

Abstract
Condensation and evaporation modify the properties and effects of atmospheric aerosol particles. We studied the evaporation of aqueous succinic acid and succinic acid/ammonium sulfate droplets to obtain insights on the effect of ammonium sulfate on the gas/particle partitioning of atmospheric organic acids. Droplet evaporation in a laminar flow tube was measured in a Tandem Differential Mobility Analyzer setup. A wide range of droplet compositions was investigated, and for some of the experiments the composition was tracked using an Aerosol Mass Spectrometer. The measured evaporation was compared to model predictions where the ammonium sulfate was assumed not to directly affect succinic acid evaporation. The model captured the evaporation rates for droplets with large organic content but overestimated the droplet size change when the molar concentration of succinic acid was similar to or lower than that of ammonium sulfate, suggesting that ammonium sulfate enhances the partitioning of dicarboxylic acids to aqueous particles more than currently expected from simple mixture thermodynamics. If extrapolated to the real atmosphere, these results imply enhanced partitioning of secondary organic compounds to particulate phase in environments dominated by inorganic aerosol.
Introduction
Atmospheric aerosol particles influence global climate directly by scattering and absorbing solar radiation and indirectly by acting as cloud condensation nuclei. Aerosols are also a major factor deteriorating air quality. All of these effects depend on particle size, composition, and concentration.
Atmospheric aerosols are complex mixtures of organic and inorganic molecules.1 During atmospheric aging the evolution of size and composition of primary particles, i.e. particles that enter the atmosphere in the condensed phase, is influenced by condensation and evaporation of vapors. For secondary particles, i.e. particles formed in the atmosphere through gas-to-particle transitions, condensational growth is a crucial step on their way to become climatically relevant, and organic vapors play a significant role in this growth.2 To quantify the climate and air quality effects of aerosols it is thus important to understand atmospheric condensation and evaporation processes.
Dicarboxylic acids are a group of water-soluble organic compounds often found in atmospheric aerosol particles.3,4 They can be classified as semi- to low-volatile,5 although the values reported for their saturation vapor pressures vary considerably depending on the measurement techniques.6−8 While there are uncertainties related to the pure-component saturation vapor pressures of organic compounds, even less experimental data is available about their interactions with inorganic aerosol constituents.
The equilibrium vapor pressures of individual compounds over a mixed particle surface are affected by the particle composition. This effect is described by the activity, i.e. the product of the activity coefficient and the molar fraction of the given compound in the particle. For aqueous solutions of single organic compounds directly measurement-based activity models9 and models based on group contribution methods, like UNIFAC,10 are available. The latter can also be applied for multicomponent mixtures. Activity models are often developed based on water equilibrium, rather than the equilibrium of the organic solute – largely due to the fact that experimental data on organic activities are extremely scarce. Also activity models for mixtures of inorganic and organic solutes have been tested with measured values of water activity11−13 – yielding information on the mixture effects on equilibrium vapor pressures of water but not directly on the activity and volatility of the organic compounds.
To our knowledge, the effect of inorganic salts on the evaporation, specifically the equilibrium vapor pressures, of dicarboxylic acids over aqueous solution droplets has so far been investigated in only two experimental studies.14,8 In both of these studies the inorganic compound was sodium chloride (NaCl). Zardini et al.14 used a Tandem Differential Mobility Analyzer (TDMA) system for submicrometer aqueous solution droplets containing succinic acid (HOOC(CH2)2COOH) and NaCl and found that the experimentally determined evaporation rate of the particles was lower than theoretically expected if NaCl did not directly affect the equilibrium vapor pressure of succinic acid. They concluded that the presence of NaCl in the droplets possibly lowers the activity coefficient of succinic acid but identified several possible uncertainties related to this conclusion and highlighted the need for direct observations of the aerosol composition. Pope et al.8 studied micrometer-sized aqueous solution droplets containing malonic (HOOC(CH2)COOH) or glutaric acid (HOOC(CH2)3COOH) and NaCl using two techniques, electrodynamic balance and optical tweezers. They did not find a clear effect of NaCl on the activity coefficient of the two dicarboxylic acids within experimental uncertainty. As the studies on the effect of inorganic compounds on the equilibrium vapor pressures of organic compounds are scarce and somewhat inconclusive, further investigations on this topic are warranted.
In this work we study, for the first time, the effect of ammonium sulfate (AS) on the equilibrium vapor pressure of succinic acid (SA) over aqueous solution droplets by investigating the evaporation rate and chemical composition of these droplets. We use a TDMA setup similar to Zardini et al.14 and Koponen et al.15 but improve the setup by coupling it to direct online measurement of the droplet composition during evaporation with an Aerosol Mass Spectrometer (AMS). We complement these studies with offline analysis of aqueous solutions using Ultra High Performance Liquid Chromatography coupled to a quadrupole Time-of-Flight mass spectrometer through an electrospray ionization inlet (UHPLC-ESI-qTOF-MS). By comparing these experimental data to predictions by an evaporation model we study the effect of AS on SA volatility in submicrometer aqueous solution droplets. We also discuss potential uncertainties related to the interpretation of the flow tube experiments, along with the influence of gas phase composition and particle phase chemistry on the evaporation.
Materials and Methods
Measurements
The evaporation of aqueous solution droplets was measured at the University of Copenhagen with a modified Tandem Differential Mobility Analyzer (TDMA) setup including a laminar flow tube. In total 22 evaporation experiments were done, and in six of them the chemical composition of the droplets during evaporation was measured with an Aerosol Mass Spectrometer (AMS, Table S1). Liquid droplets containing water, SA, and AS were studied (Table 1). Experiments with binary droplets containing water and SA were also performed to determine the subcooled liquid saturation vapor pressure of pure SA (psat,SA) under the same conditions as for the ternary droplets.
Table 1. Compounds Used in the Experiments and Their Properties: Deliquescence and Crystallization Relative Humidities (DRH, CRH) and Molar Mass (M).
The TDMA setup has been described previously,15 and only a brief summary is presented here. The liquid particles were generated with an atomizer from aqueous solutions (total solute concentrations of approximately 120 mg L–1 in experiments without AMS, and 400–500 mg L–1 in experiments with the AMS). Double deionized water purified using a Milli-Q Plus Ultrapure water system was used. A nearly monodisperse droplet population (geometric standard deviation of log-normal distribution <1.1) was selected with a Differential Mobility Analyzer (DMA, with a sheath flow of 3 L min–1) and led to a laminar flow tube where the droplets evaporated. The 3.5 m long tube allows particle residence times up to several minutes. The time evolution of droplet size was obtained by sampling the droplets along the flow tube with a Scanning Mobility Particle Sizer (SMPS).
In experiments without the AMS (experiments 1–16, Table S1) initial particle diameters were in the range 95–120 nm, and sheath air was added to the laminar flow tube to better control the gas phase and to decrease the spread in residence times. The sample and sheath flow rates in the flow tube were 0.3 and 0.6 L min–1, respectively. Number concentrations (N) of the aerosol sampled with SMPS were in the range 60–780 cm–3. In the experiments with the AMS (experiments 17–22, Table S1) some compromises were made to have enough particle mass for detection with AMS: the initial droplet sizes were increased to 120–170 nm, sample flow rate in the flow tube was 0.4 L min–1, no sheath flow was used, and N was increased to 3600–43000 cm–3.
All experiments were performed in a temperature-controlled laboratory. Relative humidity (RH) was controlled throughout the system: sheath air in the two DMA and in the laminar flow tube were humidified.15
The Aerosol Mass Spectrometer used was a High-Resolution Aerosol Mass Spectrometer (AMS; Aerodyne Research Inc., Billerica, MA, USA), which measures the particle phase chemical composition by thermal vaporization and electron impact ionization mass spectrometry.16 Size-resolved data was obtained through particle time-of-flight (PToF) measurements. With the AMS it was possible to monitor the time evolution of the particle composition during the evaporation. The AMS data were analyzed with IGOR pro 6 (Wavemetrics, USA) SQUIRREL 1.51 and PIKA 1.1. The AS concentration was deduced from the sulfate ions, using the default fragmentation patterns. Quantifying the SA mass fraction in the particles was complicated by the high abundances of H2O+ and C2H4+, which made the default treatment of organic PM incorrect. Separate experiments with high mass loadings (∼50 μg m–3) and low RH (∼8%) were performed to obtain the mass spectral fingerprint of SA (Figure S4). Thus, SA content from the AMS was calculated based on the observed fragmentation pattern of SA and selected oxygen containing marker fragments at m/z 45, 55, 56, 73, 74 and 100, contributing 16% of the mass spectra from dry SA particles (Figure S4). This information and an assumed relative ionization efficiency (RIE) of 1.4 (standard for organic particulate matter) enabled quantification of SA in the droplets.
For two experiments (experiments 17 and 18, Table S1) additional size-resolved analysis in the high resolution mode was performed with PIKA 1.11. This was done to separate singly charged particles from the aerosol size distribution and generate results comparable with those from the TDMA. The effect of multiply charged particles on the particle numbers was small (<15% of total N) but significant on the mass-based AMS measurement (50–60% of the particulate mass). Through manual inspection of the PToF distribution of AS fragments a size range was determined where singly charged particles strongly dominated the signals at each port (see Figure S5). Consequently the high end of the PToF distribution of the singly charged particles was not included. Data corresponding to very high PToF, where no particle signal was present, were used to quantify instrument background signal. The full range of PToF with particle contribution was analyzed and compared with nonsize resolved data, yielding a port-specific normalization factor of 0.9–1.1.
The water content of the particles was varied by conducting experiments at different RHs. Initial solute composition in the droplets was controlled by varying the SA to AS ratio in the atomization solution. The organic molar fraction of the total solute (Forg) is defined as
| 1 |
where nSA and nAS are the number of moles of SA and AS, respectively. The RH was varied between 60 and 80%, and the initial Forg was varied between 0.5 and 0.9 in the experiments. The temperature was approximately 294 K in all experiments (Table S1).
Off-line chemical analysis of aqueous solutions was performed using UHPLC-ESI-qTOF-MS (see the Supporting Information, SI).
Model
The evaporation of the droplets in the laminar flow tube was modeled with a dynamic evaporation model combined with a thermodynamic phase equilibrium model similarly as in Zardini et al.14 The phase equilibrium model E-AIM (Extended Aerosol Inorganic Model, http://www.aim.env.uea.ac.uk, last accessed Feb. 2013)17,18 was used for calculating the activity coefficients and water content of droplets, whereas the decrease in the size of the droplets was calculated with the evaporation model. Coagulation was not accounted for in the model, as its maximum effect on N (estimated based on the size distributions) remained below 2% for all the experiments.
In the evaporation model water and SA evaporate from the droplets and AS is assumed to be nonvolatile. Gas–liquid equilibrium was assumed for water due to the significantly shorter diffusion time scales as compared with SA. The evaporation of SA is calculated based on its mass flux from the droplet (ISA)19,20
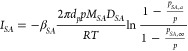 |
2 |
where βSA is the transition regime correction factor,21dp is the droplet radius, p is the total pressure, MSA is the molar mass, DSA is the diffusion coefficient of SA in air, R is the molar gas constant, T is the temperature, and pSA,a and pSA,∞ are the partial vapor pressures of SA at the droplet surface and far away from the droplet. Changes in pSA,∞ were calculated assuming that the evaporated SA accumulates in the same air parcel where the droplets are traveling through the flow tube.
The partial pressure of SA and water at the droplet surface are assumed to equal their equilibrium vapor pressures
| 3 |
where Xi is the molar fraction, vi is the molar volume, σ is the surface tension of the solution, and psat,i is the saturation vapor pressure of the pure liquid i (SA or water). The activity coefficient γi depends on the molar fractions of all compounds j.
In E-AIM the activity coefficients of organic and inorganic compounds are calculated based on purely organic or inorganic aqueous solutions11,18,22 therefore neglecting the influence of AS on γSA. The water activity is calculated as a product of water activities of the water-inorganic and water-organic solutions. The activities in water-inorganic solution are calculated with the Pitzer, Simonson, and Clegg equations.17 For activities in the SA-water solution we tested three activity models included in the E-AIM: Redlich–Kister equation,9,11 UNIFAC with a standard set of parameters,10,23−25 and UNIFAC with the parameters modified by Peng et al.26
The particles were assumed to have only aqueous phase, as the RH was above crystallization RH of both solutes (Table 1). The dissociation of SA was not taken into account in the standard model calculations, but its potential effect based on E-AIM predictions was investigated (see the SI).
The evaporation model was initialized with the droplet size and Forg at the first DMA where Forg was assumed to equal that of the atomized solution. No SA was assumed to be in the gas phase at this stage. RH and gas phase temperature were assumed constant during each experiment. The temperature of particles was assumed to be the same as that of the gas phase, which is justified for this setup.27 The mass flux of SA was calculated with 10 ms time steps, and the activity coefficients were updated from E-AIM with 5 s time steps.
The properties of SA and aqueous solutions of SA were adopted from Riipinen et al.27 and references therein. The density of the ternary solution (ρmix) was adopted from E-AIM28 (see the SI). The surface tension of the ternary solution was calculated based on the pure water surface tension (σw) and the surface tensions of the SA (σw.SA)29 and AS (σw,AS)30 aqueous solutions31 (see the SI).
Results and Discussion
Measured Evaporation Rates of SA and SA/AS Droplets
Figure 1 shows the measured (TDMA) droplet diameter (dp) as a function of the evaporation time for the ternary solution droplets of water, SA, and AS as well as the binary droplets of water and SA. Time 0 is the exit from the first DMA. The figure represents the experiments with the low particle concentrations and without AMS at 60% and 80% RH (experiments 1–16, Table S1). For each RH the evaporation rate increases with increasing initial Forg. For the same initial Forg an increase in RH slows down the evaporation due to the decrease in molar fraction of SA, but this has only a small effect on the evaporation rate. This reflects the role of SA as the controlling factor for the droplet shrinkage.
Figure 1.
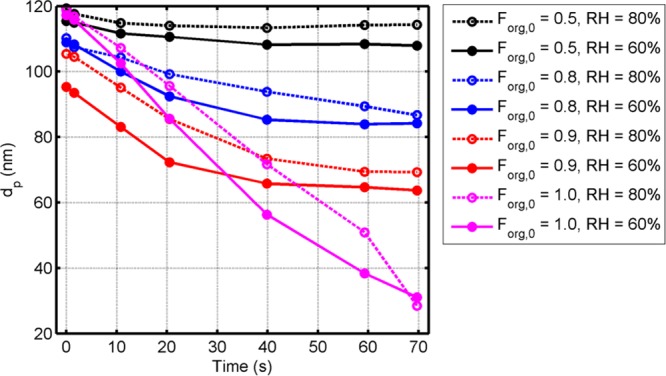
Measured particle geometric mean diameter as a function of time for SA/AS aqueous solution particles with the initial Forg of 0.5 (black), 0.8 (blue), and 0.9 (red) and for SA aqueous solution particles (Forg = 1.0) (magenta) at 60% (solid circles + solid line) and 80% RH (open circles + dashed line) (experiments 1, 4, 5, 8, 9, 12, 13, and 16, Table S1). The points refer to measurements after the first DMA (time = 0 s), before the flow tube (port 0), at ports 1–4 along the flow tube and at the end of the tube (port 5).
Binary Droplets: Subcooled Liquid Saturation Vapor Pressure of Succinic Acid
The subcooled liquid saturation vapor pressure of SA (psat,SA) has previously been determined in the laminar flow tube at an RH of approximately 65% and in the temperature range 298–301 K.15,27 For reproducibility check, we performed a series of similar experiments with binary SA aqueous solution droplets at varying RHs (experiments 13–16, Table S1). The value of psat,SA is determined by a least-squares fit between modeled and measured evolution of dp with time. Following Koponen et al.15 only the SMPS measurements from the beginning of the tube (port 0) and ports 1–4 along the tube were utilized. The same activity models as for the ternary mixtures were used (Table 2).
Table 2. Subcooled Liquid Saturation Vapor Pressures of Succinic Acid (psat,SA) Obtained from Measurements at Different RHs Using Three Different Activity Modelsa.
| expt 13 RH = 60%, T = 294.5 K | expt 14 RH = 65%, T = 294.9 K | expt 15 RH = 75%, T = 295.0 K | expt 16 RH = 80%, T = 294.6 K | mean at 298 K | |
|---|---|---|---|---|---|
| psat,SA, fitted activity eq (10–3 Pa) | 0.75 | 0.77 | 0.84 | 0.81 | 1.29 |
| psat,SA, UNIFAC Standard (10–3 Pa) | 0.68 | 0.69 | 0.75 | 0.72 | 1.15 |
| psat,SA,UNIFAC Peng (10–3 Pa) | 1.04 | 1.11 | 1.33 | 1.34 | 1.95 |
Saturation vapor pressures at 298 K where calculated assuming the temperature dependence by Koponen et al.15 obtained using activity model UNIFAC Dortmund.
To facilitate further comparisons, the psat,SA values were transformed to psat,SA(298K) (Table 2) using the temperature dependence of psat,SA by Koponen et al.15 with UNIFAC Dortmund activity model. With all activity models the psat,SA(298K) inferred from experiments at 75–80% RH was higher compared to experiments at 60–65% RH. The deviation was most pronounced and systematic when using UNIFAC with Peng et al.26 corrections. In all cases the variations with RH for a given model were smaller than the differences between the models. The psat,SA values measured here agree with the psat,SA(298K) of 1.1–1.5 · 10–3 Pa reported by Koponen et al.15 In the further investigation of the ternary droplets we used the mean psat,SA(298K) values from the binary experiments, consistently with respect to the choice of activity model, together with the temperature dependence.15
Ternary Droplets: Measured and Modeled Evaporation
The measured and modeled dp of the SA/AS aqueous solution droplets as a function of time are presented in Figure 2 for the experiments without the AMS (experiments 1–12, Table S1). Only the model predictions using the Redlich–Kister fitted activity equation are shown, but the group contribution-based activity models gave similar results (Figure S1). For the droplets with initial Forg = 0.9 the model captures the size change very well in the beginning of the evaporation (ports 0–2) but at later stages (approximately ports 3–5) overestimates it. This applies also for the droplets with smaller initial Forg, although in these cases the model overestimates the evaporation rate earlier. In general, the model captures the trends and magnitude of the evaporation, although overall the decrease in droplet size is overestimated.
Figure 2.
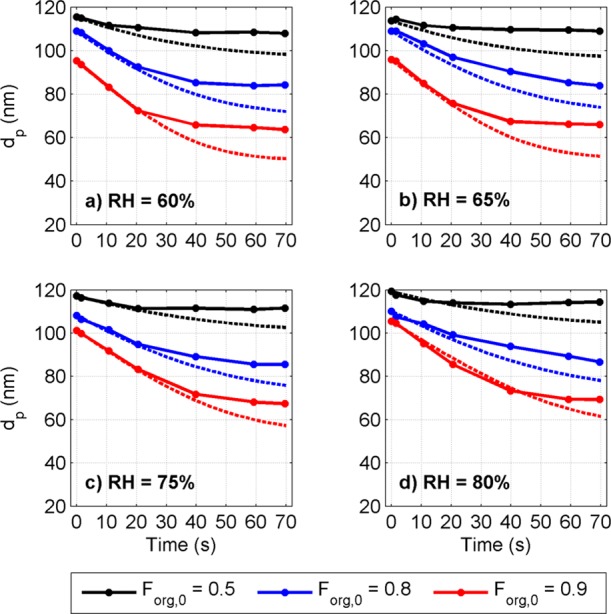
Measured (circles connected with solid line) and modeled (dashed line) diameter of SA/AS aqueous solution particles as a function of time at RHs a) 60%, b) 65%, c) 75%, and d) 80% (experiments 1–12, Table S1). Color indicates the initial Forg of droplets: 0.5 (black), 0.8 (blue), and 0.9 (red).
The difference between measured and modeled evaporation rates depends systematically on the initial Forg: with initial Forg of 0.9 the model predicted on average 21% larger total droplet volume change during the evaporation (from 0 to 70 s) compared to the measured, whereas with initial Forg of 0.5 the difference was 193%. During droplet evaporation Forg decreases and the molar fraction of AS increases. Therefore the effect of AS on the droplet evaporation becomes more significant along the evaporation, and the results suggest that the overestimation of evaporation rate is related to the presence of AS.
The evaporation rate was overestimated at the end of the flow tube at all RHs. For the highest Forg values the agreement between model and measurement was best at RH of 80%, but for Forg < 0.9 such RH effect was not seen. This suggests that the water content of the particles is not governing the difference between the model and the observations.
Zardini et al.14 observed similar overestimation of evaporation rates for droplets containing SA and NaCl and speculated on three potential reasons for this: 1) the pure SA saturation vapor pressure being lower than the value used in the model, 2) overestimating the initial Forg of the droplets, and/or 3) overestimating the SA activity coefficient. We can identify at least two other potential sources of error in the model predictions: uncertainty in the gas phase composition (saturation of SA) and particle-phase processes (e.g., SA dissociation, condensed-phase impurities, and chemical reactions).
The uncertainty in psat,SA does not explain the overestimation of evaporation rate, as lowering psat,SA enough to capture the total dp change correctly with low Forg would result in a clear underestimation of evaporation rate with high Forg (see SI, Figure S1).
The AMS data from two experiments (17 and 18, Table S1) were analyzed in detail (separation of the composition of singly charged particles) to study the time evolution of SA contents in the droplets and test the assumption of the initial Forg being equal to that of the atomized solution. In both cases Forg was 0.8 in the atomized solution and RH was 65% or 80%. The Forg derived from AMS measurements at the first DMA were 0.82 and 0.84 confirming Forg at the first DMA to be the same as in the atomization solution within measurement uncertainty. Furthermore, error in the initial composition would be expected to result in largest discrepancies between modeled and measured evaporation at the largest Forg values (Figure S1), counter to the observations. Wrong initial Forg is thus not a likely explanation for the difference between the model predictions and the experiments. The uncertainties related to experimental temperature, RH, and other thermodynamic properties of SA and AS were also small (see the SI).
Figure 3 shows the comparison between the TDMA and AMS data during the evaporation, along with the corresponding model predictions for experiments 17 and 18. The Forg values from the AMS were converted to wet particle diameters by estimating the particle water content with E-AIM. The two experimental data sets agree relatively well at ports 0–2, while at the last two ports (3–4) the dp values inferred from the AMS are lower than the mobility diameters measured by the TDMA. There are several potential reasons for this. First, the selection of the PToF range to separate the singly charged particles (see Figure S5) causes an uncertainty of maximum 5% in Forg. Second, the particulate mass concentration of SA is very low at the last ports (0.002–0.1 μg m–3), which caused some variation in the Forg (see Figures 3 c-d). Third, there is some uncertainty in the RIE of SA, although our results are not overly sensitive to it: for the conditions in Figure 3, as high as 50% uncertainty in RIE would result in an uncertainty less than 0.1 in Forg at the first port and less than 0.05 at the last port. None of these uncertainties fully explains the discrepancy between dp from the TDMA and the Forg from the AMS. The Forg from the AMS includes only the part of the spectra that resembles that of the calibration measurements with pure dry (or nearly dry) SA, thus not accounting for particle phase impurities or potential reaction products given that these are strongly transformed and not provide signal at the selected SA marker peaks. The AMS data indicated, however, that about 3–5% of the initial particle dry mass consisted of an unidentified nonvolatile organic material that was different from but correlated with initial SA mass fraction and contained mostly hydrocarbon fragments (Figure S6). The AMS also detected excess ammonium ions compared to the AS. This ammonia corresponded to 1–3% of the dry particle mass initially and was volatile – evaporating slightly faster than SA (Figure S7). Besides indicating potential contaminations in the experimental system, the presence of these unexpected impurities could also point to condensed-phase reaction products, anything that is not SA, AS, or water.
Figure 3.

Particle diameter (a,b) and Forg (c,d) for experiments 17 (a,c) and 18 (b,d) with the AMS and high aerosol loadings (see Table S1). Red solid circles in a and b: The geometric mean diameter of a log-normal mode fitted to size distribution measured with the TDMA (the error bars indicate ± one standard deviation of the mode). Red solid circles in c and d: Forg calculated based on measured geometric mean droplet diameter, initial Forg, and water content predicted with E-AIM. Black solid squares in a and b: particle diameter calculated from Forg measured with the AMS (black solid squares in c and d), initial particle dp, and water content predicted with E-AIM at the experimental RH (error bars in y-direction: ± one standard deviation of measured Forg; error bars in x-direction are due to estimated longer residence time from flow tube to AMS compared with the SMPS). Solid lines: model prediction with gas phase saturation considered (black) and by assuming p∞,SA = 0 (blue) when no impurity is taken into account. Dashed lines: model prediction assuming 5% of initial particle dry mass to consist of nonvolatile impurity. The shaded areas show model predictions with 0 to 20% impurity of initial dry mass.
A number of mechanisms forming low-volatility material in aqueous solutions containing inorganic salts and dicarboxylic acids have been reported in the literature, including reactions between NaCl and organic acids,32 formation of organosulfates and -nitrates in the presence of AS and sulfuric acid,33,34 and different self-cycling/oligomerization reactions. Many of the aforementioned processes proceed through the enol tautomer of the dicarboxylic acids, which has been proposed to be the dominant form of these acids in deliquesced (highly concentrated) aerosol, as opposed to the case of bulk aqueous chemistry.35
Since the sheath flow rate in experiments 17–18 was set to zero and the aerosol loadings were considerably higher than in the experiments without the AMS (see Table S1), the model calculations were run for two limiting assumptions about the gas phase: the base case where the SA vapor pressure pSA,∞ was updated, and a case where pSA,∞ was fixed to 0. Direct comparison between the model and measurements in these experiments proved to be challenging, as the model predictions were extremely sensitive to the gas phase composition (Figure 3). If the gas phase was allowed to saturate, the size change upon evaporation was drastically underestimated and Forg overestimated, while pSA,∞ = 0 resulted in a similar overestimation of the evaporation rate as for experiments 1–12 when compared with the TDMA data. Interestingly, the latter case resulted in an agreement between the modeled and measured Forg from the AMS, while the Forg values assessed from the measured dp (with TDMA), initial Forg, and water content from E-AIM are higher than the values measured with the AMS (Figure 3).
While the modeled evaporation was extremely sensitive to gas phase composition at high aerosol loadings (experiments 17–22), it played no role in the experiments without the AMS (experiments 1–16). Figure 4 illustrates this for typical conditions for the TDMA experiments: the modeled evaporation rate is practically the same for all N < 1000 cm–3 (<1 μg m–3), while for N > 1000 cm–3 (>1 μg m–3) it depends drastically on aerosol loadings, the effect increasing with increasing Forg. The limiting concentration for gas-phase saturation decreases with decreasing equilibrium vapor pressure of the evaporating compound (Figure S2). The assumption of pSA,∞ = 0 gave consistent model results for experiments 17–21 compared to experiments 1–12, which supports the picture of the droplets being concentrated in the center of the tube and part of the SA vapor diffusing toward the walls of the tube, thus diluting the gas phase.
Figure 4.
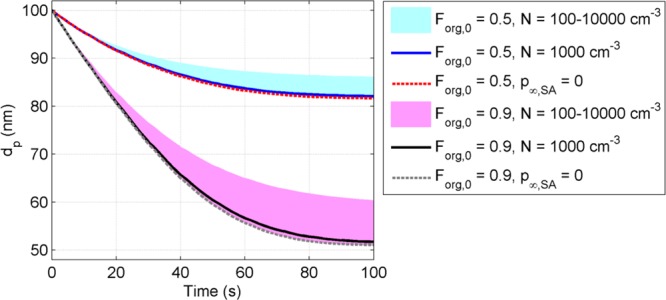
Predicted particle size evolution for initial Forg (Forg,0) of 0.5 (blue line, blue area, and red dashed line) and 0.9 (black line, magenta area, and gray dashed line) with different particle number concentrations. Colored areas present model prediction with N = 100–10000 cm-3, solid lines with N = 1000 cm–3 and dashed line with assumption of p∞,SA = 0, in which case the evaporation is insensitive to N.
We investigated the role of particle phase impurities or chemistry by introducing 5–20% nonvolatile material to the modeled particles initially (see Figure 3 and Figure S3). The evaporation slows down and the final size of the particles increases. While already 5% nonvolatile material in the initial particle dry mass affects the evaporation rate, it should have accounted for 20% or more of the initial particle dry mass to fully explain the difference in the final droplet size (Figures 3 and S3), if SA molar mass was assumed for the nonvolatile material – with lower molar mass the effect increases. Nonvolatile material whose concentration depends on the initial Forg could thus explain the discrepancy between measured and modeled particle evaporation and potentially the difference between the TDMA and AMS. In principle a reaction product of SA and AS could represent such a material, but no such compounds could be clearly identified from the AMS spectra. No evidence of such products, specifically organosulfates or organic oligomers, was visible in the UHPLC-ESI-qTOF-MS analysis run for the bulk solutions either (see the SI) – although further studies are needed to confirm this conclusion to hold also for our supersaturated droplets. From the modeling perspective the “impurity” could also refer to the dissociated fraction of SA which was predicted to increase with decreasing Forg. However, in all cases less than 5% of SA was predicted to dissociate, and the dissociated SA would be detected as SA with the AMS.
Above the mobility equivalent diameter measured with SMPS was compared to the volume equivalent diameter from the model. In principle, nonspherical shape could lead to mobility diameter being larger than the volume equivalent diameter. Nonsphericity of the particles would, however, be somewhat unexpected as the experiments were conducted above the CRHs of both AS and SA.
As a summary, the dynamic evaporation model coupled with the E-AIM thermodynamics captures the evaporation of SA from aqueous solutions containing AS well if the relative abundance of SA is larger than or equal to AS. The model and the observations start to deviate at lower organic to inorganic ratios: the model predicts larger decrease in particle size than observed with TDMA. These results suggest that the presence of AS in the particles enhances the partitioning of SA to aqueous particles more than expected based on current knowledge. This enhancement could be through lowering the activity coefficient of SA in the solution or through other interactions between AS and SA in the aqueous phase, naturally having implications for predictions of the gas-aerosol partitioning of atmospheric organic compounds. For particles with high organic fraction this effect is not strong. However, at inorganic dominated regions the partitioning of organic compounds to particulate phase can be enhanced by these interactions with the inorganic constituents. Direct composition data collected using the AMS confirmed the assumptions about the initial composition of the droplets, but neither AMS nor UHPLC-ESI-qTOF-MS results yield a conclusive chemical explanation to the suppressed evaporation observed with the TDMA. The results show a strong sensitivity of evaporation rate predictions to accurate description of the particle and gas phase composition – particularly at high aerosol loadings (larger than about 1 μg m–3 for compounds with psat < 10–3 Pa).
Acknowledgments
Financial support by the Academy of Finland Centre of Excellence program (No. 1118615), Doctoral Programme in Atmospheric Composition and Climate Change (No. 129663), Danish Agency for Science Technology and Innovation (No. 09-052140), Vetenskapsrådet (No. 2011-5120), European Research Council Grant ATMOGAIN (No. 278277), and The Swedish Research Council FORMAS (2009-615) is acknowledged. Professors Markku Kulmala and Simon Clegg are acknowledged for useful discussions.
Supporting Information Available
Sensitivity analysis of model predictions with respect to thermodynamic properties and assumptions made in model, table of experimental conditions, description of AMS calibration, and the UHPLC-ESI-qTOF-MS measurements on the offline solution composition measurements. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry, Aarhus University, Langelandsgade 140, DK-8000, Aarhus C, Denmark.
The authors declare no competing financial interest.
Supplementary Material
References
- Jimenez J. L.; Canagaratna M. R.; Donahue N. M.; Prevot A. S. H.; Zhang Q.; Kroll J. H.; DeCarlo P. F.; Allan J. D.; Coe H.; Ng N. L.; Aiken A. C.; Docherty K. S.; Ulbrich I. M.; Grieshop A. P.; Robinson A. L.; Duplissy J.; Smith J. D.; Wilson K. R.; Lanz V. A.; Hueglin C.; Sun Y. L.; Tian J.; Laaksonen A.; Raatikainen T.; Rautiainen J.; Vaattovaara P.; Ehn M.; Kulmala M.; Tomlinson J. M.; Collins D. R.; Cubison M. J.; Dunlea E. J.; Huffman J. A.; Onasch T. B.; Alfarra M. R.; Williams P. I.; Bower K.; Kondo Y.; Schneider J.; Drewnick F.; Borrmann S.; Weimer S.; Demerjian K.; Salcedo D.; Cottrell L.; Griffin R.; Takami A.; Miyoshi T.; Hatakeyama S.; Shimono A.; Sun J. Y.; Zhang Y. M.; Dzepina K.; Kimmel J. R.; Sueper D.; Jayne J. T.; Herndon S. C.; Trimborn A. M.; Williams L. R.; Wood E. C.; Middlebrook A. M.; Kolb C. E.; Baltensperger U.; Worsnop D. R. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. [DOI] [PubMed] [Google Scholar]
- Riipinen I.; Yli-Juuti T.; Pierce J. R.; Petäjä T.; Worsnop D. R.; Kulmala M.; Donahue N. M. The contribution of organics to atmospheric nanoparticle growth. Nat. Geosci. 2012, 5, 453–458. [Google Scholar]
- Zappoli S.; Andracchio A.; Fuzzi S.; Facchini M. C.; Gelencsér A.; Kiss G.; Krivácsy Z.; Molnár Á.; Mészaros E.; Hansson H.-C.; Rosman K.; Zebühr Y. Inorganic, organic and macromolecular components of fine aerosol in different areas of Europe in relation to their water solubility. Atmos. Environ. 1999, 33, 2733–2743. [Google Scholar]
- Rogge W. F.; Mazurek M. A.; Hildemann L. M.; Cass G. R.; Simoneit B. R. T. Quantification of urban organic aerosols at a molecular level: Identification, abundance and seasonal variation. Atmos. Environ. 1993, 27A, 1309–1330. [Google Scholar]
- Goldstein A. H.; Galbally I. E. Known and unexplored organic constituents in the Earth’s atmosphere. Environ. Sci. Technol. 2007, 41, 1514–1521. [DOI] [PubMed] [Google Scholar]
- Huisman A. J.; Krieger U. K.; Zuend A.; Marcolli C.; Peter T. Vapor pressures of substituted polycarboxylic acids are much lower than previously reported. Atmos. Chem. Phys. Discuss. 2013, 13, 1133–1177. [Google Scholar]
- Soonsin V.; Zardini A. A.; Marcolli C.; Zuend A.; Krieger U. K. The vapor pressure and activities of dicarboxylic acids reconsidered: the impact of the physical state of the aerosol. Atmos. Chem. Phys. 2010, 10, 11753–11767. [Google Scholar]
- Pope F. D.; Tong H.-J.; Dennis-Smither B. J.; Griffiths P. T.; Clegg S. L.; Reid J. P.; Cox R. A. Studies of single aerosol particles containing malonic acid, glutaric acid, and their mixtures with sodium chloride. II. Liquid-state vapor pressures of the acids. J. Phys. Chem. A 2010, 114, 10156–10165. [DOI] [PubMed] [Google Scholar]
- Redlich O.; Kister T. Algebraic representation on thermodynamic properties and the classification of solutions. Ind. Eng. Chem. 1948, 40, 345–348. [Google Scholar]
- Fredenslund A.; Jones R. L.; Prausnitz J. M. Group-contribution estimation of activity coefficinets in nonideal liquid mixtures. AIChE J. 1975, 21, 1086–1099. [Google Scholar]
- Clegg S. L.; Seinfeld J. H. Thermodynamic models of aqueous solutions containing inorganic electrolytes and dicarboxylic acids at 298.15 K. I. The acids as non-dissociating components. J. Phys. Chem. A 2006, 110, 5692–5717. [DOI] [PubMed] [Google Scholar]
- Hanford L.; Mitchem L.; Reid J. P.; Clegg S. L.; Topping D. O.; McFiggans G. B. Comparative thermodynamic studies of aqueous glutaric acid, ammonium sulfate and sodium chloride aerosol at high humidity. J. Phys. Chem. A 2008, 112, 9413–9422. [DOI] [PubMed] [Google Scholar]
- Pope F. D.; Dennis-Smitter B. J.; Griffiths P. T.; Clegg S. L.; Cox R. A. Studies of single aerosol particles containing malonic acid, glutaric acid, and their mixtures with sodium chloride. I. Hygroscopic growth. J. Phys. Chem. A 2010, 114, 5335–5341. [DOI] [PubMed] [Google Scholar]
- Zardini A. A.; Riipinen I.; Koponen I. K.; Kulmala M.; Bilde M. Evaporation of ternary inorganic/organic aqueous droplets: Sodium chloride, succinic acid and water. J. Aerosol Sci. 2010, 41, 760–770. [Google Scholar]
- Koponen I. K.; Riipinen I.; Hienola A.; Kulmala M.; Bilde M. Thermodynamic properties of, malonic, succinic, and glutaric acids: Evaporation rates and saturation vapor pressures. Environ. Sci. Technol. 2007, 41, 3926–3933. [DOI] [PubMed] [Google Scholar]
- DeCarlo P F.; Kimmel J. R.; Trimborn A.; Northway M. J.; Jayne J. T.; Aiken A. C.; Gonin M.; Fuhrer K.; Horvath T.; Docherty K. S.; Worsnop D. R.; Jimenez J. L. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [DOI] [PubMed] [Google Scholar]
- Clegg S. L.; Brimblecombe P.; Wexler A. S. A thermodynamic model of the system H+ - NH4+ - SO42– - NO3– - H2O at tropospheric temperatures. J. Phys. Chem. A 1998, 102, 2137–2154. [Google Scholar]
- Clegg S. L.; Seinfeld J. H.; Brimblecombe P. Thermodynamic modelling of aqueous aerosols containing electrolytes and dissolved organic compounds. J. Aerosol Sci. 2001, 32, 713–738. [Google Scholar]
- Kulmala M.; Vesala T. Condensation in the continuum regime. J. Aerosol Sci. 1991, 22, 337–346. [Google Scholar]
- Vesala T.; Kulmala M.; Rudolf R.; Vrtala A.; Wagner P. E. Models for condensational growth and evaporation of binary aerosol particles. J. Aerosol Sci. 1997, 28, 565–598. [Google Scholar]
- Fuchs N. A.; Sutugin A. G.. Highly Dispersed Aerosols; Ann Arbor Science Publisher: London, U.K., 1970. [Google Scholar]
- Clegg S. L.; Seinfeld J. H. Thermodynamic models of aqueous solutions containing inorganic electrolytes and dicarboxylic acids at 298.15 K. II. Systems including dissociation equilibria. J. Phys. Chem. A 2006, 110, 5718–5734. [DOI] [PubMed] [Google Scholar]
- Hansen H. K.; Rasmussen P.; Fredenslund A.; Schiller M.; Gmehling J. Vapor-liquid equilibria by UNIFAC group contribution. 5. Revision and extension. Ind. Eng. Chem. Res. 1991, 30, 2352–2355. [Google Scholar]
- Wittig R.; Lohmann J.; Gmehling J. Vapor-liquid equilibria by UNIFAC group contribution. 6. Revision and extension. Ind. Eng. Chem. Res. 2003, 42, 183–188. [Google Scholar]
- Balslev K.; Abildskov J. UNIFAC parameters for four new groups. Ind. Eng. Chem. Res. 2002, 41, 2047–2057. [Google Scholar]
- Peng C.; Chan M. N.; Chan C. K. The hygroscopic properties of dicarboxylic and multifunctional acids: Measurements and UNIFAC predictions. Environ. Sci. Technol. 2001, 35, 4495–4501. [DOI] [PubMed] [Google Scholar]
- Riipinen I.; Svenningsson B.; Bilde M.; Gaman A.; Lehtinen K. E. J.; Kulmala M. A method for determining thermophysical properties of organic material in aqueous solutions: Succinic acid. Atmos. Res. 2006, 82, 579–590. [Google Scholar]
- Semmler M.; Luo B. P.; Koop T. Densities of liquid H+/NH4+/SO42-/NO3–/H2O solutions at tropospheric temperatures. Atmos. Environ. 2006, 40, 467–483. [Google Scholar]
- Hyvärinen A.-P.; Lihavainen H.; Gaman A.; Vairila L.; Ojala H.; Kulmala M.; Viisanen Y. Surface tensions and densities of oxalic, malonic, succinic, maleic, malic, and cis-pinonic acids. J. Chem. Eng. Data 2006, 51, 255–260. [Google Scholar]
- Chen J.-P. Theory of deliquescence and modified Köhler curves. J. Atmos. Sci. 1994, 51, 3505–3516. [Google Scholar]
- Topping D. O.; McFiggans G. B.; Kiss G.; Varga Z.; Facchini M. C.; Decesari S.; Mircea M. Surface tensions of multi-component mixed inorganic/organic aqueous systems of atmospheric significance: measurements, model predictions and importance for cloud activation predictions. Atmos. Chem. Phys. 2007, 7, 2371–2398. [Google Scholar]
- Laskin A.; Moffet R. C.; Gilles M. K.; Fast J. D.; Zaveri R. A.; Wang B.; Pascal N.; Shutthanandan J. Tropospheric chemistry of internally mixed sea salt and organic particle: Surprising reactivity of NaCl with weak organic acids. J. Geophys. Res. 2012, 117, D15303. [Google Scholar]
- Lim Y. B.; Tan Y.; Perri M. J.; Seitzinger S. P.; Turpin B. J. Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Atmos. Chem. Phys. 2010, 10, 10521–10539. [Google Scholar]
- Ngueyen T. B.; Lee P. B.; Updyke K. M.; Bones D. L.; Laskin J.; Laskin A.; Nizkorodov A. Formation of nitrogen- and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols. J. Geophys. Res. 2012, 117, D01207. [Google Scholar]
- Ghorai S.; Laskin A.; Tivanski A. V. Spectroscopic evidence of keto-enol tautomerism in deliquesced malonic acid particles. J. Phys. Chem. A 2011, 115, 4373–4380. [DOI] [PubMed] [Google Scholar]
- Zardini A. A.; Sjogren S.; Marcolli C.; Krieger U. K.; Gysel M.; Weingartner E.; Baltensperger U.; Peter T. A combined particle trap/HTDMA hygroscopicity study of mixed inorganic/organic aerosol particles. Atmos. Chem. Phys. 2008, 8, 5589–5601. [Google Scholar]
- Sjogren S.; Gysel M.; Weingartner E.; Baltensperger U.; Cubison M. J.; Coe H.; Zardini A. A.; Marcolli C.; Krieger U. K.; Peter T. Hygroscopic growth and water uptake kinetics of two-phase aerosol particles consisting of ammonium sulfate, adipic and humic acid mixtures. J. Aerosol Sci. 2007, 38, 157–171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.