

Abstract
Five new sesquiterpenes, neurolobatin A (1), neurolobatin B (2), 5β-hydroxy-8β-isovaleroyloxy-9α-hydroxycalyculatolide (3), 3-epi-desacetylisovaleroylheliangine (4), and 3β-acetoxy-8β-isovaleroyloxyreynosin (5), were isolated from the aerial parts of Neurolaena lobata. The structures were established by means of a combined spectroscopic data analysis, including ESIMS, APCI-MS, and 1D- and 2D-NMR techniques. Neurolobatin A (1) and B (2) are unusual isomeric seco-germacranolide sesquiterpenes with a bicyclic acetal moiety, compounds 3 and 4 are unsaturated epoxy-germacranolide esters, and compound 5 is the first eudesmanolide isolated from the genus Neurolaena. The isolated compounds (1–5) were shown to have noteworthy antiproliferative activities against human tumor cell lines (A2780, A431, HeLa, and MCF7). The anti-inflammatory effects of 1–5, evaluated in vitro using LPS- and TNF-α-induced IL-8 expression inhibitory assays, revealed that all these compounds strongly down-regulated the LPS-induced production of IL-8 protein, with neurolobatin B (2) and 3-epi-desacetylisovaleroylheliangine (4) being the most effective.
Neurolaena lobata (L.) R. Br. ex Cass. (Asteraceae) is a herbaceous plant distributed widely in Central America and northwestern parts of South America. In Caribbean traditional medicine, the leaves of this plant have been used for the treatment of different types of cancer, ulcers, inflammatory skin disorders, diabetes, and pain of various origins. In some regions, N. lobata is also used to treat or prevent a variety of parasitic ailments, such as malaria, fungus, ringworm, and amebic and intestinal parasites.1−3
Previous investigations of the crude leaf extract proved its antiulcerogenic,4 antinociceptive,5 antiparasitic,6,7 and antiviral activities.8 Moreover, of particular relevance to the present study, a N. lobata leaf extract was found to exert anti-inflammatory activity in a carrageenan-induced mouse paw edema test.9 It was reported recently that the 80% EtOH extract of N. lobata leaves reduces LPS-stimulated TNF-α production in THP-1 monocytes, and isolated sesquiterpene lactones were identified as active principles of the extract.10 The cytotoxic activities of an aqueous extract and sesquiterpene lactones of N. lobata have been examined in vitro against GLC4 human small cell lung carcinoma and COLO 320 human colorectal cancer cells.11 Antineoplastic properties of the dichloromethane extract have also been investigated in HL-60 cells and in anaplastic large cell lymphoma cell lines of human or mouse origin. The extract reproducibly demonstrated antiproliferative and proapoptotic effects. Furthermore, it was shown that the extract down-regulated the expression of oncogenes, induced tumor suppressors, inhibited cell proliferation, and triggered the apoptosis of malignant cells.12
Phytochemical studies on N. lobata have led to the isolation of sesquiterpene lactones of the germacranolide (neurolenins A–F and lobatin A) and furanoheliangolide (lobatins B and C, 8β-isovalerianyloxy-9α-hydroxycalyculatolide and 8β-isovalerianyloxy-9α-acetoxycalyculatolide) types.13−15N. lobata has also been investigated for pyrrolizidine alkaloids, and tussilagine, isotussilagine, and the methyl ester of their possible biosynthetic precursor, 2-pyrrolidineacetic acid, have been detected in the MeOH leaf extract.16 Moreover, 12 6-hydroxy- and 6-methoxy flavonoids have been described from this plant.17
The present study was designed to isolate new promising bioactive metabolites from N. lobata. Reported herein are the isolation and structure elucidation of five new sesquiterpene lactones, 1–5, including two unusual seco derivatives with a bicyclic acetal moiety (1 and 2), from the CH2Cl2 extract of the dried herb. The isolated compounds were evaluated for their antiproliferative activities against four human cancer cell lines and for their effects on TNF-α- and LPS-induced IL-8 expression in vascular endothelial cells.

Results and Discussion
Five sesquiterpenes (1–5) were isolated from the CH2Cl2-soluble phase of the MeOH extract prepared from the aerial parts of N. lobata by a combination of different chromatographic methods, including column chromatography (CC), vacuum liquid chromatography (VLC), rotation planar chromatography (RPC), and preparative TLC. The structure elucidation was carried out by extensive spectroscopic analysis, using one- and two-dimensional (1D- and 2D-) NMR (1H–1H COSY, HSQC, HMBC, and NOESY) spectroscopic and HRESIMS experiments.
Compounds 1 and 2 were isolated as amorphous solids with [α]27D −89 (c 0.2, CHCl3) and [α]27D −32 (c 0.2, CHCl3), respectively. HRESIMS and detailed 1D- and 2D-NMR spectroscopic studies led to the determination of two isomeric seco-sesquiterpene lactones (1 and 2) with a bicyclic acetal moiety on the basis of the following arguments.
HRESIMS suggested that the two compounds are stereoisomers with the molecular composition, C20H26O8, according to the quasimolecular ion peaks at m/z 417.1525 (1) and 417.1530 (2) [M + Na]+ (calcd for C20H26O8Na, 417.1525). On APCIMS, the quasimolecular ion was detected at m/z 395 [M + H]+, with the sequential loss of H2O and a saturated C5 acid (m/z 377 and 293). Analysis of the 1H and 13C NMR spectra confirmed that the two compounds are stereoisomers, exhibiting closely comparable spectroscopic features. The 1H and 13C NMR spectra of 1 and 2 revealed the presence of an isovaleroyl group in the molecule [1, δH 2.16 m, 2.01 m, 0.92 and 0.93 d, δC 172.5, 42.9, 25.6, and 2 × 22.3; 2, δH 2.20 m, 2.04 m, 0.93 and 0.92 d, δC 172.2, 43.0, 25.6, and 2 × 22.3]. Informative signals for 1/2 at δH 4.91/4.71 (H-6), 3.45/3.23 (H-7), 5.38/5.47 (H-13a), and 6.26/6.25 (H-13b) and δC 73.7/74.1 (C-6), 46.3/46.2 (C-7), 134.1/134.7 (C-11), 169.2 (C-12), and 121.3 (C-13) additionally indicated the presence of a trans-fused α-methylene-γ-lactone ring with H-6 in the β position.14,18 With this partial structure as starting point, consecutive analysis of the 1H and 13C NMR spectra and 2D homo- (1H–1H COSY) and heteronuclear correlation data (HSQC and HMBC) resulted in complete 1H and 13C NMR assignments (Table 1). The determined chemical shifts could be interpreted only with the constitutions depicted in structural formulas 1 and 2. In the HMBC spectra, no correlations were detected between H-9 and C-10 or between H-15 and C-9, indicating fission of the C-9–C-10 bond in both compounds. On the other hand, an HMBC correlation was observed between H-9 and C-4, suggesting that the bond fission was followed by ring closure between C-4 and C-9. In view of the molecular composition determined, the chemical shift of C-9 (98.8 in 1 and 102.0 in 2) revealed the presence of a bicyclic acetal moiety in both compounds. The HMBC cross-peak of the carbonyl carbon signal (isovaleroyl CO) with the H-8 signal demonstrated the presence of the ester group at C-8.
Table 1. NMR Spectroscopic Data [CDCl3, 800 MHz (1H), 200 MHz (13C), δ (ppm)] for Neurolobatins A (1) and B (2).
| neurolobatin
A (1) |
neurolobatin
B (2) |
|||||||
|---|---|---|---|---|---|---|---|---|
| position | δH (J in Hz) | δC | HMBCa | NOESY | δH (J in Hz) | δC | HMBCa | NOESY |
| 1 | 194.8 | 194.7 | ||||||
| 2a | 2.77 dd (16.4, 3.2) | 34.5 | C-1, C-3 | H-3, H-5a, H-15, H-6 | 2.81 dd (16.9, 2.5) | 35.9 | C-1, C-3 | H-3, H-9 |
| 2b | 3.27 dd (16.4, 9.9) | C-1, C-3 | H-3, H-5a, H-6 | 3.22 dd (16.9, 10.1) | C-1, C-3 | H-3, H-9 | ||
| 3 | 4.07 dd (9.7, 2.7) | 79.0 | C-1, C-4, C-5, C-15 | H-2a, H-2b, H-9, H-14, H-15 | 4.14 dd (10.1, 2.6) | 80.7 | C-1, C-2, C-4, C-5, C-15 | H-2a, H-2b, H-9, H-14, H-15 |
| 4 | 81.9 | 79.0 | ||||||
| 5a | 2.40 dd (14, 4.1) | 40.7 | C-6, C-7 | H-2a, H-2b, H-6, H-15 | 2.45 dd (13.8, 5.1) | 38.5 | C-3, C-4, C-6, C-7, C-15 | H-6 |
| 5b | 1.84 dd (14.0, 11.8) | C-3, C-6, C-7, C-15 | H-6, H-15 | 2.01 dd (13.8, 11.8) | C-6, C-7 | |||
| 6 | 4.91 ddd (11.8, 9.4, 3.9) | 73.7 | C-7 | H-2a, H-2b, H-5b | 4.71 td (10.5, 5.1) | 74.1 | C-7 | H-5a, H-9, H-15 |
| 7 | 3.45 dddd (9.0, 4.1, 3.7, 3.1) | 46.3 | C-5, C-6, C-12 | H-5a, H-8 | 3.23 dq (9.9, 3.1) | 46.2 | C-5, C-6, C-12 | H-13a |
| 8 | 5.40 t (4.1) | 68.4 | C-6, C-1′ | H-9 | 5.43 t (2.5) | 67.1 | C-6, C-1′ | |
| 9 | 5.62 d (4.1) | 98.9 | C-4, C-7 | H-3, H-8, H-15 | 5.35 d (1.9) | 102.0 | C-4, C-7, C-8 | H-2a, H-2b, H-3, H-6 |
| 10 | 196.4 | 196.4 | ||||||
| 11 | 134.1 | 134.7 | ||||||
| 12 | 169.2 | 169.2 | ||||||
| 13a | 5.38 d (3.1) | 121.3 | C-7, C-11 | 5.47 d (3.4) | 121.3 | C-7, C-11 | H-7 | |
| 13b | 6.26 d (3.7) | C-7, C-11, C-12 | 6.25 d (3.4) | C-7, C-11, C-12 | ||||
| 14 | 2.37 s | 23.5 | C-10 | H-3 | 2.40 s | 23.5 | C-10 | H-3 |
| 15 | 1.40 s | 23.0 | C-3, C-4, C-5 | H-2a, H-3, H-5a, H-5b, H-9 | 1.48 s | 26.0 | C-3, C-4, C-5 | H-3, H-6 |
| 1′ | 172.5 | 172.2 | ||||||
| 2′ | 2.16 m | 42.9 | C-3′, C-4′, C-5′ | 2.20 m | 43.0 | C-3′, C-4′, C-5′ | ||
| 3′ | 2.01 m | 25.6 | C-2′, C-4′, C-5′ | 2.04 m | 25.6 | C-2′, C-4′, C-5′ | ||
| 4′ | 0.92 d (6.6) | 22.3 | C-2′, C-3′ | 0.93 d (6.7) | 22.3 | C-2′, C-3′ | ||
| 5′ | 0.93 d (6.6) | 22.3 | C-2′, C-3′ | 0.92 d (6.7) | 22.3 | C-2′, C-3′ | ||
HMBC correlations are from proton(s) stated to the indicated carbon.
To determine the stereochemistry of 1 and 2, the coupling constants and nuclear Overhauser effects detected in a NOESY experiment (Table 1) were analyzed. The relative configuration of the lactone ring and C-8 were similar to those determined in sesquiterpenes isolated from N. lobata earlier.14,18 Thus, H-6 was in a β orientation, while H-7 and H-8 were in α positions in compounds 1 and 2. The coupling constants (4.1 and 1.9 Hz in 1 and 2, respectively) observed between H-8 and H-9 suggested that these protons are in equatorial positions in both compounds. The most significant difference in the NOE correlations was observed for H-6. In the case of 1, a strong NOE correlation was detected between H-6 and the two H-2 protons, while a strong correlation between H-6 and the H-15 methyl protons was detected instead of this in 2. These data indicated a seco-germacranolide structure of 1 and 2 for the two stereoisomers, named neurolobatin A (1) and neurolobatin B (2). The oxygen in the eight-membered ring in 1 and in the seven-membered ring in 2 are in the β position. It was assumed that the two isomers have a common origin: the compounds may be formed from germacranolide-type sesquiterpenes after ring-opening between C-9 and C-10, followed by formation of the bicyclic acetal structure.
Compound 3 was isolated as an amorphous solid with [α]27D +53 (c 0.2, CHCl3). Its HRESIMS displayed a quasimolecular ion peak at m/z 395.1701 [M + H]+, indicating the molecular formula, C20H26O8. Characteristic fragment ions at m/z 311, 293, 275, and 257 indicated the sequential loss of H2O and a saturated C5 acid. In accordance with this, the 1H and 13C NMR spectra of 3 confirmed the presence of an isovaleroyl group (Table 2). Additionally, the 1D- and 2D-NMR spectra exhibited resonances for a carbonyl group (δC 210.2), an α-methylene-γ-lactone ring (δH 4.41 dd, 3.74 dt, 6.36 and 5.84 d, δC 74.3, 43.9, 139.1, 168.6, and 125.1), a quaternary carbon (δC 91.2), a trisubstituted olefin (δH 5.67 d, δC 104.9 and 191.9), four methines (δH 3.39 dq, 4.49 t, 5.03 and 4.09 d, δC 38.0, 73.7, 77.3, and 72.7), and one secondary (δH 1.33 d, δC 9.0) and one tertiary methyl group (δH 1.48 s, δC 18.4) (Table 2). The chemical shifts and coupling constants of 3 were closely related to those of the 1-keto-furanoheliangolide derivative 8β-isovaleroyloxy-9α-hydroxycalyculatolide,14 with the only difference being the absence of the signals of a methylene and the appearance of the signal of a methine group (δH 4.49 t, δC 73.7). This methine was assigned as C-5 with regard to the HMBC correlations between C-5 and H-6, C-5 and H-7, and C-5 and H-15 (Figure 1). The relative configuration of compound 3 was studied by means of a NOESY experiment. This was deduced by starting from the β orientation of H-6 and α orientation of H-7, indicated by the coupling constant J6,7 = 4.9 Hz and found in all sesquiterpenes isolated previously from N. lobata. Cross-peaks between H-6 and H-15 proved the β orientation of the 15-methyl group, while NOE effects observed between H-7 and H-5, H-5 and H-4, H-7 and H-13b, and H-13b and H-8 dictated the α orientation of these protons. All of the above evidence supported the structure of this compound as 5β-hydroxy-8β-isovaleroyloxy-9α-hydroxycalyculatolide (3).
Table 2. NMR Spectroscopic Data [CDCl3, 500 MHz (1H), 125 MHz (13C), δ (ppm)] for Compounds 3–5.
|
3a |
4 |
5b |
||||
|---|---|---|---|---|---|---|
| position | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC |
| 1 | 210.2 | 2.57 dd (9.6, 5.0) | 59.7 | 3.61 dd (11.5, 4.5) | 76.4 | |
| 2 | 5.67 d (0.7) | 104.9 | 2.36 dt (14.8, 4.9) | 34.5 | 2.20 m | 36.8 |
| 1.68 ddd (14.8, 11.9, 1.6) | 1.63 m | |||||
| 3 | 191.9 | 4.96 dd (11.9, 5.0) | 66.8 | 5.18 dd (11.5, 5.5) | 70.5 | |
| 4 | 3.39 dq (6.8, 6.8) | 38.0 | 141.9 | 139.9 | ||
| 5 | 4.49 t (8.0, 6.9) | 73.7 | 5.28 d (11.1) | 124.6 | 2.22 m | 50.9 |
| 6 | 4.41 dd (8.1, 4.9) | 74.3 | 5.53 dd (11.1, 2.2) | 72.9 | 4.56 t (11.0) | 74.6 |
| 7 | 3.74 dt (1.6, 4.9) | 43.9 | 2.88 s | 48.7 | 2.83 dd (11.0, 2.3) | 52.2 |
| 8 | 5.03 d (4.9) | 77.3 | 5.21 t (2.4) | 75.5 | 5.75 dd (2.3, 4.0) | 65.8 |
| 9 | 4.09 d (4.9) | 72.7 | 2.71 dd (15.2, 2.4) | 43.8 | 2.32 dd (15.5, 1.5) | 40.6 |
| 1.29 dd (15.2, 2.4) | 1.58 dd (15.5, 3.0) | |||||
| 10 | 91.2 | 57.1 | 42.9 | |||
| 11 | 139.1 | 137.1 | 134.5 | |||
| 12 | 168.6 | 169.1 | 169.9 | |||
| 13a | 6.36 d (3.1) | 125.1 | 6.40 d (2.0) | 125.5 | 6.17 d (3.0) | 120.1 |
| 13b | 5.84 d (2.8) | 5.79 d (2.0) | 5.47 d (3.0) | |||
| 14 | 1.48 s | 18.4 | 1.52 s | 19.0 | 0.99 s | 13.9 |
| 15 | 1.33 d (6.9) | 9.0 | 1.88 s | 17.3 | 5.20 s | 108.6 |
| 5.09 s | ||||||
| 1′ | 171.7 | 172.0 | 172.5 | |||
| 2′ | 2.11 m (2H) | 42.8 | 2.18 m (2H) | 43.7 | 2.18 d (7.5) (2H) | 43.8 |
| 3′ | 1.98 sept (6.8) | 25.2 | 2.04 m | 25.8 | 2.06 m | 25.9 |
| 4′ | 0.91 d (6.7) | 22.3 | 0.90 d (6.6) | 22.5 | 0.94 d (6.7) | 22.7 |
| 5′ | 0.90 d (6.7) | 22.3 | 0.91 d (6.6) | 22.5 | 0.94 d (6.7) | 22.7 |
OH groups: δ 4.23 brs, 3.31 brs.
1-OH group: δ 2.04 brs, 3-O-acetate group: δH 2.14 s, δC 170.2, 21.4).
Figure 1.

Key COSY (—) and HMBC correlation of compound 3.
Compound 4 was obtained as a colorless gum. From the molecular ion peak at m/z 365.1961 [M + H]+, (calcd for 365.1959) in the positive-ion HRESIMS, its molecular formula was determined to be C20H28O6. The 1H NMR spectrum showed the presence of signals due to an isovaleroyl side chain (Table 2). Further, the JMOD spectrum suggested that the skeleton consists of 15 carbons, including two methyls, three methylenes, six methines, and four quaternary carbons. From the HSQC spectrum, the chemical shifts of the protonated carbons were assigned, and the proton–proton connectivities were then studied. The 1H–1H COSY spectrum was used to define two structural fragments with correlated protons: −CHR-CH2-CH– (A) (δH 2.57, 2.36, 1.68, and 4.96) (C-1–C-3) and =CH–CHR-CHR-CHR-CH2– (B) (δH 5.28, 5.53, 2.88, 5.21, 2.71, and 1.29) (C-5–C-9). These structural parts and tertiary methyls and quaternary carbons were connected by inspection of the long-range C–H correlations observed in the HMBC spectrum. The two- and three-bond correlations between the quaternary carbon C-4 and H-6, H-15, and H2-2 and between the quaternary C-10 and H-8, H2-2, H-14, and H2-9 signals revealed that structural fragment A together with C-10, C-4, and the 14- and 15-methyl groups forms a germacrane skeleton. The lactone ring connected to the macrocycle in positions C-6, C-7 was evident from the HMBC cross-peaks between C-12 and H2-13, C-12 and H-6, C-11 and H2-13, and C-11 and H-7. The position of the ester group was proved by the long-range correlation between the ester carbonyl group (δC 172.0) and H-8 (δH 5.21 t). The remaining epoxy and hydroxy groups, which were elucidated from the molecular composition, were placed at C-10–C-1 and C-3, respectively, with regard to the 13C NMR chemical shifts (δC-10 57.1, δC-1 59.7, and δC-3 66.8) and literature data for similar epoxy germacranolides.19 The relative configuration of the chiral centers was studied by NOESY measurements (Figure 2). Diagnostic NOE correlations were detected between H-6 and H-3 and H-6 and H-14, demonstrating the β orientation of these protons. Furthermore, NOESY cross-peaks were observed between H-1 and H-2a, H-2b and H-3, H-14 and H-9a, and H-7 and H-8, indicating the α-oriented H-1, H-2a, H-7, H-8, and H-9b and the β orientation of H-9a and H-2b. All of the above evidence was used to propose the structure of this compound as depicted in structural formula 4. Compound 4 is the 3-epimer of desacetylisovaleroylheliangine, reported from Calea megacephala (Asteraceae) earlier. Accordingly, significant differences can be observed in their 1H NMR chemical shifts and coupling contants of H-3.19
Figure 2.

Key NOESY correlation of compound 4.
Compound 5 was isolated as a colorless oil with [α]27D +5 (c 0.1, CHCl3). It was shown by HRESIMS to have the molecular formula C22H30O7m/z 407.2065 [M + H]+, (calcd for 407.2064). The 1H and 13C NMR spectra of 5 revealed the presence of one acetate (δH 2.14 s; δC 170.2 and 21.4) and one isovaleroyl group in the molecule (δH 2.18 d, 2.06 m, and 2 × 0.94 d; δC 172.5, 43.8, 25.9, and 2 × 22.7). Additionally, the 13C and JMOD spectra suggested a sesquiterpene skeleton consisting of one methyl, four methylenes, six methines, and four quaternary carbons (Table 2). From the HSQC spectrum, the chemical shifts of the protonated carbons were assigned, and the 1H–1H COSY spectrum was then evaluated, which indicated two structural elements: −CHR-CH2-CHR– (A) (δH 3.61, 2.20, 1.63, and 5.18) (C-1–C-3) and −CHR-CHR-CHR-CHR-CH2– (B) (δH 2.22, 4.56, 2.83, 5.75, 2.32, and 1.58) (C-5–C-9). Their connectivities were determined from the HMBC spectrum observed between the quaternary carbons and protons of the structural fragments A and B (C-4 and H-3, H-5, H-6, and H-2b; C-10 and H-1, H-6, H-8, H-9b, and H-15; C-11 and H-13a, H-6, and H-7; C-12 and H-13a, and 13b). The positions of the ester groups were also established via the HMBC experiment on the basis of the 3JC,H couplings of the acetyl and isovaleroyl CO (δC 170.2 and 172.5) and skeletal protons H-3 (δH 5.18) and H-8 (δH 5.75), respectively.
The relative configuration of 5 was studied by means of a NOESY experiment. Starting from the α orientation of H-5, characteristic cross-peaks between H-5 and H-7, H-7 and H-8, H-9b and H-1, and H-1 and H-3 confirmed the β orientation of these protons, while the NOE interactions of H-6 and H-14, H-14 and H-2b, and H-14 and H-9a revealed the α orientation of H-6 and the 14-methyl group. All of the above evidence proved the eudesmanolide structure 3β-acetoxy-8β-isovaleroyloxyreynosin (5) for this compound. Eudesmanolides occur widely in the family Asteraceae,20 but this is the first such isolation from the genus Neurolaena.
The isolated compounds were tested for antiproliferative activities against a set of human adherent cell lines (A2780, A431, HeLa, and MCF7). All compounds except for 3 inhibited the proliferation of A431 and A2780 cells and were less active against MCF7, while HeLa cells demonstrated the lowest sensitivity (Table 3). The IC50 values of 2, 4, and 5 against A431 and MCF7 cells were comparable to those of the reference agent cisplatin. Neurolobatin A (1) and 5β-hydroxy-8β-isovaleroyloxy-9α-hydroxycalyculatolide (3) exhibited relatively low potencies on all tested cell lines, and were completely ineffective against HeLa and MCF7 cells at the concentrations used.
Table 3. Antiproliferative Effects of the Isolated Compounds (1–5) on Different Human Tumor Cell Lines.
| calculated
IC50 (μM) ± SEM |
||||
|---|---|---|---|---|
| compd | HeLa | A431 | A2780 | MCF7 |
| neurolobatin A (1) | >10 | >10 | 9.8 ± 1.0 | >10 |
| neurolobatin B (2) | >10 | 6.8 ± 0.56 | 5.4 ± 0.67 | >10 |
| 5β-hydroxy-8β-isovaleroyloxy-9α-hydroxy-calyculatolide (3) | >10 | >10 | >10 | >10 |
| 3-epi-desacetylisovalerylheliangine (4) | >10 | 7.2 ± 0.99 | 4.7 ± 0.19 | 7.2 ± 0.57 |
| 3β-acetoxy-8β-isovaleroyloxyreynosin (5) | >10 | 5.3 ± 0.47 | 7.2 ± 0.42 | >10 |
| cisplatin | 5.7 ± 0.84 | 8.8 ± 0.97 | 0.86 ± 0.12 | 8.0 ± 1.1 |
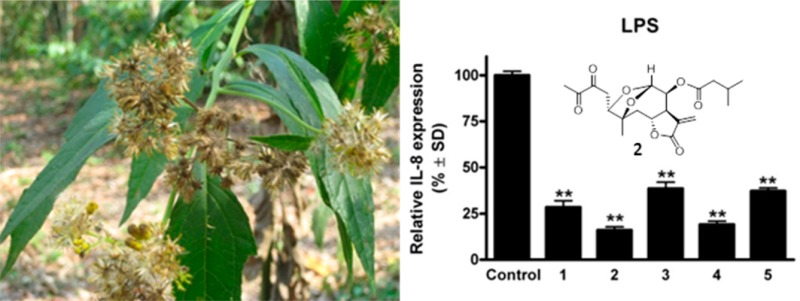
To investigate the anti-inflammatory effects of the compounds, the extent of inhibition of LPS- and TNF-α-induced IL-8 expression was determined in HUVEC-tert endothelial cells. Treatment with the sesquiterpene lactones decreased the LPS-induced secretion of the cytokine IL-8 (Figure 3). All the compounds down-regulated the LPS-induced production of IL-8 protein, with neurolobatin B (2) and 3-epi-desacetylisovalerylheliangine (4) being the most effective. However, the test compounds (1–5) did not significantly influence the production of IL-8 in TNF-α-treated endothelial cells. Moreover, in contrast to the positive control BAY (decrease of IL-8 expression 23.7 ± 4% in case of TNF-α, and 12.7 ± 4.2% in case of LPS), the isolated sesquiterpenes were selective as they inhibited only the LPS-induced IL-8 production.
Figure 3.

Action of sesquiterpene lactones (1–5) on IL-8 production in HUVEC-tert cells stimulated by LPS (100 ng/mL, left panel) or TNF-α (30 ng/mL, right panel). Effects of LPS and TNF-α were analyzed within the same experiment. Results of a representative experiment are shown (** indicates p < 0.01 as compared with the control condition).
Experimental Section
General Experimental Procedures
Optical rotations were determined in chloroform by using a Perkin-Elmer 341 polarimeter. The NMR spectra of 1 and 2 were recorded in CDCl3 (Merck, Darmstadt, Germany) in a Shigemi sample tube at 298 K, using a Varian 800 MHz NMR spectrometer equipped with a 1H{13C/15N} triple resonance 13C enhanced salt tolerant cold probe operating at 799.9 and 201 MHz for 1H and 13C nuclei, respectively. Chemical shifts were referenced to TMS (1H) or to residual solvent resonances. Pulse sequences (1H,1H-Presat, GHSQCAD, GHMBCAD, and NOESY-1D) were taken from the VNMRJ-3.2 software library without any modification. The NMR spectra of 3–5 were recorded in CDCl3 on a Bruker Avance DRX 500 spectrometer at 500 MHz (1H) and 125 MHz (13C). 2D-NMR data were acquired and processed with standard Bruker software. In the COSY, HSQC, and HMBC experiments, gradient-enhanced versions were used. HRMS analyses were performed on an LTQ FT Ultra (Thermo Fisher Scientific, Bremen, Germany) system. The ionization method was ESI and operated in positive ion mode. For CID experiments, helium was used as the collision gas, and normalized collision energy (expressed as a percentage), which is a measure of the amplitude of the resonance excitation RF voltage applied to the end-caps of the linear ion trap, was used to bring about fragmentation. The protonated molecular ion peaks were fragmented by CID at a normalized collision energy of 35%. The samples were dissolved in MeOH. Data acquisition and analysis were accomplished with Xcalibur software version 2.0 (Thermo Fisher Scientific). APCIMS measurements were performed on API 2000 triple quadrupole mass spectrometer equipped with an atmospheric-pressure chemical ionization interface (AB SCIEX, Framingham, MA, USA). The samples were dissolved in acetonitrile. Column chromatography (CC) was carried out on polyamide (ICN), vacuum liquid chromatography (VLC) on silica gel G (15 mm, Merck), preparative thin-layer chromatography (preparative TLC) on silica gel 60 F254 and RP-18 F254 plates (Merck), and rotation planar chromatography (RPC) on silica gel 60 GF254 with a Chromatotron instrument (model 8924, Harrison Research).
Plant Material
Neurolaena lobata (L.) R. Br. ex Cass. was collected 0.5 km NNW of San José in the area of the Chakmamantok-rock formation (16 59′16″ N, 89 53′45″ W), near the northwestern shore of Lago Petén Itzá, Departamento Petén, Guatemala, and within the botanical garden of the Institute for Ethnobiology, San José, Guatemala (Unger et al., 201312). Voucher specimens (leg. R. Diaz, det. R. O. Frisch 03. 02. 2011/a,b, Chakmamantok) were archived at the herbarium of the Institute for Ethnobiology, San Jose, Guatemala. A voucher specimen (no. 813) has also been deposited at the Herbarium of the Department of Pharmacognosy, University of Szeged, Szeged, Hungary. The fresh plant material (the aerial plant parts, leaves, caulis, and florescence) of N. lobata was air-dried in Guatemala, sent to Austria, and stored deep-frozen until subsequent extraction.
Extraction and Isolation
Dried and ground aerial parts of the plant (3.00 kg), which was stored at −70 °C before processing, was crushed in a blender and then percolated with MeOH (50 L) at room temperature. The crude extract was concentrated in vacuo and subjected to solvent–solvent partitioning with 5 × 1 L petroleum ether (A), then with 5 × 1 L of CH2Cl2 (B), and finally with 5 × 1 L of EtOAc (C). The CH2Cl2 phase (95.4 g) was chromatographed on a polyamide column (287 g) with mixtures of MeOH and H2O (1:4, 2:3, 3:2, and 4:1, 3 L of each) as eluents. Fractions with similar compositions were combined according to TLC monitoring, affording fractions BI–BVII. Fraction BII (33.6 g), obtained with MeOH–H2O (1:4), was subjected to silica gel VLC, using a gradient system of cyclohexane–EtOAc–EtOH (from 30:10:0 to 30:30:6). The CC fractions were combined after TLC monitoring into nine fractions (BII/1-BII/9). Fraction BII/7 was separated by VLC with a CH2Cl2–acetone gradient system (from 99:1 to 4:1 and finally MeOH). The subfraction eluted with CH2Cl2–acetone 19:1 was subjected to preparative TLC, using a system of cyclohexane–EtOAc–EtOH (60:40:1), to give compounds 1 (3.2 mg) and 2 (2.8 mg). Moreover, the subfraction obtained with CH2Cl2–acetone 80:20 was separated by RPC with a gradient system of cyclohexane–EtOAc–EtOH (from 60:20:0.5 to 60:60:1) and then further purified by preparative TLC and RP-TLC on silica gel with the mobile phase cyclohexane–EtOAc–EtOH (30:30:1) and MeOH–H2O (7:3), affording the isolation of compound 4 (1.8 mg). The subfraction eluted with MeOH was further fractionated by VLC, using a mobile phase of cyclohexane–EtOAc–EtOH (from 30:10 to 30:30:1) and finally subjected to preparative RP-TLC and NP-TLC, using MeOH–H2O 7:3 and CH2Cl2–MeOH (18:1), to yield compound 5 (3.5 mg). Finally, a subsequent subfraction obtained with MeOH was purified by preparative RP-TLC on silica gel with MeOH–H2O (7:3) and then by preparative TLC, using cyclohexane–EtOAc–EtOH (30:30:1) as developing system on silica gel, resulting in the isolation of compound 3 (7.8 mg).
Neurolobatin A (1)
Colorless gum; [α]27D −89 (c 0.2, CHCl3); 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 449.1784 [M + MeOH + Na]+ (calcd for C21H30O9Na, 449.1788); 417.1525 [M + Na]+ (calcd for C20H26O8Na, 417.1525); APCIMS m/z 395 [M + H]+, 377 [M + H – H2O]+, 293 [M + H – 102]+.
Neurolobatin B (2)
Colorless gum; [α]27D −32 (c 0.2, CHCl3); 1H NMR and 13C NMR data, see Table 1; HRESIMS m/z 449.1788 [M + MeOH + Na]+ (calcd for C21H30O9Na, 449.1788); 417.1530 [M + Na]+ (calcd for C20H26O8Na, 417.1525); APCIMS m/z 395 [M + H]+, 377 [M + H – H2O]+, 293 [M + H – 102]+.
5β-Hydroxy-8β-isovaleroyloxy-9α-hydroxycalyculatolide (3)
Amorphous solid; [α]27D +53 (c 0.2, CHCl3); 1H NMR and 13C NMR data, see Table 2; HRESIMS m/z 395.1701 [M + H]+ (calcd for C20H27O8, 395.1700); ESIMS-MS m/z 395.1703 [M + H]+; 377 ([M + H – H2O]+, 33), 359 ([M + H – 2 × H2O]+, 5); 311 ([M + H – C5H8O]+, 45), 293 ([M + H – C5H8O – H2O]+, 100), 275 ([M + H – C5H8O – 2 × H2O]+, 77), 257 ([M + H – C5H8O – 3 × H2O]+, 11), 231([C14H15O3]+, 9), 213 ([C14H13O2]+, 12); 203 ([C13H15O2]+, 5); 185 ([C13H13O]+, 5).
3-Epi-desacetylisovaleroylheliangine (4)
Amorphous solid; [α]27D −33 (c 0.1, CHCl3); 1H NMR and 13C NMR data, see Table 2; HRESIMS m/z 365.1961 [M + H]+ (calcd for C20H29O6; 365.1959); ESIMS-MS m/z 365.2035 [M + H]+; 347 ([M + H – H2O]+, 3); 295 ([C15H19O6]+, 10); 277 ([C15H17O5]+, 32), 263 ([C15H19O4]+, 64), 261 ([C15H17O4]+, 35), 245 ([C15H17O3]+, 100), 227 ([C15H15O2]+, 41), 219 ([C13H15O3]+, 21); 217 ([C14H17O2]+, 20), 201 ([C13H13O2]+, 15), 199 ([C14H15O]+, 19), 179 ([C10H11O3]+, 5), 163 ([C10H11O2]+, 8).
3β-Acetoxy-8β-isovaleroyloxyreynosin (5)
Colorless gum; [α]27D +5 (c 0.1, CHCl3); 1H NMR and 13C NMR data, see Table 2; HRESIMS m/z 407.2065 [M + H]+(calcd for C22H31O7; 407.2064). ESIMS-MS m/z 407.2135 [M + H]+; 329 ([M + H – H2O – CH3COOH]+, 2); 305 ([M + H – C5H10O2]+, 3); 245 ([M + H – C5H10O2 – CH3COOH], 50); 227 ([M + H – C5H10O2 – CH3COOH – H2O]+, 100); 217 ([C14H17O2]+, 11); 209 ([M + H – C5H10O2 – CH3COOH – H2O – H2O]+, 9); 199 ([C14H15Og, 17); 181 ([C14H13], 33); 171 ([C13H15], 6).
Antiproliferative Assay
Antiproliferative effects of the isolated compounds were determined in vitro using four human cell lines (A2780, A431, HeLa, and MCF7, isolated from ovarian cancer, skin epidermoid carcinoma, cervical, and breast cancers, respectively) by means of a MTT ([3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]) assay, as described previously.21 Briefly, a limited number of human cancer cells (5000/well) were seeded onto a 96-well microplate and became attached to the bottom of the well overnight. On the second day of the procedure, the original medium was removed and 200 μL of new medium containing the test substances was added. After an incubation period of 72 h, the living cells were assayed by the addition of 20 μL of 5 mg/mL MTT solution. After a 4 h incubation, the medium was removed and the precipitated formazan was dissolved in 100 μL of DMSO during a 60 min period of shaking. Finally, the reduced MTT was assayed at 545 nm, using a microplate reader. Untreated cells were taken as the negative control, and cisplatin (Ebewe Pharma GmbH, Unterach, Austria) was used as a reference active compound. All cell lines were purchased from the European Collection of Cell Cultures (Salisbury, UK). Stock solutions (10 mM) of the tested compounds were prepared with DMSO. The highest DMSO concentration (0.3%) of the medium did not have any substantial effect on the cell proliferation.15 All in vitro experiments were carried out on two 96-well dishes with at least five parallel wells. Sigmoidal dose–response curves were fitted to the measured points, and the IC50 values were calculated by means of GraphPad Prism 4.0 (GraphPad Software; San Diego, CA, USA).
Anti-inflammatory Assay
Immortalized human umbilical vein endothelial cells were grown in medium 199 containing 20% FBS, 1% PSF (penicillin, streptomycin, fungizone; Lonza), 1% glutamine, and 0.4% ECGS/H (Endothelial Cell Growth Supplement/Heparin; PromoCell). Compounds 1–5 were tested in sixtuplicates. The medium was aspirated and 100 μL of test compounds (1.5× final concentration in medium containing 5% FBS) were added. After 15 min, 50 μL of TNF-α or LPS (3× final concentration) were added to provide final concentrations of 30 μM N. lobata compounds, 30 ng/mL of TNF-α, or 100 ng/mL of LPS. After 6 h, cell culture media were collected and analyzed for IL-8 protein using DuoSet ELISA Development kit (R&D Systems) and the TMB 2-Component Microwell Peroxidase Substrate Kit (VWR International). BAY (Sigma–Aldrich, St. Louis, MO) was used as positive control. Statistical significance of differences was calculated using ANOVA test.
Acknowledgments
Financial support of the Hungarian Scientific Research Fund (OTKA K109846) is gratefully acknowledged. This research was supported by the European Union and the State of Hungary, cofinanced by the European Social Fund in the framework of TÁMOP 4.2.4.A/2-11-1-2012-0001 “National Excellence Program” and TÁMOP-4.2.2.A-11/1/KONV-2012-0035. The financial support of the Austrian Science Foundation (FWF) grant NFN S10713-B13 is gratefully acknowledged by Valery Bochkov.
Supporting Information Available
NMR spectra of compounds 1–5. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Dedication
Dedicated to Prof. Dr. Otto Sticher, of ETH-Zurich, Zurich, Switzerland, for his pioneering work in pharmacognosy and phytochemistry.
Supplementary Material
References
- Giron L. M.; Freire V.; Alonz A.; Caceres A. J. Ethnopharmacol. 1991, 34, 173–187. [DOI] [PubMed] [Google Scholar]
- Hartwell J. L. J. Nat. Prod. 1968, 31, 71–170. [Google Scholar]
- Amiguet V. T.; Arnason J. T.; Maquin P.; Cal V.; Vindas P. S.; Poveda L. Econ. Bot. 2005, 59, 29–42. [Google Scholar]
- Gracioso J. S.; Hiruma-Lima C. A.; Souza Brito A. R. M. Phytomedicine 2000, 7, 283–289. [DOI] [PubMed] [Google Scholar]
- Gracioso J. S.; Paulo M. Q.; Hiruma-Lima C. A.; Souza Brito A. R. J. Pharm. Pharmacol. 1998, 50, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Fujimaki Y.; Kamachi T.; Yanagi T.; Caceres A.; Maki J.; Aoki Y. J. Helminthol. 2005, 79, 23–28. [DOI] [PubMed] [Google Scholar]
- Chinchilla M.; Valerio I.; Sanchez R.; Mora V.; Bagnarello V.; Martinez L.; Gonzalez A.; Vanegas J. C.; Apestegui A. Rev. Biol. Trop. 2012, 60, 881–891. [DOI] [PubMed] [Google Scholar]
- Bedoya L. M.; Alvarez A.; Bermejo M.; Gonzalez N.; Beltran M.; Sanchez-Palomino S.; Cruz S. M.; Gaitan I.; del Olmo E.; Escarcena R.; Garcia P. A.; Caceres A.; Feliciano A. S.; Alcami J. Phytomedicine 2008, 15, 520–524. [DOI] [PubMed] [Google Scholar]
- de Las Heras B.; Slowing K.; Benedi J.; Carretero E.; Ortega T.; Toledo C.; Bermejo P.; Iglesias I.; Abad M. J.; Gomez-Serranillos P.; Liso P. A.; Villar A.; Chiriboga X. J. Ethnopharmacol. 1998, 61, 161–166. [DOI] [PubMed] [Google Scholar]
- Walshe-Roussel B.; Choueiri C.; Saleem A.; Asim M.; Caal F.; Cal V.; Rojas M. O.; Pesek T.; Durst T.; Arnason J. T. Phytochemistry 2013, 92, 122–127. [DOI] [PubMed] [Google Scholar]
- Francois G.; Passreiter C. M.; Woerdenbag H. J.; van Looveren M. Planta Med. 1996, 62, 126–129. [DOI] [PubMed] [Google Scholar]
- Unger C.; Popescu R.; Giessrigl B.; Laimer D.; Heider S.; Seelinger M.; Diaz R.; Wallnofer B.; Egger G.; Hassler M.; Knofler M.; Saleh L.; Sahin E.; Grusch M.; Fritzer-Szekeres M.; Dolznig H.; Frisch R.; Kenner L.; Kopp B.; Krupitza G. Int. J. Oncol. 2013, 42, 338–348. [DOI] [PubMed] [Google Scholar]
- Manchand P. S.; Blount J. F. J. Org. Chem. 1978, 43, 4352–4354. [Google Scholar]
- Passreiter C. M.; Wendisch D.; Gondol D. Phytochemistry 1995, 39, 133–137. [Google Scholar]
- Passreiter C. M. Phytochem. Anal. 1998, 9, 67–70. [Google Scholar]
- Passreiter C. M. Biochem. Syst. Ecol. 1998, 26, 839–843. [Google Scholar]
- Kerr K. M.; Mabry T. J.; Yoser S. Phytochemistry 1981, 20, 791–794. [Google Scholar]
- Passreiter C. M.; Sandoval-Ramirez J.; Wright C. W. J. Nat. Prod. 1999, 62, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Ober A. G.; Urbatsch L. E.; Fischer N. H. Phytochemistry 1987, 26, 1204–1206. [Google Scholar]
- Wu Q. X.; Shi Y. P.; Jia Z. J. Nat. Prod. Rep. 2006, 23, 699–734. [DOI] [PubMed] [Google Scholar]
- Mossmann T. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.