

Abstract
Objective
To investigate the relationship between BRCA mutation status and response to taxane-based chemotherapy, since BRCA mutation carriers with prostate cancer appear to have worse survival than non-carriers and docetaxel improves survival in patients with castration-resistant prostate cancer.
Patients and Methods
We determined BRCA mutation prevalence in 158 Ashkenazi Jewish (AJ) men with castration-resistant prostate cancer. Clinical data were collected as part of an institutional prostate cancer research database and through additional medical record review.
Clinical records and DNA samples were linked through a unique identifier, anonymizing the samples before genetic testing for the AJ BRCA1/2 founder mutations.
Response to taxane-based therapy was defined by the prostate-specific antigen nadir within 12 weeks of therapy.
Results
In all, 88 men received taxane-based treatment, seven of whom were BRCA carriers (three BRCA1, four BRCA2; 8%). Initial response to taxane was available for all seven BRCA carriers and for 69 non-carriers.
Overall, 71% (54/76) of patients responded to treatment, with no significant difference between carriers (57%) and non-carriers (72%) (absolute difference 15%; 95% confidence interval −23% to 53%; P = 0.4).
Among patients with an initial response, the median change in prostate-specific antigen was similar for BRCA carriers (−63%, interquartile range −71% to −57%) and non-carriers (−60%, interquartile range −78% to −35%) (P = 0.6).
At last follow-up, all seven BRCA carriers and 49 non-carriers had died from prostate cancer. One BRCA2 carrier treated with docetaxel plus platinum survived 37 months.
Conclusion
In this small, hypothesis-generating study approximately half of BRCA carriers had a prostate-specific antigen response to taxane-based chemotherapy, suggesting that it is an active therapy in these individuals.
Keywords: castration-resistant prostate cancer, BRCA1, BRCA2, taxane, docetaxel, response
Introduction
Germline BRCA mutations are associated with an increased risk of prostate cancer and appear to predict aggressive disease with carriers showing earlier progression through the prostate cancer clinical states model and increased prostate-cancer-specific mortality than non-carriers [1–5]. The inferior outcome in BRCA carriers with prostate cancer contrasts with the improved outcome reported in BRCA-associated ovarian cancer [6–9] and with the similar outcomes seen in carriers and non-carriers with breast cancer [10].
BRCA1 and BRCA2 are implicated in many pathways that contribute to DNA repair [11], a function that may explain the differential outcome for carriers and non-carriers. It has been proposed that double-strand DNA breaks, acting through the androgen receptor, produce the TMPRSS2–ERG gene fusion that is found in 50% of prostate cancer [12,13]. Androgen signalling appears to induce proximity of the TMPRSS2 and ERG genomic loci, and in one study radiation-induced DNA double-strand breaks then facilitated the TMPRSS2–ERG gene fusion [12]. This gene fusion has been associated with aggressive disease, and it is possible that, in the absence of BRCA, accumulating double-strand breaks facilitate this translocation. A pharmacogenetic effect whereby germline BRCA mutation alters the efficacy of some agents but not of others may also explain the opposing prognostic reports of BRCA status in cancers treated with different agents.
Preclinical and clinical data consistently show increased platinum sensitivity among BRCA1 and BRCA2 mutation carriers with ovarian cancer [14], and a similar effect has been reported in breast cancer [15]. It has been proposed that response to taxanes requires functional BRCA1 which activates the mitotic checkpoint and induces apoptosis [16–20]; however, the chemo-modulatory effect of BRCA status on taxane sensitivity is not known. Inhibition of BRCA1 in ovarian and breast cancer cell lines has been reported to decrease the apoptotic response to docetaxel and paclitaxel leading to increased chemo-resistance [17]. In contrast, two ovarian cancer cell line studies suggest that increased BRCA1 expression confers increased sensitivity to paclitaxel [21,22]. Clinical data in this setting are very limited with only two small breast cancer studies suggesting that BRCA1-associated breast cancer is less responsive to taxanes [23,24]. An association between BRCA2 mutation status and response to taxanes has not been reported.
Docetaxel is the only treatment that leads to an improvement in survival in men with castration-resistant prostate cancer (CRPC) [25]. We investigated whether BRCA1/2 mutation status conferred resistance or sensitivity to taxane-based therapy in patients with advanced prostate cancer.
Patients and Methods
The case records of 832 Ashkenazi Jewish men treated for localized prostate cancer at Memorial Sloan-Kettering Cancer Center (MSKCC) between June 1988 and December 2007 were reviewed to identify a subset of this population who developed metastatic CRPC. Castration-resistant metastatic disease was defined as progression of disease following androgen deprivation therapy. In all, 158 patients were identified and blood samples provided by these men were genotyped for germline BRCA1 and BRCA2 mutations. The inclusion of only Ashkenazi Jewish men allowed for targeted genotyping for founder mutations with all blood samples genotyped for the two major Ashkenazi BRCA founder mutations BRCA1*185delAG and BRCA2*6174delT. Genotyping was not performed for the BRCA1*5382insC founder mutation. The TaqMan™ (fluorogenic 5′ nuclease) assay was used for SNP genotyping. The primers and probes were obtained from Applied Biosystems (Foster City, CA, USA). PCR was conducted with both primers and probes added in the ABI 9700 thermocycler, and the endpoint results were scored using the ABI 7900HT Sequence Detection System (version 2.3). In each 384-well plate, one reference sample and one negative control were included for quality control.
Cases' medical records and pathology reports were collected as part of an institutional prostate cancer research database using standardized questionnaires and chart abstraction forms, created and maintained by the prostate cancer disease management team at MSKCC. In addition, each chart was reviewed by a single physician to confirm existing data and gather additional information regarding disease progression and response to treatment. Clinical records and DNA samples were linked through a unique identifier. In accordance with an institutional research board approved protocol, the link between patient information and the identifier was destroyed once clinical data were abstracted, thereby anonymizing the samples before germline analysis. For men with CRPC, data were collected regarding parameters at the start of taxane-based therapy including age, PSA, lactate dehydrogenase, haemoglobin, alkaline phosphatase, albumin, Karnofsky performance status and year of treatment. At the time of diagnosis clinical stage and biopsy score in addition to initial treatment were recorded. Analysis was based on PSA values within 12 weeks of treatment and cause of death. Response to taxane-based therapy was assessed using the maximum decline within a 12-week period after initiation of treatment.
Statistical Methods
The proportion of patients with an initial PSA response to taxane-based therapy was compared between carriers and non-carriers using Fisher's exact test. In the subgroup of patients with an initial response to taxane-based therapy, the percentage change in PSA (calculated as 100 times the lowest PSA within 12 weeks of treatment start, divided by PSA at treatment start) was compared between carriers and non-carriers using the Mann–Whitney test. Survival from the start of treatment was summarized descriptively. Survival was not compared formally between carriers and non-carriers because of the limited number of BRCA carriers. Statistical analyses were conducted using Stata 10.1 (StataCorp LP, College Station, TX, USA).
Results
After a median follow-up in excess of 8 years, 156 men from the original cohort of 832 individuals with localized prostate cancer developed castration-resistant metastatic disease, and 76 of these patients were evaluable for response to taxane-based therapy. Treatment regimens for these 76 patients included single-agent docetaxel (n = 33 patients), docetaxel + estramustine (17 patients), docetaxel + samarium (five patients), docetaxel + bevacizumab (four patients), docetaxel + 17AAG (two patients), docetaxel + DN-101 (two patients), docetaxel + carboplatin (one patient), docetaxel + traztusumab (one patient), docetaxel + cyclophosphamide (one patient), paclitaxel (three patients), paclitaxel + carboplatin + estramustine (six patients) and paclitaxel + estramustine (one patient). Seven of 88 patients were BRCA carriers (three BRCA1, four BRCA2; 8%).
Patient characteristics according to BRCA carrier status are shown in Table 1. At the time of initiating taxane-based therapy, BRCA carriers had higher PSA, lactate dehydrogenase and alkaline phosphatase levels. The BRCA carriers also had higher biopsy Gleason scores at diagnosis; however, primary treatment approaches were similar in both groups.
Table 1. Patient characteristics for men treated with taxane-based chemotherapy for CRPC.
| BRCA carrier | ||
|---|---|---|
|
| ||
| Yes, N = 7 | No, N = 81 | |
| Characteristics at start of docetaxel treatment | ||
| Age (median, range) | 72 (59, 82) | 75 (69, 79) |
| PSA level | 199 (3, 811) | 73 (18, 262) |
| Haemoglobin | 12 (11, 13) | 12 (10, 13) |
| LDH | 259 (196, 480) | 191 (155, 231) |
| Alkaline phosphatase | 272 (132, 446) | 91 (64, 169) |
| Albumin | 4.1 (3.8, 4.4) | 4.2 (3.9, 4.4) |
| KPS | ||
| ≤70 | 2 (28%) | 6 (8%) |
| ≥80 | 5 (72%) | 68 (92%) |
| Characteristics of initial prostate cancer diagnosis | ||
| Clinical stage | ||
| T1 | 1 (14%) | 11 (14%) |
| T2 | 3 (43%) | 21 (26%) |
| T3 | 2 (29%) | 22 (27%) |
| T4 | 0 (0%) | 3 (4%) |
| Unknown | 1 (14%) | 24 (30%) |
| Biopsy Gleason score | ||
| ≤6 | 1 (14%) | 13 (16%) |
| 7 | 3 (43%) | 28 (35%) |
| 8 | 0 (0%) | 19 (23%) |
| 9 | 3 (43%) | 19 (23%) |
| Unknown | 0 (0%) | 2 (2%) |
| Primary treatment | ||
| Prostatectomy | 3 (43%) | 39 (48%) |
| Radiotherapy alone | 1 (14%) | 22 (27%) |
| Radiotherapy + ADT | 2 (29%) | 12 (15%) |
| Hormones alone | 1 (14%) | 7 (9%) |
| Watchful waiting | 0 (0%) | 3 (4%) |
| Chemotherapy | 0 (0%) | 1 (1%) |
| BRCA carrier status | ||
| BRCA1+ | 3 (43%) | 0 (0%) |
| BRCA2+ | 4 (57%) | 0 (0%) |
LDH, lactate dehydrogenase; KPS, Karnofsky performance status; ADT, androgen deprivation score.
Initial PSA response to taxanes was available for all seven BRCA carriers and for 69 non-carriers. Overall, 71% (54/76) of patients responded to taxanes (Table 2). Four (one BRCA1, three BRCA2) out of seven BRCA carriers (57%) had a PSA response. In the subgroup of patients who responded, the median change in PSA level among four BRCA carriers was −63%; all these patients had a >50% decrease in PSA. Among non-carriers, the proportion of patients who responded to docetaxel was similar (73%, 50/69), as was the median change in PSA level in the subgroup of 50 non-carriers who responded (−60%, interquartile range −78% to 35%). There was no significant difference in the proportion of responders among BRCA carriers and non-carriers (P = 0.4), nor in the percentage change in PSA level among responders between the groups (P = 0.6). After a median follow-up of 8 years, all the seven BRCA carriers had died from prostate cancer, compared with 49 non-carriers (Table 3). One BRCA2 carrier treated with docetaxel plus platinum survived 37 months. Among the 81 non-carriers, only 10 (12%) were alive and followed past 36 months.
Table 2. PSA response to taxane-based treatment.
| BRCA carrier | |||
|---|---|---|---|
|
| |||
| Yes, N = 7 | No, N = 69 | P | |
| Initial response to taxane (n = 76) | |||
| Yes (>50% decline) | 4 (57%) | 28 (42%) | 0.5 |
| Yes (31–50% decline) | 0 (0%) | 14 (20%) | |
| Yes (1–30% decline) | 0 (0%) | 8 (12%) | |
| No | 3 (43%) | 19 (28%) | |
| Subgroup with initial response (n = 54) | |||
| PSA level at treatment start | 44, 1.8, 250, 811* | 86 (23, 262) | |
| Lowest PSA level within 12 weeks of treatment start | 20, 0.7, 88, 179* | 25 (7, 109) | |
| Percentage change in PSA level | −53, −62, −64, −78* | −60 (−78, −35) | 0.6 |
Actual values for the four BRCA carriers with an initial response. The median percentage change in PSA level for the four BRCA carriers was −63%.
Table 3. Treatment details for the seven BRCA carriers studied.
| Patient | BRCA | Age (years) | Treatment | Response | PSA level at treatment start | PSA nadir within 12 weeks | % change in PSA level | Treatment to death (months) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 77 | D | No | 3 | 8 | 220 | 4.1 |
| 2 | 1 | 82 | D | Yes | 44 | 20 | −53 | 16.9 |
| 3 | 1 | 88 | D | No | 199 | 294 | 48 | 2.1 |
| 4 | 2 | 59 | D | Yes | 811 | 179 | −78 | 8.1 |
| 5 | 2 | 64 | D+carboplatin | Yes | 2 | 1 | −62 | 37 |
| 6 | 2 | 78 | D+estramustine | No | 1110 | 1115 | 0.5 | 10.1 |
| 7 | 2 | 72 | D | Yes | 250 | 88 | −65 | 17.4 |
D, docetaxel.
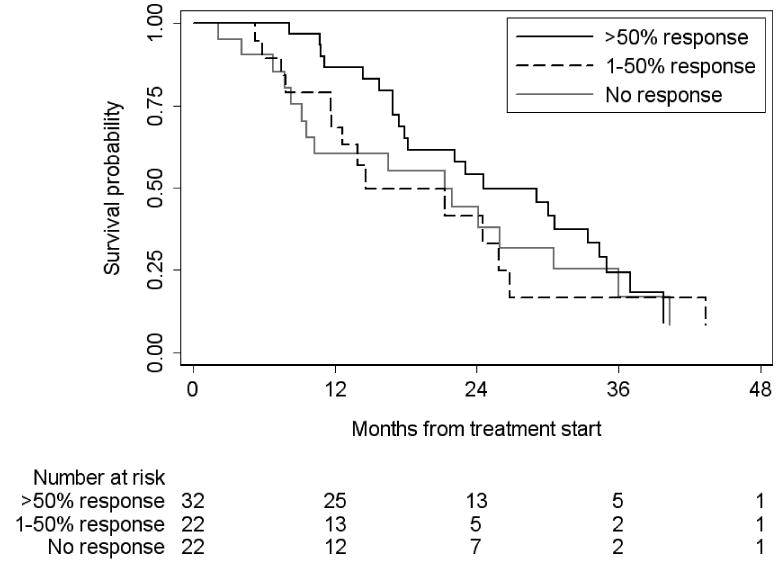
There were 60 deaths overall, and 53 in the subgroup of 76 evaluable patients for initial response to docetaxel. Kaplan–Meier survival probabilities according to response status are given in Fig. 1. Median survival was 25 months for patients with >50% response, 15 months for patients with a 1–50% response and 21 months for patients without an initial response (P = 0.4 by log-rank test).
FIG. 1. Overall survival stratified by initial response to docetaxel (n = 76).
Discussion
In this cohort of men with CRPC, BRCA mutation carriers had similar rates of PSA response to treatment and similar absolute reductions in PSA value to non-carriers. Despite preclinical data suggesting that BRCA (particularly BRCA1) mutation acts as a modulator of response to chemotherapy in ovarian and breast cancer cell lines [16–20], BRCA mutation did not appear to confer resistance to taxanes in men with CRPC. A low rate of pathological complete response was recently observed in female BRCA1 mutation carriers with breast cancer treated with neoadjuvant doxorubicin plus docetaxel (response rate 8%) [15]. In contrast, another study including 12 women treated with neoadjuvant cisplatin showed an 83% response rate, suggesting that the modulatory effect of BRCA mutation status on response to chemotherapy may be agent specific. Docetaxel (75 mg/m2 every 3 weeks) plus prednisolone (5 mg twice a day) is the standard of care for the treatment of CRPC based on two randomized trials showing a significant improvement in overall survival for the combination compared with mitoxantrone plus prednisolone [25,26]. Despite preclinical data suggesting that the presence of a BRCA1 mutation may predict resistance to taxanes, the present small study does not support the hypothesis that BRCA mutation status overall acts as a modulator of response to these agents.
Our use of PSA decline as an endpoint in the present study was mandated by its retrospective design. A decline of 50% was previously suggested as a measure of efficacy [27]; however, the subsequent Prostate Cancer Clinical Trials Working Group clearly outlined the inadequacy of early changes in PSA as the sole endpoint in trials investigating novel therapies for CRPC, and emphasized the additional importance of time-to-event endpoints which more meaningfully reflect the clinical impact of treatment. A retrospective review of men with androgen-independent prostate cancer treated with either docetaxel/estramustine or mitoxantrone/prednisone on Southwest Oncology Group Protocol 99-16 suggested that PSA decline is a surrogate for survival [28,29]. Three-month PSA level declines of 20–40%, a 2-month PSA decline of 30%, and PSA velocity at 2 and 3 months were all surrogates for survival in this retrospective cohort. PSA declined by >50% in all four BRCA mutation carriers treated with docetaxel in the present study, and we could therefore only assess a 50% decline as a measure of PSA response. Our survival analysis was limited by small numbers but greater PSA decline was not significantly associated with improved survival (P = 0.4), again emphasizing the recommendations of the Prostate Cancer Clinical Trials Working Group and the importance of time-to-event endpoints.
BRCA-associated prostate cancer represents a genetically defined subset of the disease that may be more sensitive to platinum-based chemotherapy. Platinum agents, such as cisplatin and carboplatin, have moderate single-agent activity in CRPC [30–33]. Carboplatin has been combined with estramustine and a taxane (docetaxel or paclitaxel) in a number of phase II studies with PSA decline ≥50% seen in 60–100% of patients and objective response rates from 45–65% in patients with measurable disease, although it is difficult to separate the effect of carboplatin from that of the taxane used in these studies [34–40]. In a recent phase III study, the oral platinum agent satraplatin delayed progression of disease and pain in patients with metastatic CRPC experiencing progression after initial chemotherapy [41]. An overall survival benefit was not shown in this study although tailoring therapy to a group more likely to respond, such as BRCA mutation carriers, may have resulted in a different outcome. BRCA mutation status appears to predict response to platinum-based therapy for patients with ovarian and breast cancer and may do the same for men with prostate cancer [14,15,24]. The one BRCA2 carrier treated with docetaxel plus carboplatin in our study survived 37 months, more than twice the survival of other mutation carriers. Using the MSKCC nomogram for patients with hormone refractory disease which does not include BRCA status as a variable, this man's expected median overall survival was 20 months. It is possible that the use of carboplatin overcame the reported poor prognostic association between BRCA mutation and prostate-cancer-specific survival, suggesting a potential role for carboplatin in this setting.
Inhibitors of poly-ADP ribose polymerase 1 (PARP1) offer an additional therapeutic option for BRCA carriers with reported efficacy in BRCA-associated breast and ovarian cancer [42,43]. PARP1 is a constitutively expressed nuclear enzyme important in base excision repair of single-stranded DNA breaks [44]. In its absence excessive single-strand breaks accumulate, leading to collapsed DNA replication forks and conversion of single-strand breaks to double-strand breaks. BRCA1 and BRCA2 proteins repair double-strand DNA breaks by homologous recombination, and BRCA-defective cells are unable to repair these double-strand breaks, resulting in tumour cell death [45,46]. Inhibition of PARP1 in BRCA-deficient cells uses the concept of synthetic lethality whereby a mutation in either of two genes individually has no effect on cell survival but combining the mutations leads to cell death. Loss of the tumour suppressor gene phosphatase and tensin homologue (PTEN) also results in impaired homologous recombination, and tumours deficient in PTEN may also be sensitive to PARP1 inhibition [47]. Prostate cancer is not a recognized manifestation of Cowden syndrome, an autosomal dominant condition caused by germline mutations in PTEN, and common genomic variation at PTEN does not appear to increase risk of prostate cancer, but studies of somatic mutations in prostate cancer report PTEN abnormalities. In one study 63% of radical prostatectomy specimens had heterozygous loss of PTEN using fluorescent in situ hybridization [48], although studies looking at PTEN mutation report lower proportions. Homozygous deletions of PTEN have been detected in up to 15% of locally confined cancers and up to 30% of metastases. Heterozygous loss has been reported in 13% of locally confined cancers and up to 39% of metastases [49–53]. A phase II study of a PARP inhibitor with temozolomide for the treatment of prostate cancer recently opened, and PARP inhibition of BRCA-associated prostate cancer and possible PTEN-deficient disease is worth further investigation.
BRCA1 and BRCA2 mutation carriers have a reported threefold increased risk of developing prostate cancer [1,2]. Given the estimated 0.0069 BRCA2 mutation frequency in the population, this translates to ≈2% of the 180 000 prostate cancer cases in the USA attributable to BRCA2 mutations, or 3600 cases each year which could benefit from treatments tailored to BRCA2 status. Treatment of BRCA-associated prostate cancer with PARP inhibition would represent a first step towards the personalized management of this disease. As emphasized by three recent studies of PARP inhibition in BRCA-associated cancers a cooperative multi-institution approach facilitated rapid accrual [42,43,54]. Of the 171 participants in these studies, only 12% (20 patients) were male and it is likely that male family members of female probands in families with the hereditary breast ovarian cancer syndrome would willingly participate in future studies investigating targeted therapy for BRCA-associated prostate cancer.
As this is a small study of only seven BRCA mutation carriers, it must be viewed as hypothesis generating. We grouped BRCA1 and BRCA2 mutation carriers together despite the absence of preclinical data suggesting that BRCA2 predicts resistance to taxanes. Nonetheless, our results illustrate that both BRCA1 and BRCA2 mutation carriers may respond to taxane-based therapy. The retrospective design of the present study limits interpretation of the results, not only because of the nature of the endpoints evaluated but also because we included different taxane-based regimens in our analysis. It is unknown if combination regimens could alter the predictive potential of BRCA1, although all BRCA1 carriers in the present study received single-agent docetaxel.
BRCA-associated prostate cancer represents a genetically defined subset of the disease associated with worse outcome [55]. As such BRCA mutation may be considered a biomarker for aggressive prostate cancer. We report that it is possible for BRCA-associated prostate cancer to respond to taxane-based therapy, with no suggestion of resistance based on BRCA status. One BRCA mutation carrier achieved prolonged survival with platinum-based treatment. Platinum chemotherapy and PARP inhibition are active in BRCA-associated breast and ovarian cancer and are worth investigation in BRCA-associated prostate cancer.
Abbreviations
- CRPC
castration-resistant prostate cancer
- MSKCC
Memorial Sloan-Kettering Cancer Center
- PARP1
poly-ADP ribose polymerase 1
- PTEN
phosphatase and tensin homologue
References
- 1.Kirchhoff T, Kauff ND, Mitra N, et al. BRCA mutations and risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res. 2004;10:2918–21. doi: 10.1158/1078-0432.ccr-03-0604. [DOI] [PubMed] [Google Scholar]
- 2.Gallagher DJ, Pal P, Kirchhoff T, et al. Correlation of BRCA2 mutation and prostate cancer phenotype. Proc Am Soc Clin Oncol Genitourinary Symposium. 2009 abstr 20. [Google Scholar]
- 3.Thompson D, Easton DF Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–65. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 4.Struewing JP, Hartge P, Wacholder S, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997;336:1401–8. doi: 10.1056/NEJM199705153362001. [DOI] [PubMed] [Google Scholar]
- 5.Gayther SA, de Foy KA, Harrington P, et al. The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 2000;60:4513–8. [PubMed] [Google Scholar]
- 6.Boyd J, Sonoda Y, Federici MG, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA. 2000;283:2260–5. doi: 10.1001/jama.283.17.2260. [DOI] [PubMed] [Google Scholar]
- 7.Rubin SC, Blackwood MA, Bandera C, et al. BRCA1, BRCA2, and hereditary nonpolyposis colorectal cancer gene mutations in an unselected ovarian cancer population: relationship to family history and implications for genetic testing. Am J Obstet Gynecol. 1998;178:670–7. doi: 10.1016/s0002-9378(98)70476-4. [DOI] [PubMed] [Google Scholar]
- 8.Kauff ND. Is it time to stratify for BRCA mutation status in therapeutic trials in ovarian cancer? J Clin Oncol. 2008;26:9–10. doi: 10.1200/JCO.2007.14.0244. [DOI] [PubMed] [Google Scholar]
- 9.Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol. 2008;26:20–5. doi: 10.1200/JCO.2007.11.6905. [DOI] [PubMed] [Google Scholar]
- 10.Rennert G, Bisland-Naggan S, Barnett-Griness O, et al. Clinical outcomes of breast cancer in carriers of BRCA1 and BRCA2 mutations. N Engl J Med. 2007;357:115–23. doi: 10.1056/NEJMoa070608. [DOI] [PubMed] [Google Scholar]
- 11.Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–74. doi: 10.1038/sj.onc.1209874. [DOI] [PubMed] [Google Scholar]
- 12.Mani RS, Tomlins SA, Callahan K, et al. Induced chromosomal proximity and gene fusions in prostate cancer. Science. 2009;326:1230. doi: 10.1126/science.1178124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luedeke M, Linnert CM, Hofer MD, et al. Predisposition for TMPRSS2–ERG fusion in prostate cancer by variants in DNA repair genes. Cancer Epidemiol Biomarkers Prev. 2009;18:3030–5. doi: 10.1158/1055-9965.EPI-09-0772. [DOI] [PubMed] [Google Scholar]
- 14.Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case–control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26:5530–6. doi: 10.1200/JCO.2008.16.1703. [DOI] [PubMed] [Google Scholar]
- 15.Byrski T, Gronwald J, Huzarski T, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28:375–9. doi: 10.1200/JCO.2008.20.7019. [DOI] [PubMed] [Google Scholar]
- 16.Chabalier C, Lamare C, Racca C, Privat M, Valette A, Larminat F. BRCA1 downregulation leads to premature inactivation of spindle checkpoint and confers paclitaxel resistance. Cell Cycle. 2006;5:1001–7. doi: 10.4161/cc.5.9.2726. [DOI] [PubMed] [Google Scholar]
- 17.Quinn JE, Kennedy RD, Mullan PB, et al. BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res. 2003;63:6221–8. [PubMed] [Google Scholar]
- 18.Fedier A, Steiner RA, Schwarz VA, Lenherr L, Haller U, Fink D. The effect of loss of Brca1 on the sensitivity to anticancer agents in p53-deficient cells. Int J Oncol. 2003;22:1169–73. [PubMed] [Google Scholar]
- 19.Mullan PB, Quinn JE, Gilmore PM, et al. BRCA1 and GADD45 mediated G2/M cell cycle arrest in response to antimicrotubule agents. Oncogene. 2001;20:6123–31. doi: 10.1038/sj.onc.1204712. [DOI] [PubMed] [Google Scholar]
- 20.Tassone P, Blotta S, Palmieri C, et al. Differential sensitivity of BRCA1-mutated HCC1937 human breast cancer cells to microtubule-interfering agents. Int J Oncol. 2005;26:1257–63. [PubMed] [Google Scholar]
- 21.Zhou C, Smith JL, Liu J. Role of BRCA1 in cellular resistance to paclitaxel and ionizing radiation in an ovarian cancer cell line carrying a defective BRCA1. Oncogene. 2003;22:2396–404. doi: 10.1038/sj.onc.1206319. [DOI] [PubMed] [Google Scholar]
- 22.Sylvain V, Lafarge S, Bignon YJ. Dominant-negative activity of a Brca1 truncation mutant: effects on proliferation, tumorigenicity in vivo, and chemosensitivity in a mouse ovarian cancer cell line. Int J Oncol. 2002;20:845–53. [PubMed] [Google Scholar]
- 23.Kurebayashi J, Yamamoto Y, Kurosumi M, et al. Loss of BRCA1 expression may predict shorter time-to-progression in metastatic breast cancer patients treated with taxanes. Anticancer Res. 2006;26:695–701. [PubMed] [Google Scholar]
- 24.Byrski T, Gronwald J, Huzarski T, et al. Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat. 2008;108:289–96. doi: 10.1007/s10549-007-9600-1. [DOI] [PubMed] [Google Scholar]
- 25.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 26.Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008;26:242–5. doi: 10.1200/JCO.2007.12.4008. [DOI] [PubMed] [Google Scholar]
- 27.Bubley GJ, Carducci M, Dahut W, et al. Eligibility and response guidelines for phase II clinical trials in androgen-independent prostate cancer: recommendations from the Prostate-Specific Antigen Working Group. J Clin Oncol. 1999;17:3461–7. doi: 10.1200/JCO.1999.17.11.3461. [DOI] [PubMed] [Google Scholar]
- 28.Petrylak DP, Ankerst DP, Jiang CS, et al. Evaluation of prostate-specific antigen declines for surrogacy in patients treated on SWOG 99-16. J Natl Cancer Inst. 2006;98:516–21. doi: 10.1093/jnci/djj129. [DOI] [PubMed] [Google Scholar]
- 29.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 30.Miglietta L, Cannobbio L, Boccardo F. Assessment of response to carboplatin in patients with hormone-refractory prostate cancer: a critical analysis of drug activity. Anticancer Res. 1995;15:2825–8. [PubMed] [Google Scholar]
- 31.Canobbio L, Guarneri D, Miglietta L, Decensi A, Oneto F, Boccardo F. Carboplatin in advanced hormone refractory prostatic cancer patients. Eur J Cancer. 1993;29A:2094–6. doi: 10.1016/0959-8049(93)90040-m. [DOI] [PubMed] [Google Scholar]
- 32.Jungi WF, Bernhard J, Hürny C, et al. Effect of carboplatin on response and palliation in hormone-refractory prostate cancer. Swiss Group for Clinical Cancer Research (SAKK) Support Care Cancer. 1998;6:462–8. doi: 10.1007/s005200050195. [DOI] [PubMed] [Google Scholar]
- 33.Oh WK, Tay MH, Huang J. Is there a role for platinum chemotherapy in the treatment of patients with hormone-refractory prostate cancer? Cancer. 2007;109:477–86. doi: 10.1002/cncr.22439. [DOI] [PubMed] [Google Scholar]
- 34.Oh WK, Halabi S, Kelly WK, et al. A phase II study of estramustine, docetaxel, and carboplatin with granulocyte-colony-stimulating factor support in patients with hormone-refractory prostate carcinoma: Cancer and Leukemia Group B 99813. Cancer. 2003;98:2592–8. doi: 10.1002/cncr.11829. [DOI] [PubMed] [Google Scholar]
- 35.Oh WK, Hagmann E, Manola J, et al. A phase I study of estramustine, weekly docetaxel, and carboplatin chemotherapy in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2005;11:284–9. [PubMed] [Google Scholar]
- 36.Papandreou CN, Daliani DD, Thall PF, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol. 2002;20:3072–80. doi: 10.1200/JCO.2002.12.065. [DOI] [PubMed] [Google Scholar]
- 37.Berry W, Friedland D, Fleagle J, et al. A phase II study of weekly paclitaxel/estramustine/carboplatin in hormone-refractory prostate cancer. Clin Genitourin Cancer. 2006;5:131–7. doi: 10.3816/CGC.2006.n.029. [DOI] [PubMed] [Google Scholar]
- 38.Kelly WK, Curley T, Slovin S, et al. Paclitaxel, estramustine phosphate, and carboplatin in patients with advanced prostate cancer. J Clin Oncol. 2001;19:44–53. doi: 10.1200/JCO.2001.19.1.44. [DOI] [PubMed] [Google Scholar]
- 39.Urakami S, Igawa M, Kikuno N, et al. Combination chemotherapy with paclitaxel, estramustine and carboplatin for hormone refractory prostate cancer. J Urol. 2002;168:2444–50. doi: 10.1016/S0022-5347(05)64164-X. [DOI] [PubMed] [Google Scholar]
- 40.Solit DB, Morris M, Slovin S, et al. Clinical experience with intravenous estramustine phosphate, paclitaxel, and carboplatin in patients with castrate, metastatic prostate adenocarcinoma. Cancer. 2003;98:1842–8. doi: 10.1002/cncr.11754. [DOI] [PubMed] [Google Scholar]
- 41.Sternberg CN, Petrylak DP, Sartor O, et al. Multinational, double-blind, phase III study of prednisone and either satraplatin or placebo in patients with castrate-refractory prostate cancer progressing after prior chemotherapy: the SPARC trial. J Clin Oncol. 2009;27:5431–8. doi: 10.1200/JCO.2008.20.1228. [DOI] [PubMed] [Google Scholar]
- 42.Tutt A, Robson M, Garber JE, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J Clin Oncol. 2009;27(suppl) abstr CRA501. [Google Scholar]
- 43.Audeh MW, Penson RT, Friedlander M, et al. Phase II trial of the oral PARP inhibitor olaparib (AZD2281) in BRCA-deficient advanced ovarian cancer. J Clin Oncol. 2009;27(suppl) abstr 5500. [Google Scholar]
- 44.McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 45.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 46.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 47.Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–22. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshimoto M, Cutz JC, Nuin PA, et al. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet Cytogenet. 2006;169:128–37. doi: 10.1016/j.cancergencyto.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 49.Cairns P, Okami K, Halachmi S, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- 50.Dong JT, Sipe TW, Hyytinen ER, et al. PTEN/MMAC1 is infrequently mutated in pT2 and pT3 carcinomas of the prostate. Oncogene. 1998;17:1979–82. doi: 10.1038/sj.onc.1202119. [DOI] [PubMed] [Google Scholar]
- 51.Verhagen PC, van Duijn PW, Hermans KG, et al. The PTEN gene in locally progressive prostate cancer is preferentially inactivated by bi-allelic gene deletion. J Pathol. 2006;208:699–707. doi: 10.1002/path.1929. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki H, Freije D, Nusskern DR, et al. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998;58:204–9. [PubMed] [Google Scholar]
- 53.Yoshimoto M, Cunha IW, Coudry RA, et al. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br J Cancer. 2007;97:678–85. doi: 10.1038/sj.bjc.6603924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fong PC, Boss DS, Carden CP, et al. AZD2281 (KU-0059436), a PARP (poly ADP-ribose polymerase) inhibitor with single agent anticancer activity in patients with BRCA deficient ovarian cancer: results from a phase I study. J Clin Oncol. 2008;26(suppl) abstr 5510. [Google Scholar]
- 55.Gallagher DJ, Gaudet MM, Pal P, et al. Germline BRCA mutations denote a clinicopathologic subset of prostate cancer. Clin Cancer Res. 2010;16:2115–21. doi: 10.1158/1078-0432.CCR-09-2871. [DOI] [PMC free article] [PubMed] [Google Scholar]