

Abstract
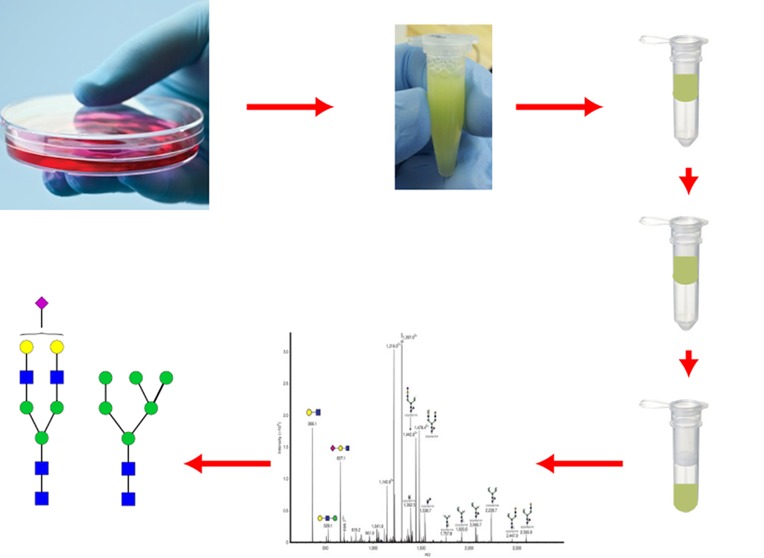
We have developed a simple method for the release and isolation of glycoprotein N-glycans from whole-cell lysates using less than a million cells, for subsequent implementation with mass spectrometric analysis. Cellular protein extracts prepared using SDS solubilization were sequentially treated in a membrane filter device to ultimately release glycans enzymatically using PNGase F in the volatile buffer ammonium bicarbonate. The released glycans are recovered in the filtrate following centrifugation and typically permethylated prior to mass spectrometric analysis. We call our method “filter-aided N-glycan separation” and have successfully applied it to investigate N-glycan profiles of wild-type and mutant Chinese hamster ovary cells. This method is readily multiplexed and, because of the small numbers of cells needed, is compatible with the analysis of replicate samples to assess the true nature of glycan variability in tissue culture samples.
Keywords: cultured mammalian cells, SDS solubilization, N-glycans, filter-aided sample preparation, MALDI-MS, permethylation
Introduction
Glycosylation is the most common posttranslational modification of secreted and membrane proteins. Different monosaccharides are linked to each other to form oligosaccharides, and one or more of the resulting glycan chains may be attached to the polypeptide backbone to form a glycoprotein. Glycans are important modulators of protein function but have functions of their own in cell/tissue structure and signaling.1 Glycans are usually highly heterogeneous branched oligomers, although often very little functional information is available for specific glycan species. One of the major bottlenecks in such functional analysis is the difficulty of complete structural analysis of the full complement of glycans in a system, the glycome. However, this is somewhat ameliorated for glycan subclasses whose biosynthesis has been well studied, such as mammalian N-glycans, in which the oligosaccharide chain is attached to an Asn side chain (reviewed in ref (2)). The consequence of such detailed biosynthetic knowledge is that the resulting glycan structures are well established and their limited repertoire is well accepted.
Mass spectrometry (MS) is well developed for glycan profiling3−6 and for the study of glycan structures,7−9 and so it is accepted as a key tool in glycoprotein structure–function studies, although reliable medium-throughput methods for establishing the glycome remain scarce. The simplest approaches to glycoprotein structural studies involve the release of the glycans from the polypeptide chain for subsequent analysis. The most widely adopted approach for N-glycan release uses the enzyme peptide-N4-(acetyl-β-glucosaminyl)-asparagine amidase from Elizabethkingia meningoseptica (previously Flavobacterium meningosepticum) (PNGase F) to cleave the amide bond linking the glycan to the Asn side-chain of glycoproteins in solution and thereby release intact N-glycans for analysis. For MS analysis, involatile reagents and detergents, both of which are detrimental to MS analysis, are generally avoided. This simple approach works well for handling soluble glycoproteins, and the literature contains reports of a very wide variety of different protocols for release and isolation of N-glycans from such soluble glycoprotein samples. However, the majority of glycoproteins are membrane proteins and so demand the use of detergents and other aggressive reagents for effective solubilization, which must then be removed before MS analysis. This inherent reagent incompatibility presents a significant challenge – how to effectively solubilize and sample all glycoproteins from a cell or tissue specimen and yet process them efficiently for sensitive MS analysis.
Adherent cells grown in culture are the workhorse of cell- and glycobiologists studying the effects of mutations, transformations, or drug treatments on physiological states under controlled experimental conditions. They offer a significant advantage over attempting to use human or animal tissue samples such as serum or biopsies, which are much more difficult to manipulate. However, recent studies using adherent cells for deep glycan profiling have used between 10 and 100 million cells10,11 prohibiting anything but low-throughput studies. Most importantly, the large numbers of cells needed precludes the production of replicates. This results in the inability to report the difference between inherent sample-to-sample variability and the important variances between biologically distinct samples. There are surprisingly few reports of studies that have attempted to address this significant methodological challenge. Nakano et al. worked not on whole cells but on isolated membranes and starting from 107 cells of a leukemia cell line carried out an involved, multistep procedure to release N- and O-glycans for mass spectrometric analysis.12 They carried out fluorescent labeling and analyzed the resulting labeled glycans using LC–MS. This approach was well-suited to the aims of that study (detailed glycome analysis for drug-resistant and susceptible cells) but would not be readily applicable to the medium-throughput glycome profiling of cells in primary cell culture or when expensive reagents such as siRNA are used, due to the complex sample handling and the scale of the cultures used. Nagahori et al. released and isolated N-glycans (and all other major types of glycoconjugate glycans) from mouse embryonic fibroblast cells in culture,13 initially using cells from three 10 cm dishes and, very recently, reducing this to cell pellets from one 10 cm dish ((1 to 2) × 106 cells).14 Their aims also included complete glycome analysis and consequently their sampling and sample handling were tailored to that goal; the result is a very complex process with many individual steps. Moreover, they did not permethylate their released glycans but instead methyl-esterified the sialic acids and carried out absolute quantitation using an internal standard, making this an inappropriate approach for medium throughput studies (and indeed that was not their goal). A similar study, also aimed at complete glycomic analysis, in this case after RNAi knockdown of two Golgi stacking factors, was published by Xiang et al.;15 this study uses a similarly complex, multistep protein extraction and isolation procedure, tailored to preparing lipid linked and free oligosaccharide fractions as well as glycoprotein glycans, and does not indicate the number of cells that were used, although N-glycan release used 2 mg protein powder. N-glycan identification following permethylation was achieved using nanospray-MS, and quantitation made use of external calibration; this approach is well suited to deep glycomic profiling and absolute quantitation but is not ideal for medium-throughput analysis of a range of cultures for comparative analyses, as is our aim. The group of Jaatinen has analyzed the N-glycome of cultured human embryonic stem cells.16 Although they do not describe how the N-glycoproteins are solubilized from the cells, the released N-glycan isolation and purification for mass spectrometric analysis is elaborate, involving many individual steps including precipitation, extraction, and several SPE steps that purify and ultimately separate sialylated from neutral species, for separate analyses, because they do not permethylate. Although this approach is not convenient for medium throughput and quantitation is very involved, it is reported that it is possible to work with 100 000 cells derived from cell cultures. Because of the lack of an approach appropriate for medium-throughput, quantitative, glycome profiling (rather than in depth glycomic analysis) that is amenable to generation of biological replicates, we have developed a novel sensitive glycan profiling approach and used it to compare wild-type (WT) and mutant Chinese hamster ovary (CHO) cells to explain the qualitative Golgi trafficking differences previously observed17,18 using glycomic data.
Our method makes use of very efficient SDS solubilization of glycoproteins from whole cell lysates, combined with membrane filter devices for sample handling and N-glycan release. This approach is ideal for medium-throughput analysis of cultured cells because it requires only 200–500 thousand cells, the equivalent of one or two wells of a six-well culture dish. Our method should make routine MS glycan profiling of cultured cell mutants feasible and will also be easily adaptable for tissue samples where material is limited, such as fruit flies, worms, or small biopsies. We have found that our protocol is simple and efficient. Because it makes use of the filter-aided sample preparation (FASP) approach for SDS removal,19,20 we have called it filter-aided N-glycan separation (FANGS).
Experimental Procedures
Cell Culture
HeLa cells were selected with puromycin for clones stably expressing an shRNA directed against COG4.21 HeLa cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum. Where indicated, swainsonine at a final concentration of 10 μg/mL was added to the medium. Chinese hamster ovary (CHO) cells were cultured in Ham’s F12 medium supplemented with 5% fetal bovine serum.
Cell Lysis
For cellular (glyco)protein extraction, the cells were washed six times with 5 mL of phosphate-buffered saline (PBS). After washing, 1 mL of fresh PBS was added onto a 10 cm tissue culture dish of cells, and the cells were dislodged using a scraper and transferred to a 15 mL plastic tube. The tissue culture dish was rinsed with 1 mL of PBS, and this was also transferred to the tube. When necessary, 10 μL of the cell suspension was removed for counting to determine the total number of cells used. The remaining cell suspension was transferred to 2 mL plastic microfuge tubes for centrifugation at 14 000g for 5 min. After centrifugation, the pellet was resuspended in lysis buffer (4% (w/v) SDS, 100 mM Tris/HCl pH 7.6, 0.1 M dithiothreitol) using a 1:10 pellet/lysis buffer volume ratio. The sample was heated to 95 °C for 5 min. The lysate was centrifuged at 14 000g for 10 min, and the supernatant was collected and kept at −80 °C.
FANGS
The supernatant collected after cell lysis was transferred to a 1.5 mL plastic microfuge tube. Urea solution (8 M in 100 mM Tris/HCL pH 8.5) was added to the sample in a ratio of 10:1 urea solution to sample solution by volume. An aliquot of this mixture (250–300 μL) was transferred to an ultrafiltration device (Amicon Ultra-0.5, Ultracel-30 membrane, nominal mass cutoff 30 kDa, Millipore) and centrifuged at 15 000g for 10 min, when a further 300 μL was transferred and centrifugation was repeated until all of the liquid had passed through the ultrafiltration device. The sample retained above the filter membrane was washed twice by the addition of 250 μL of the urea solution and centrifugation at 15 000g for 10 min.
Freshly prepared 40 mM iodoacetamide in urea solution (300 μL) was added to the ultrafiltration device and mixed well. The device was placed in the dark at room temperature for 15 min before being centrifuged for 10 min. Urea solution (250 μL, 8 M) was added to the ultrafiltration device and centrifuged for 10 min at 15 000g for 5 min. The sample retained above the filter membrane was washed four times by the addition of 250 μL of 50 mM ammonium bicarbonate (pH 7.5 to 8) and centrifugation at 15 000g for 10 min. The ultrafiltration device was transferred to a new collection tube, and 100 μL of 50 mM ammonium bicarbonate solution was added, followed by 8 U (8 μL of 1000 U/mL solution in 5 mM potassium phosphate, pH 7.5) of PNGase F from Elizabethkingia miricola (Sigma). The ultrafiltration device was sealed with Parafilm and incubated at 37 °C for 16 h. After incubation, the device was centrifuged for 15 min at 15 000g and washed twice with 250 μL of water (HPLC grade) followed by centrifugation for 10 min at 15 000g. The released N-glycans in solution were retrieved from the collection tube.
Permethylation
The N-glycan solution from FANGS was transferred to a glass screw-capped tube and dried in a vacuum centrifuge. The sample was redissolved in ∼600 μL of dimethyl sulfoxide (DMSO) and agitated manually. Two pellets of sodium hydroxide were crushed in a mortar, and ∼25 mg of the powder was added to the sample. The sample was manually agitated once before repeated additions of iodomethane as follows: ∼375 μL was added and left for 10 min; then, ∼375 μL was added and left for a further 10 min; finally, ∼750 μL was added and left for a further 20 min. Approximately 1.5 mL of 100 mg/mL sodium thiosulfate solution was added to the sample and vortexed to quench the reaction. Immediately ∼1.5 mL of dichloromethane was added and the tube was vortexed. After separation of the phases, the upper layer was removed and the retained organic layer was washed sequentially 15 times, each with ∼1.5 mL of water (HPLC grade). The organic layer was taken to complete dryness under vacuum.
MALDI-MS Analysis
Two μL of a solution of 20 mg/mL 2,5-dihydroxybenzoic acid (DHB) in 70% methanol was mixed with 1 μL of 0.5 M NaNO3 and 2 μL of the permethylated glycan solution (total volume of glycan solution was 20 μL in methanol), and 2 μL of this mixture was spotted onto a MALDI target plate and allowed to air-dry.
TOF–MS
A Bruker Daltonics ultraflex III TOF/TOF mass spectrometer equipped with a Smartbeam laser was used in positive-ion mode. The mass spectrometer was calibrated externally using a Bruker Peptide Mix II MALDI standard preparation. Mass spectra were recorded over the m/z range 800–4000 using a total of 4000 shots in steps of 800, which were summed to give one spectrum. One spectrum was recorded from each sample spot. The laser power setting was varied over the range 40–65%.
FT-ICR–MS
A 9 T solariX FT mass spectrometer (Bruker Daltonics) was operated in MALDI positive ion mode. The mass spectrometer was calibrated externally using a Bruker Peptide Mix II MALDI standard preparation. Mass spectra were recorded over the m/z range 500–6000, acquiring 32 scans, with each scan being derived from 30 laser shots. One spectrum was recorded from each sample spot. The laser power was set at 35%.
For quantification of relative glycan abundance, seven batches of WT, five batches of ldlB, and four batches of ldlC cells from a 10 cm dish (1.5 to 2.5 × 106 cells each) were processed using FANGS, and MALDI-TOF–MS data from each batch were collected. The Bruker FlexAnalysis software was used to smooth the data (Savitzky-Golay). Following smoothing, all glycan signal intensities assigned a S:N > 3 by the software were selected, and those belonging to the same species (same isotopic envelope) were summed to generate a total signal intensity for each glycan species. The total intensities of the isotopic envelope signals for the glycan species common to all three cell lines were summed within each spectrum. This sum of intensities was used to normalize the intensity of the signal for each glycan within a given spectrum. To generate Figure 3c, the normalized intensities of signals for individual glycan species from either the WT or the mutant data sets were averaged and plotted using Excel; the error bars indicate standard error of the mean.
Figure 3.

(a,b) MALDI-TOF mass spectra of permethylated FANGS-released N-glycans from (a) WT CHO and (b) ldlB cells. Cellulose oligomer contamination peaks are indicated with *. (c) Bar chart representing the relative intensities of the MALDI-TOF-MS signals for the different N-glycan species detected in WT, ldlB, and ldlC cells. The glycan intensities in each individual spectrum are normalized to the sum of intensities in a given spectrum for the subset of glycans that are common to all three cell lines. Standard errors of the mean are shown for WT CHO cells (n = 7), ldlB cells (n = 5), and ldlC cells (n = 4). Statistically significant differences between relative glycan peak intensities are indicated: *** for p < 0.001, ** for p < 0.01, and * for p < 0.05. None of ldlB- and ldlC-derived glycan samples showed a significant difference when compared with each other. The differences between WT and the two mutants showed similar significance, except for the Hex3HexNAc4 and the Fuc1Hex4HexNAc4 species. For both of these species, the ldlC-derived glycans showed differences of lower significance when compared with WT than for the ldlB compared with WT. For the Fuc1Hex4HexNAc4 species, the difference between WT and ldlC was not significant.
Statistical significance for the differences between relative glycan abundance was determined using one-way ANOVA analysis and a Tukey–Cramer multiple comparisons post hoc test. Each glycan was individually evaluated to test for differences between the glycans from the three cell lines.
Results and Discussion
A simple and rapid protocol has been developed for glycoprotein solubilization from whole cell lysates, followed by N-glycan release in a high-molecular-weight cutoff (30 kDa) filter device (Figure 1) and N-glycan isolation for MS analysis. Whole cells are solubilized by boiling in buffered SDS solution. SDS can be efficiently removed and exchanged for a suitable buffer using spin filter devices;19,20 we make use of a similar approach to remove SDS from our cell lysates. The solubilized cell lysate in buffered SDS solution is placed in the spin filters; SDS is exchanged with urea solution; then, reduced disulfide bonds in the proteins are chemically modified using iodoacetamide. Subsequently the urea solution is exchanged for volatile ammonium bicarbonate buffer. In our procedure, the membrane filtration unit acts as a small reactor, in which PNGase F digestion is used to release the N-glycans (Figure 1). Although some (but not all) commercial PNGase F protocols recommend the use of the enzyme on tryptic glycopeptides rather than on intact glycoproteins, boiling the samples in SDS presumably denatures the glycoproteins sufficiently to allow the effective glycan release we show here from undigested glycoproteins. After overnight incubation with PNGase F, the released glycans are retrieved from the collection tube following centrifugation, because once released from the polypeptide, the N-glycans readily pass through the membrane. We have analyzed these glycans following permethylation to exploit the excellent MS response of permethylated glycans.22 Positive-mode MALDI-MS was then used to analyze the samples obtained, taking advantage of the relative quantitation shown to be possible from relative MALDI signal intensities of permethylated glycans.23
Figure 1.
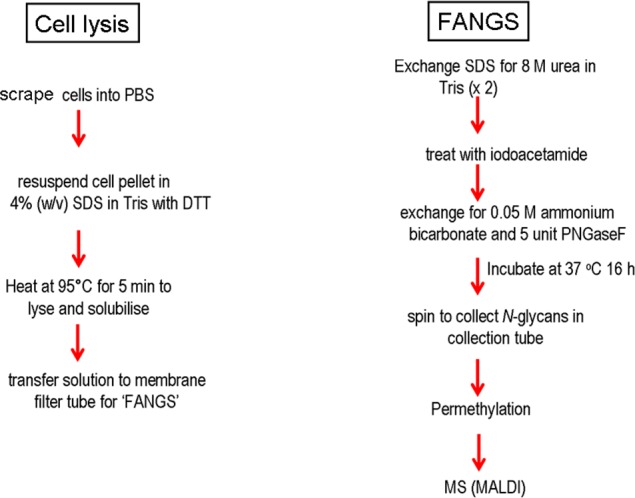
Schematic of FANGS protocol for cell lysate preparation from cultured cells, N-glycan release, and sample handling in membrane filter devices. (a) Extraction of glycoproteins from whole cells and (b) release and permethylation of N-glycans.
Validation of FANGS Using Well-Characterized Systems
To validate the FANGS protocol, we analyzed two standard samples using this approach. First, a soluble glycoprotein, fetuin, was chosen as a very well-characterized, commercially available standard, described in the literature to bear bi- and triantennary sialylated N-glycan structures.24 Second, to test applicability with cultured cells, we grew HeLa cells and (glyco)proteins were solubilized from the whole cells. Both the fetuin and the HeLa cells were processed using the FANGS protocol, and the released N-glycans from the two samples were permethylated for MALDI-MS analysis.
In Figure 2a–c, MALDI mass spectra of permethylated FANGS-released N-glycans from fetuin are presented. Monoisotopic [M+Na]+ signals are observed at m/z 2792.53 (NeuAc2Hex5HexNAc4), 3241.85 (NeuAc2Hex6HexNAc5), and 3603.01 (NeuAc3Hex6HexNAc5), with a final signal centered around m/z 3965.71 (NeuAc4Hex6HexNAc5) in Figure 2a and at m/z 3963.92/3963.88 in Figure 2b,c. The signal at m/z 3965.71 corresponds to a species incorporating two 13C atoms – the monoisotopic signal for this species is weak in this spectrum due to the glycan’s high molecular mass. Our spectra are very similar to that reported by Kang et al., who used fetuin as a standard glycoprotein in the development of their solid-phase permethylation procedure (see figure 5 in ref (25)). These data also highlight the reproducibility of FANGS, given the similarity of the spectra in Figure 2a–c, even though we have chosen to present data from two different instruments (FT-ICR in Figure 2a and reflectron TOF in Figures 2b,c).
Figure 2.

(a–c) Positive-mode MALDI mass spectra of permethylated FANGS-released N-glycans from 2.5 μg fetuin: (a) 1/10th of the N-glycan sample loaded onto the MALDI plate for analysis by FT-ICR and (b,c) 1/20th of the N-glycan sample loaded onto the MALDI plate for analysis by TOF-MS. (d–f) MALDI-TOF mass spectra of permethylated FANGS-released N-glycans from (d) WT HeLa cells, (e) HeLa cells treated with 10 μg/mL swainsonine, and (f) COG4KD cells. Each spectrum is derived from cells collected off one confluent 10 cm dish, approximately 2 to 3 × 106 cells. Peaks derived from contaminating cellulose oligomers are indicated with *. N-Glycan structures are denoted following the conventional symbols described in ref (36). Peak intensities in each spectrum are normalized to the most intense signal in the spectrum.
The MALDI mass spectrum of the permethylated N-glycans from WT HeLa cells (Figure 2d) contains [M+Na]+ glycan signals for oligomannose glycans at m/z 1579.91 (Hex5HexNAc2), 1784.00 (Hex6HexNAc2), 1988.11 (Hex7HexNAc2), 2192.21 (Hex8HexNAc2), and 2396.33 (Hex9HexNAc2) and at m/z 2600.48 for the glucosylated analogue of the Man9 species (Hex10HexNAc2). Processed oligo-mannose species, with and without fucose, give rise to the signals at m/z 1141.69 (FucHex2HexNAc2), 1171.71 (Hex3HexNAc2), 1345.79 (FucHex3HexNAc2), and 1375.83 (Hex4HexNAc2), while hybrid glycans yield the signals at m/z 2028.98 (Hex6HexNAc3) and 2390.13 (NeuAcHex6HexNAc3). Fucosylated complex glycan signals were observed at m/z 2244.29 (Fuc1Hex5HexNAc4) and 2693.24 (Fuc1Hex6HexNAc5), with sialylated species detected at m/z 2605.48 (NeuAc1Fuc1Hex5HexNAc4), 2792.32 (NeuAc2Hex5HexNAc4), and 2966.92 (NeuAc2Fuc1Hex5HexNAc4). This spectrum is similar to that reported in a recent analysis of HeLa cell N-glycans released in solution following tryptic digestion (see figure 4A in ref (26)); the major signals observed in the two studies are the same, and the relative intensities of the signals for those species are very comparable. Having validated the FANGS protocol and shown that it is applicable for both soluble glycoproteins and cultured cells, we then went on to use the protocol for N-glycan profiling of differently treated as well as mutant cell lines.
Application of FANGS to the Comparison of WT and Treated HeLa Cells
The N-glycan structures produced upon different treatments in a given cell line can be used to address the mechanism of action of a drug or biological pathway influencing glycosylation. We therefore decided to test whether FANGS is suitable for revealing the differences expected in N-glycosylation of wild-type and differently treated HeLa cells. Swainsonine, a well-characterized α-mannosidase II inhibitor that prevents the formation of complex glycans but promotes hybrid glycan formation,27 was used. In addition, we tested HeLa cells stably expressing a Cog4 shRNA, to deplete a subunit of the conserved oligomeric Golgi (COG) complex.21 COG is responsible for maintaining the proper vesicle-mediated sorting of glycosylation enzymes to their cisternal locations.28,29
Treated HeLa cells were grown to confluence in 10 cm dishes, the N-glycans were obtained using the FANGS protocol, and MALDI N-glycan profiling was performed following permethylation, in the same way as for the WT HeLa cells. Representative spectra are shown in Figure 2e,f. The MALDI mass spectrum of the permethylated N-glycans from WT HeLa cells treated with swainsonine contains intense [M+Na]+ signals (Figure 2e) for oligomannose glycans (m/z 1579.92, 1753.97 (FucHex5HexNAc2), 1783.10, 1988.06, 2192.16, and 2396.24). The signal at m/z 2600.21 corresponds to a monoglucosylated Man9GlcNAc2 species. Hybrid glycans were observed (m/z 2028.93 (Hex6HexNAc3), 2377.28 (Fuc2Hex6HexNAc3), 2390.13 (NeuAcHex6HexNAc3), and 2564.35 (FucNeuAcHex6HexNAc3)), as well as a very small amount of the fully trimmed fucosylated glycan core at m/z 1345.60 (FucHex3HexNAc2). Swainsonine treatment thus had a marked effect on the distribution of N-glycans; complex glycans were no longer detected, while hybrid N-glycans predominated in the sample. Thus FANGS was readily able to provide a readout of the effect of swainsonine treatment by revealing the switch from complex to hybrid N-glycans.
Cog4 shRNA-treated (COG4KD) cells gave a complement of [M+Na]+ signals (Figure 2f), similar to those observed from the WT HeLa cells, with signals at m/z 1141.58, 1171.59, 1345.67, 1375.67, 1579.79, 1783.89, 1987.99, 2192.08, 2396.18, 2600.30, 2605.28, and 2966.43. The signal at m/z 2244.29 (Fuc1Hex5HexNAc4) seen in the spectrum from WT samples is not observed, while an ungalactosylated triantennary glycan at m/z 2081.99 (FucHex3HexNAc5) as well as the hybrid glycan at m/z 2390.27 (NeuAcHex6HexNAc3) were detected in the spectra from the COG4KD cell samples. The relative intensities of some glycans have also been altered; in particular, there is a reduction in the relative abundance of the Man5 compared with the larger oligomannose species in the Cog4KD cells. In addition, an increase in the hybrid glycan signals and a decrease or absence of sialylated glycan signals is evident in the knock-down compared with the WT cells. These observations are in line with the effects expected from a COG defect, which is the mistargeting of enzymes throughout the Golgi leading to less efficient glycan processing.28
Thus FANGS allows the detection of qualitative differences between different cell lines, such as the differences in trimming efficiency between WT and COG4KD HeLa cells, or the switch from complex to hybrid glycans upon swainsonine treatment. The main advantage of our new method in this case is the possibility to investigate a multitude of conditions, indicating that FANGS can be used to study both biological and chemical effects upon the N-glycosylation process. The ultimate challenge, though, is to obtain glycan profiles in which small but significant changes in the levels of individual glycans can be determined in a statistically robust way. To test if FANGS is applicable for this sort of analysis, we chose to investigate three Chinese hamster ovary (CHO) cell lines.
Application of FANGS to the Study of WT and Mutant CHO Cells
By studying the N-glycan structures produced by WT and mutant CHO cell lines, fundamental questions about the biosynthesis of these glycans can be answered.30 LdlB cells are a mutant CHO cell-line-deficient in Cog1,31 a subunit of the COG complex.29 Studying these cells also makes it possible to address questions surrounding congenital glycosylation disorders that result from COG mutations.32
WT and mutant CHO cells were grown in culture in 10 cm dishes, and the N-glycans were obtained using the FANGS protocol. MS N-glycan profiling was performed following permethylation (data summarized in Table 1). Such experiments have been carried out very many times; here we present examples of those data, choosing typical FT-ICR and TOF spectra.
Table 1. N-Glycan Profiles from WT and Mutant ldlB CHO Cellsa.
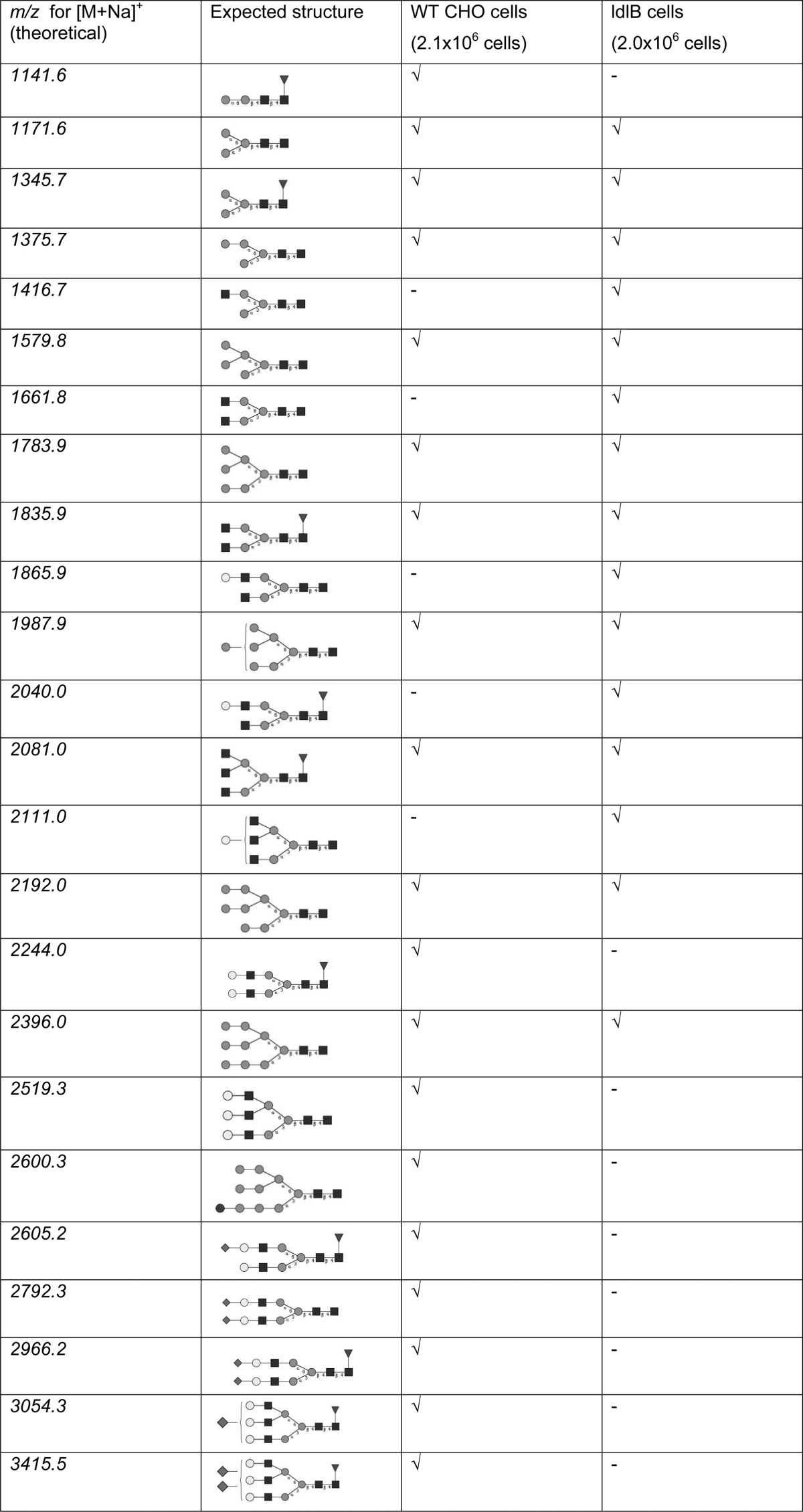
√ = observed; - = not observed.
The MALDI mass spectrum of the permethylated N-glycans from WT CHO cells contains [M+Na]+ signals (Figure 3a) for complex glycans (m/z 1835.98, 2081.07, 2244.29, 2519.29, 2605.48, 2792.35, 2966.92, 3054.51, and 3415.63), oligomannose glycans (m/z 1579.83, 1783.93, 1988.04, 2192.21, and 2396.33), a glucosylated oligomannose glycan (m/z 2600.29), and fucosylated and/or trimmed oligomannose glycans (m/z 1141.62 (Fuc1Hex2HexNAc2), 1171.63 (Hex3HexNAc2), 1345.72 (Fuc1Hex3HexNAc2), and 1375.73 (Hex4HexNAc2)), as previously reported in a glycomic profiling study of CHO cells.11
Mutant ldlB cells gave a complement of [M+Na]+ signals similar to that from the WT cells, with signals at m/z 1171.56 (Hex3HexNAc2), 1345.66 (Fuc1Hex3HexNAc2), 1375.67 (Hex4HexNAc2), 1835.89 (Fuc1Hex3HexNAc4), 2081.00 (triantennary Fuc1Hex3HexNAc5), and m/z 1579.77, 1783.86, 1987.94, 2192.04, and 2396.14 (for oligomannose glycans). In addition, signals at m/z 1416.68 (Hex3HexNAc3), 1661.82 (Hex3HexNAc4), 1865.90 (Hex4HexNAc4), 2039.97 (Fuc1Hex4HexNAc4), and 2111.01 (triantennary Hex4HexNAc5), corresponding to incompletely processed complex glycans, were detected (Figure 3b). These signals were not detected in the spectra of the WT samples. In addition, the NeuAc1Fuc1Hex5HexNAc4 glycan (at m/z 2605.37) detected in the WT sample was not observed in the spectrum from the ldlB mutant cell sample. Moreover, the relative intensities of the signals of several N-glycan species were observed to change (Figure 3c). To assess the fine details of the inherent differences between the WT and mutant glycan profiles, we have made use of the possibility that FANGS offers of collecting several independent sample repeats, thereby assessing the biological variability.
Consequently, seven separate cultures of WT CHO cells, five different cultures of mutant ldlB cells, and four different cultures of ldlC cells were grown, their N-glycans were prepared using FANGS, and the relative intensities of the signals from the permethylated glycans were determined using MALDI-TOF-MS (Figure 3c). We have taken the approach of comparing relative signal intensities of permethylated glycans using MALDI-MS16,33 to determine relative quantitative information. We are able to assess biological variability by studying biological replicates and whether differences are significant by virtue of having standard deviation information. Such an approach of studying replicates was not feasible when working at the 107 to 108 cell levels11 that were necessary for the purposes of deep-glycan profiling. Deep profiling is not required for our experiments that aim not to exhaustively define the glycome of a single cell line, but instead to observe sufficient glycans to draw conclusions about the nature of the differences between glycan profiles of WT and mutant cell lines.
Using our approach, we can see, for example, that oligomannose glycans are apparently trimmed more efficiently in the mutant cells because the Hex5 species is more prominent in the ldlB and ldlC cell samples, whereas the Hex6-Hex9 species are all more prominent in the samples from the WT cells. This may represent a lack of efficient GlcNAc-1-phosphotransferase processing in the mutant, which channels the oligomannose glycans into the lysosomal sorting pathway and thereby prevents further trimming.34 Furthermore, in addition to the absence of signals for sialylated glycans in the spectra from ldlB cell samples and the detection of incompletely processed complex glycans (such as FucHex3HexNAc5) from ldlB cells that are not detected from CHO cells, significant differences between other partially processed glycans can also be observed. Signals for the Hex3HexNAc3 and FucHex3HexNAc4 species are both much more prominent in the spectra from the mutant samples (Figure 3c), pointing to a delay in the very early processing of complex glycans. The comparative glycan profiling approach we present here can therefore, by showing small but significant differences in related glycan structures, point to alterations in biosynthetic enzyme functions that simple enzymatic rate measurements could not recapitulate, mainly due to the importance of enzyme localization during glycan biogenesis.
We can therefore conclude that in our experiments WT ldlB and ldlC mutant CHO cells all produce oligomannose glycans, but the complex biantennary glycans produced by the WT cells are not detected from the mutant cells; instead, truncated biantennary species are observed. Our results are therefore in line with previous qualitative assessments of WT CHO, ldlB, and ldlC cells.17 It is known that the COG complex has a significant impact on enzyme recycling in the Golgi and therefore guides glycosylation maintenance.28 The presence of these truncated structures is thus likely to be due to the mislocalization or degradation of the glycosylation enzymes in the mutant cells, as reported for mannosidase II in ldlB and ldlC cells.18 Our results suggest that the most severely affected processing enzymes are galactosyl- and fucosyltransferases because it is the widespread absence of completed complex N-glycans that is the most striking difference between the glycans identified from the WT and mutant cells, but N-acetylglucosaminyl transferases are probably affected too. Detailed systems-level modeling of the glycosylation machinery will be necessary to describe all of the alterations in glycosyltransferase levels in the mutant cells. Although such modeling is beyond the scope of this study, the precision and throughput with which differences in glycan profiles can be measured with our new method will be important for developing precise computational models.
Glycan profiling has previously been performed on serum-derived N-glycans of a Cog1-deficient patient.35 Although that profile also shows a reduction in the levels of completed complex glycans in the absence of functional Cog1, the defects are generally less severe because not all of the most highly processed forms are missing. This is probably due to the allele in the patient causing a partial loss of Cog1 function as opposed to the full loss of the subunit in ldlB cells. It will be interesting to see how expression of a Cog1 transgene carrying the patient mutation will influence the glycan profile of ldlB cells, an experiment now rendered straightforward using our FANGS glycan profiling approach.
Cell Number Limits of Applicability of the FANGS Method
Although working with 10 cm dishes is clearly compatible with the analysis of several repeats of several different cell lines, it can be prohibitive when culturing difficult primary cells or cultures that require expensive media. We therefore sought to further miniaturize our sample handling by reducing the number of cells used for N-glycan profiling using the FANGS approach. WT CHO cells were processed from one well of a six-well plate, providing a six-fold smaller surface area than a 10 cm plate and consequent reduction in cell and reagent quantities.
A typical mass spectrum obtained from the permethylated CHO cell N-glycans from a sample derived from 3.8 × 105 cells is shown in Figure 4. The spectrum showed a very similar set of glycans to those detected from samples derived from a 10 cm dish (typically six times more cells), including the expected oligomannose type and complex glycans (Table 2). Detection of fucosylated and fully sialylated bi- and triantennary complex glycans well above m/z 3000 indicates that 400 000 cells are sufficient for the reliable detection of the major N-glycans from CHO cells using the FANGS sample preparation approach. Given the quality of this spectrum, it is possible that cell numbers could be even further reduced. However, it is likely that to detect very minor glycan components, especially if these are of very high mass, half a million cells or more will be needed.
Figure 4.

MALDI-TOF mass spectrum of permethylated FANGS-released N-glycans from 3.8 × 105 WT CHO cells (one well of a six-well plate). Peak intensities are normalized to the most intense signal in the spectrum.
Table 2. WT CHO Cell N-Glycans Detected from 3.8 × 105 Cells (One Well of a Six-Well Plate).
| m/z | composition |
|---|---|
| 967.4 | Hex2HexNAc2 |
| 1141.5 | Fuc1Hex2HexNAc2 |
| 1171.5 | Hex3HexNAc2 |
| 1345.6 | Fuc1Hex3HexNAc2 |
| 1375.6 | Hex4HexNAc2 |
| 1416.6 | Hex3HexNAc3 |
| 1579.7 | Hex5HexNAc2 |
| 1661.8 | Hex3HexNAc4 |
| 1783.8 | Hex6HexNAc2 |
| 1835.8 | Fuc1Hex3HexNAc4 |
| 1987.9 | Hex7HexNAc2 |
| 2039.9 | Fuc1Hex4HexNAc4 |
| 2069.9 | Hex5HexNAc4 |
| 2192.0 | Hex8HexNAc2 |
| 2244.0 | Fuc1Hex5HexNAc4 |
| 2396.1 | Hex9HexNAc2 |
| 2605.1 | NeuAc1Fuc1Hex5HexNAc4 |
| 2693.1 | Fuc1Hex6HexNAc5 |
| 2792.3 | NeuAc2Hex5HexNAc4 |
| 2966.2 | NeuAc2Fuc1Hex5HexNAc4 |
| 3054.3 | NeuAc1Fuc1Hex6HexNAc5 |
| 3415.5 | NeuAc2Fuc1Hex6HexNAc5 |
| 3776.7 | NeuAc3Fuc1Hex6HexNAc5 |
The FANGS protocol was developed to allow solubilization, release, and isolation of N-glycans from glycoproteins in cell lysates in a miniaturized approach for medium throughput analyses of cultured cell lines. It has proved to be a very sensitive tool for preparing samples for N-glycan profiling of WT, treated, and mutant cells, allowing the precise comparison of differences in cellular glycan repertoires. This method should be invaluable for determining small quantitative differences in N-glycan profiles, thereby allowing the potential functional contributions of individual N-glycans to cellular phenotypes to be assessed.
Acknowledgments
S.A.R. gratefully acknowledges her Ph.D. Scholarship from the Ministry of Health, Malaysia. The York Centre of Excellence in Mass Spectrometry was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative. D.U. was supported by a Marie Curie IRG (#201098), while C.J.W. received an internship from the C2D2 initiative of the University of York and the Wellcome Trust, granted to D.U. and J.T.-O. The authors are grateful to Monty Krieger from MIT for the ldlB and ldlC cells.
The authors declare no competing financial interest.
References
- Ohtsubo K.; Marth J. D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 1265855–867. [DOI] [PubMed] [Google Scholar]
- Ungar D. Golgi linked protein glycosylation and associated diseases. Semin. Cell Dev. Biol. 2009, 20, 762–769. [DOI] [PubMed] [Google Scholar]
- Babu P.; North S. J.; Jang-Lee J.; Chalabi S.; Mackerness K.; Stowell S. R.; Cummings R. D.; Rankin S.; Dell A.; Haslam S. M. Structural characterisation of neutrophil glycans by ultra sensitive mass spectrometric glycomics methodology. Glycoconjugate J. 2009, 268975–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C. S.; Ninonuevo M. R.; Clowers B. H.; Perkins P. D.; An H. J.; Yin H.; Killeen K.; Miyamoto S.; Grimm R.; Lebrilla C. B. Profile of native N-linked glycan structures from human serum using high performance liquid chromatography on a microfluidic chip and time-of-flight mass spectrometry. Proteomics 2009, 971939–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell A.; Oates J. E.; Morris H. R.; Egge H. Structure Determination of Carbohydrates and Glycosphingolipids by Fast Atom Bombardment Mass-Spectrometry. Int. J. Mass Spectrom. Ion Processes 1983, 46Jan415–418. [Google Scholar]
- Ruhaak L. R.; Miyamoto S.; Kelly K.; Lebrilla C. B. N-Glycan Profiling of Dried Blood Spots. Anal. Chem. 2012, 841396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell A.; Carman N. H.; Tiller P. R.; Thomas-Oates J. E. Fast atom bombardment mass spectrometric strategies for characterizing carbohydrate-containing biopolymers. Biomed. Environ. Mass Spectrom. 1988, 161–1219–24. [DOI] [PubMed] [Google Scholar]
- Domon B.; Costello C. E. A Systematic Nomenclature for Carbohydrate Fragmentations in Fab-Ms Ms Spectra of Glycoconjugates. Glycoconjugate J. 1988, 54397–409. [Google Scholar]
- Harvey D. J. Matrix-assisted laser desorption ionisation mass spectrometry of oligosaccharides and glycoconjugates. J. Chromatogr., A 1996, 7201–2429–446. [DOI] [PubMed] [Google Scholar]
- Bateman A. C.; Karamanska R.; Busch M. G.; Dell A.; Olsen C. W.; Haslam S. M. Glycan analysis and influenza A virus infection of primary swine respiratory epithelial cells: the importance of NeuAc{alpha}2–6 glycans. J. Biol. Chem. 2010, 2854434016–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North S. J.; Huang H. H.; Sundaram S.; Jang-Lee J.; Etienne A. T.; Trollope A.; Chalabi S.; Dell A.; Stanley P.; Haslam S. M. Glycomics profiling of Chinese hamster ovary cell glycosylation mutants reveals N-glycans of a novel size and complexity. J. Biol. Chem. 2010, 28585759–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M.; Saldanha R.; Gobel A.; Kavallaris M.; Packer N. H. Identification of Glycan Structure Alterations on Cell Membrane Proteins in Desoxyepothilone B Resistant Leukemia Cells. Mol. Cell. Proteomics 2011, 10111–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahori N.; Yamashita T.; Amano M.; Nishimura S. I. Effect of Ganglioside GM3 Synthase Gene Knockout on the Glycoprotein N-Glycan Profile of Mouse Embryonic Fibroblast. ChemBioChem 2013, 14173–82. [DOI] [PubMed] [Google Scholar]
- Fujitani N.; Furukawa J.; Araki K.; Fujioka T.; Takegawa Y.; Piao J.; Nishioka T.; Tamura T.; Nikaido T.; Ito M.; Nakamura Y.; Shinohara Y. Total cellular glycomics allows characterizing cells and streamlining the discovery process for cellular biomarkers. Proc. Natl. Acad. Sci. U.S.A. 2013, 11062105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Zhang X.; Nix D. B.; Katoh T.; Aoki K.; Tiemeyer M.; Wang Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomaa T.; Heiskanen A.; Mikkola M.; Olsson C.; Blomqvist M.; Tiittanen M.; Jaatinen T.; Aitio O.; Olonen A.; Helin J.; Hiltunen J.; Natunen J.; Tuuri T.; Otonkoski T.; Saarinen J.; Laine J. The N-glycome of human embryonic stem cells. BMC Cell Biol. 2009, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley D. M.; Kozarsky K. F.; Segal M.; Krieger M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 10251576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T.; Ungar D.; Hughson F. M.; Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 2004, 1552423–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manza L. L.; Stamer S. L.; Ham A. J. L.; Codreanu S. G.; Liebler D. C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 571742–1745. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zielinska D. F.; Mann M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal. Biochem. 2011, 4102307–309. [DOI] [PubMed] [Google Scholar]
- Richardson B. C.; Smith R. D.; Ungar D.; Nakamura A.; Jeffrey P. D.; Lupashin V. V.; Hughson F. M. Structural basis for a human glycosylation disorder caused, in part, by mutation of the COG4 gene. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13329–13334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell A.; Morris H. R.; Egge H.; Vonnicolai H.; Strecker G. Fast-Atom-Bombardment Mass-Spectrometry for Carbohydrate-Structure Determination. Carbohydr. Res. 1983, 115Apr41–52. [Google Scholar]
- Wada Y.; Azadi P.; Costello C. E.; Dell A.; Dwek R. A.; Geyer H.; Geyer R.; Kakehi K.; Karlsson N. G.; Kato K.; Kawasaki N.; Khoo K. H.; Kim S.; Kondo A.; Lattova E.; Mechref Y.; Miyoshi E.; Nakamura K.; Narimatsu H.; Novotny M. V.; Packer N. H.; Perreault H.; Peter-Katalinic J.; Pohlentz G.; Reinhold V. N.; Rudd P. M.; Suzuki A.; Taniguchi N. Comparison of the methods for profiling glycoprotein glycans--HUPO Human Disease Glycomics/Proteome Initiative multi-institutional study. Glycobiology 2007, 174411–22. [DOI] [PubMed] [Google Scholar]
- Baenziger J. U.; Fiete D. Structure of the complex oligosaccharides of fetuin. J. Biol. Chem. 1979, 2543789–95. [PubMed] [Google Scholar]
- Kang P.; Mechref Y.; Klouckova I.; Novotny M. V. Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun. Mass Spectrom. 2005, 19233421–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokrovskaya I. D.; Willett R.; Smith R. D.; Morelle W.; Kudlyk T.; Lupashin V. V. COG complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21121554–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross V.; Tran-Thi T. A.; Vosbeck K.; Heinrich P. C. Effect of swainsonine on the processing of the asparagine-linked carbohydrate chains of alpha 1-antitrypsin in rat hepatocytes. Evidence for the formation of hybrid oligosaccharides. J. Biol. Chem. 1983, 25864032–6. [PubMed] [Google Scholar]
- Miller V. J.; Ungar D. Re’COG’nition at the Golgi. Traffic 2012, 137891–7. [DOI] [PubMed] [Google Scholar]
- Ungar D.; Oka T.; Brittle E. E.; Vasile E.; Lupashin V. V.; Chatterton J. E.; Heuser J. E.; Krieger M.; Waters M. G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 1573405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik S. K.; Stanley P. Lectin-resistant CHO glycosylation mutants. Methods Enzymol. 2006, 416, 159–82. [DOI] [PubMed] [Google Scholar]
- Chatterton J. E.; Hirsch D.; Schwartz J. J.; Bickel P. E.; Rosenberg R. D.; Lodish H. F.; Krieger M. Expression cloning of LDLB, a gene essential for normal Golgi function and assembly of the ldlCp complex. Proc. Natl. Acad. Sci. U.S.A. 1999, 963915–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeevaert R.; Foulquier F.; Jaeken J.; Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol. Genet. Metab. 2008, 93115–21. [DOI] [PubMed] [Google Scholar]
- Guillard M.; Morava E.; van Delft F. L.; Hague R.; Korner C.; Adamowicz M.; Wevers R. A.; Lefeber D. J. Plasma N-Glycan Profiling by Mass Spectrometry for Congenital Disorders of Glycosylation Type II. Clin. Chem. 2011, 574593–602. [DOI] [PubMed] [Google Scholar]
- Kollmann K.; Pohl S.; Marschner K.; Encarnacao M.; Sakwa I.; Tiede S.; Poorthuis B. J.; Lubke T.; Muller-Loennies S.; Storch S.; Braulke T. Mannose phosphorylation in health and disease. Eur. J. Cell Biol. 2010, 891117–23. [DOI] [PubMed] [Google Scholar]
- Foulquier F.; Vasile E.; Schollen E.; Callewaert N.; Raemaekers T.; Quelhas D.; Jaeken J.; Mills P.; Winchester B.; Krieger M.; Annaert W.; Matthijs G. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. U.S.A. 2006, 103103764–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey D. J.; Merry A. H.; Royle L.; Campbell M. P.; Dwek R. A.; Rudd P. M. Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics 2009, 9153796–3801. [DOI] [PubMed] [Google Scholar]