 2010. Structure and mechanism of the S component of a bacterial ECF transporter. Nature. 468:717–720 10.1038/nature09488 [DOI] [PubMed] [Google Scholar]