

Abstract
Background
Salmonella typhi, Bacillus anthracis, and Yersinia pestis are some serious human pathogens, which their early diagnosis is of great importance. Salmonella typhi, Bacillus anthracis, and Yersinia pestis cause typhoid fever, anthrax, and plague respectively. These bacteria can be used to make biologic weapons.
Objectives
In this study, we designed a new and rapid diagnostic method based on Uniplex and Multiplex PCR method.
Materials and Methods
Uniplex and multiplex Polymerase Chain Reaction (PCR) were conducted on virulent genes of hp and invA of Salmonella typhimurium, Pa and chr of Bacillus anthracis, and pla of Yersinia pestis. A genome from other bacteria was used to study the specificity of the primer and the PCR test.
Results
Standard strains used in this study showed that primers were specific. As for sensitivity, it was shown that this method can diagnose 1-10 copies of the genome, or 1-10 Colony Forming Units (CFU) for each of the bacteria. All pieces except anthrax were sequenced in PCR to validate the product. DNA fragment resulted from Bacillus anthracis was confirmed by restriction enzyme digestions.
Conclusion
The designed methods are accurate, rapid, and inexpensive to find and differentiate these bacteria from similar bacteria. They can be applied for rapid diagnosis of these agents in different specimens, and bioterrorism cases.
Keywords: Salmonella typhi, Bacillus anthracis, Yersinia pestis, Multiplex PCR
1. Background
Salmonella typhi, Bacillus anthracis, and Yersinia pestis are known as group A pathogens, which have a high potential for human pathogenicity. To deal with these pathogens, rapid detection and treatment are very important. Salmonella typhi causes typhoid, with a morbidity of more than 16 million people worldwide, and a mortality of about 600 thousand deaths (1). All salmonellas are pathogenic for humans, mammals and birds except Salmonella typhi, which causes diseases in humans and higher primates only. Therefore, it is transmitted through water, food, feces and urine of infected humans, mammals and birds (2). Anthrax is a zoonotic infectious disease, caused by Bacillus anthracis. This disease is found worldwide, and its bacteria can be transmitted to humans indirectly from contaminated livestock products or directly by the spores of Bacillus anthracis. Four forms of anthrax include cutaneous, pulmonary, gastrointestinal, and meningitis. Due to its special features, sporogenesis, high virulence, and ease of mass production and application, this bacterium tops the biological and bioterrorism weapons (3). Plague is a dangerous and deadly disease caused by bacterium Yersinia pestis in most rodents and humans (4). The gram-negative coco-Bacillus does not have spores and movement, which is normally transmitted in enzootic form among rodents such as mice (5). The disease agent can be transmitted to humans through infected ectoparasites bite, particularly fleas (6, 7). Three clinical manifestations of disease include bubonic, septicemic, and pneumonic. Its pneumonic type causes the most severe clinical manifestation, entails high mortality, and is easily transmitted from person to person (8). Blood culture, bone marrow, rectal swabs, stool cultures, urine cultures, and other tests such as ELISA and immune-florescent are used for laboratory diagnosis (9, 10). Identifying the pathogenic agent of anthrax is difficult and time consuming due to its similarity to nonpathogenic bacilli in soil, and just advanced and reference laboratories can identify it accurately. Meanwhile, in the conventional methods, it is essential to determine the virulence in laboratory animals, which requires special biosafety facilities and equipment for dealing with hazardous agents, in addition to be time consuming, and also exposes health workers in this field at risk (11). Biochemical tests, susceptibility to specific bacteriophages, inoculation to laboratory animals, and culture in specific media are used for the diagnosis of plague agent. Furthermore, samples can be dried and contaminated or its bacteria can die due to the lack of appropriate means of transferring samples from infected areas to diagnostic centers (12). All the usual bacteriological methods like culture and biochemical tests as gold standard for identification of infectious disease agents are time-consuming and useless for rapid and accurate diagnosis of the disease (13). Meanwhile in molecular methods, the virulent infectious agent can directly and rapidly be diagnosed without the need for such investigations. Thus, conventional methods are worthless in biological and bioterrorism attacks, and we need rapid diagnostic methods which can help apply therapeutic and preventive measures in few hours. These methods are also widely used in clinical laboratories. Inability to quickly identify these biological agents means that they are life-threatening as nuclear weapons, and even more dangerous, because they can be transmitted to other areas and even the whole world (14).
2. Objectives
In this study, we designed a new and rapid diagnostic method based on Uniplex and Multiplex PCR method.
3. Materials and Methods
The standard strains were purchased from the Reference Laboratory of the Ministry of Health and Pasteur Institute in Tehran. Due to hazards of working with virulent strains, nonvirulent Bacillus anthracis strains, known as Stern or livestock vaccine strain was used, which has no difference with the wild strain regarding microbiologic features. The original strains of Staphylococcus aureus , Shigella sonnei , Escherichia coli , Enterococcus faecalis, Pseudomonas aeruginosa , Citrobacter freundii , Serratia marcescens, and Klebsiella pneumoniae were used as negative control agents, which were purchased from laboratory of Pasteur Institute in Tehran. Used strains are shown in Table 1. Chemical materials were purchased from Merck Co., MgCl 2 , buffer, nucleotides, and Taq polymerase enzyme were purchased from Cinnagen Co. In this research, Mastercycler gradient (Eppendorf Co., Germany) was used for thermal cycling. Small horizontal electrophoresis (Paya Pajouhesh Co., Mashhad, IR Iran), and its power supply with the buffer TBE 0.5 were used for electrophoresis. Gel was studied by UVIdoc (UVItec UK Co.). Standard molecular marker of 100 bp was purchased from Fermentas Co.
Table 1. List of Control Agent Bacteria Used in This Study for Specificity Analysis .
| Strain No. | Microorganism No. | Product Place | |
|---|---|---|---|
| Staphylococcus aureus | ATCC | 25923 | IPRa |
| Shigella sonnei | ATCC | 9290 | IPRa |
| Escherichia coli | ATCC | 25922 | IPRa |
| Enterococcus faecalis | ATCC | 29212 | IPRa |
| Pseudomonas aeruginosa | ATCC | 27853 | IPRa |
| Citrobacter freundii | PTCC | 1600 | IPRa |
| Serratia marcescens | PTCC | 1111 | IPRa |
| Klebsiella pneumonia | ATCC | 7881 | IPRa |
a Pasteur Institute of Iran.
3.1. Primer Designing
The primers were designed for Salmonella typhi , based on two principle segments of Salmonella typhi genome, invA and hp. The primers for Bacillus anthracis were designed based on two principle segments of organism genome, pA and chr. The primers for Yersinia pestis were designed based on one principle segment of plague bacterium genome, pla. All primers were designed and made by Cinnagen Company. DNASIS software (HITACHI, TOKYO, Japan) Blast version 6.71, and OLIGO were applied to investigate sequence analysis and primer design. Also, the loci of primers on the genome, the size of the amplified fragment, and the target genes are shown in Table 2. The sequence of the primers used in this study is shown in Table 3. The primers were diluted according to manufacturer’s guidelines at concentration of 100 pm using injection distilled water, and then diluted to a concentration of 20 pm and maintained at -20°C.
Table 2. Used Primers Profile .
| Protein | Product Size, bp | Gene Statute, Plasmid/ chromosome | Primer No. | |
|---|---|---|---|---|
| Salmonella typhi | invA | 373 | chromosome | S12-S13 |
| Salmonella typhi | Hp | 489 | chromosome | - |
| Bacillus anthracis | pA | 1083 | Plasmid (PXO1) | 125/126 |
| Bacillus anthracis | Chr | 164 | chromosome | 12-130 |
| Yersinia pestis | Pla | 520 | Plasmid (pST1) | - |
Table 3. Used Primers Sequence .
| Primer No | Sequence of Primers | |
|---|---|---|
| - | S12F | gta ttg ttg att aat gag atc cg |
| - | S13R | ata tta cgc acg gaa aca cgt t |
| Salmonella typhi | TF | tgt ccg ctg tct gaa gtc at |
| Salmonella typhi | TR | atc tca ggc aaa ctc aca agg g |
| Bacillus anthracis | 125 | tta atg cga ttg tct acg at |
| Bacillus anthracis | 126 | gat caa ttg cga ccg tac ttg aa |
| Bacillus anthracis | 129 | ccc agg ggg aca aac gat agc tcc |
| Bacillus anthracis | 130 | aac gat agc tcc tac att tgg ag |
| Yersinia pestis | Yer F | tgg act tgc agg cca gta tcg |
| Yersinia pestis | Yer R | cca tgc ctg aaa gac gtg gag |
3.2. Cultivation and Extraction of DNA
Master bacteria were inoculated on solid culture and Luria liquid media, and cultivated at 37°C for 18-24 hours. In addition to studying the shape and structure of the colonies and especially the colony characteristics, gram-staining, and biochemical tests were conducted. Then, DNA genome was extracted by conventional method from 1.5 to 5 mL of liquid culture (15).
3.3. Performing Multiplex PCR
PCR was conducted to assess target genes of invA and hp for Salmonella typhi , Pa and chr for Bacillus anthracis, and pla for Yersinia pestis in standard reaction volume of 25 µL. One µL of genomic sample preparation was added to the tube containing the compounds listed in Table 4, and placed in Master Cycler (Eppendorf) using the following program: The first step included a cycle of 7 min at 94°C, and then 30 cycles of three steps, 94°C for 1 min, 55°C junction primers for 1 min, 72°C for 1 min, and a final step at 72°C for 7 min. Before performing multiplex PCR, each of the materials and procedures of PCR were optimized. After obtaining the most appropriate amount of material and the optimum conditions for multiplex PCR, the amounts listed in Table 4 were obtained. After completing amplification reaction, 0.5 X TBE buffer was run on electrophoresis with a voltage of 100 V. Finally, to see the band, agarose gel was stained with ethidium bromide, and after rinsing, it was studied in the UV system. Reactions for all three bacteria were performed both in uniplex and multiplex ways.
Table 4. Quantities and Amounts of Compounds Used in Multiplex PCR Process .
| Materials | Amount, µL | |
|---|---|---|
| 1 | Distilled Water | 17.6 |
| 2 | Buffer 10X | 2.5 |
| 3 | MgCL2, 50 mM | 1 |
| 4 | dNTPmix, 100mM each | 0.5 |
| 5 | Primer F | 1 |
| 6 | Primer R | 1 |
| 7 | DNA | 1 |
| 8 | Taq | 0.4 |
| Total | 25 |
3.4. Determining Specificity
PCR was performed under the same conditions on the extracted genome of negative control bacteria to obtain the specificity of gene primers invA and hp of Salmonella typhi, pA and chr of Bacillus anthracis, and pla of Yersinia pestis, and also detecting the orientation of the bacteria.
3.5. Determining Sensitivity
Isolated colonies of bacteria causing typhoid and plague, and nonvirulent strain of Bacillus anthracis were inoculated in a flask containing 50 mL of broth, and placed in shaker incubator for 24 hours at 37°C. The next day, 10 tubes were prepared containing 900 µL of sterile broth, and 100 µL of each bacterial culture was added to the first tube (dilution of 10-1), and this dilution continued to the tube number 10. Simultaneous with preparing dilutions, 200 µL of each dilution was added to solid medium plates, and it was evenly spread on the media. The plates were kept at 37°C during the night. After complete growth, plates with 30 to 300 colonies were counted and measured. The experiment was repeated 3 times to increase the accuracy. Simultaneous with preparing dilutions, 2 µL of each tube was added to PCR tubes containing necessary compounds, and was tested at binding temperature of 55°C.
3.6. Studying Sensitivity With Concentration of Genome
The concentration of extracted genome was measured at 260 and 280 nm using spectrophotometer. Then, it was performed at various concentrations of PCR genome.
3.7. Verification of PCR Product
This was performed by restriction digestion of PCR product or its sequencing. For Bacillus anthracis PCR products we used HindIII digestion. Five uL of PCR product was added to the tube containing 1 µL of HindIII enzyme and enzyme buffer. Sterile distilled water was added to reach the final volume of 20 µL, and this mixture was maintained for 2 hours at 37°C. Then, 10 µL of this compound was investigated on agarose gel, and the other amplified products along with their front and rear primers were sent to the relevant company for sequencing.
3.8. Bioinformatic Analysis of Sequence
Incoming records containing sequences of primers F and R for each segment were investigated regarding accuracy and overlap with each other. Due to the small size of the parts, it was expected to have much overlap. DNASIS Ver 2.6 (Hitachi Co.) was used to identify overlapping. To this end, text file of primer F was compared with reverse complement sequences of R primer. By using the program ALIGN, they were attached together and edited to make the original sequence for each segment. Obtained sequences were compared with gene banks to find similarities and differences by using BLAST 2.2.9 (16). The DNASIS software was used to compare two sequences resulted from this study with reference sequences to identify the similarities and differences between the nucleotides.
4. Results
4.1. Uniplex and Multiplex PCR of Standard Strains
As seen in Figure 1, result of comparing the samples with four pairs of primers showed that the primers created 4 bands in uniplex and multiplex forms with sizes 164, 489, 520, and 1083 bp with primers 125 and 129 of Bacillus anthracis , primer T of Salmonella typhi, and primer F of Yersinia pestis . Figure 2 shows samples with four pairs of primers, which created 4 bands in uniplex and multiplex forms with sizes 164, 373, 520, and 1083 bp with primers 125 and 129 of Bacillus anthracis , primer S12 of Salmonella typhi, and primer F of Yersinia pestis .
Figure 1. Result of Uniplex and Multiplex PCR.
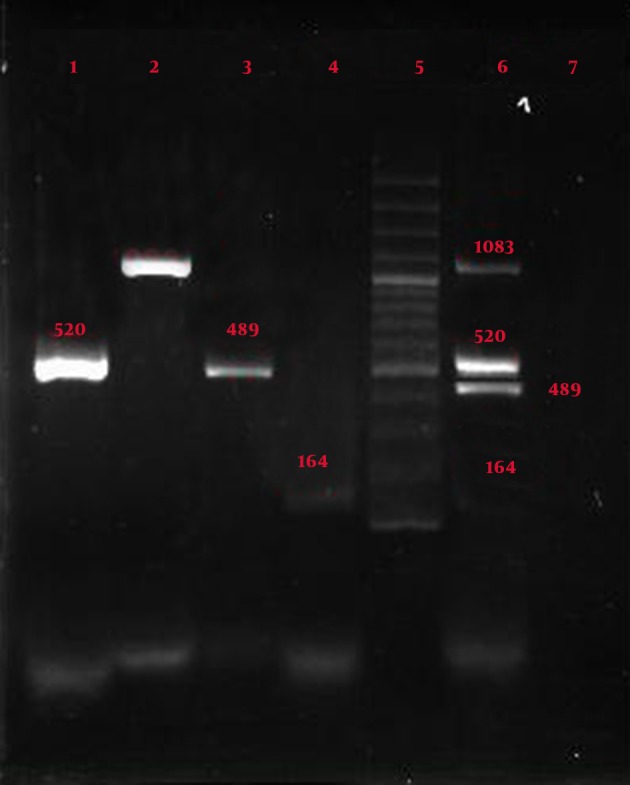
Lane1: Yersinia pestis by primer Yer (size 520 bp); Lane2: Bacillus anthracis by primer 125 (size 1083 bp); Lane3: Salmonella typhi by primer T (size 489 bp); Lane 4: Bacillus anthracis by primer 129 (size 164 bp); Lane 5: Molecular marker (100, 200, …..500, …1000, 1100, 1200, 1300, 1400); Lane 6: Multiplex PCR with the 4 pairs special primers, Yersinia pestis by primer Yer, Bacillus anthracis by primer 125, salmonella pestis by primer T, bacillus anthracis by primer 129; Lane 7: Negative control
Figure 2. Result of Uniplex and Multiplex PCR with the 4 Pairs Special Primers.
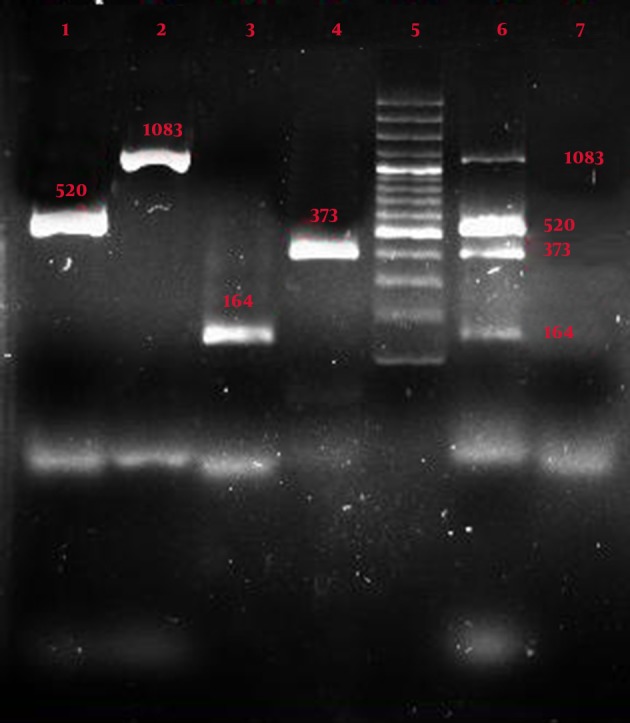
Lane 1: Yersinia pestis by the primer Yer (size 520 bp); Lane 2: Bacillus anthracis by the primer 125 (size 1083 bp); Lane 3: Bacillus anthracis by the primer 129 (size 164 bp); Lane 4: Salmonella typhi by the primer S12; Lane 5: Molecular marker; Lane 6: Multiplex PCR by the 4 pairs special primers: Bacillus anthracis( primer 129), Salmonella typhi(primer S12), Yersinia pestis (primer Yer), Bacillus anthracis (primer 125); Lane 7: Negative control
Figure 3 shows multiplex PCR with all three standard strains of bacteria, conducted with 5 pairs of specific primer PCR, and shows the presence of all expected components.
Figure 3. Result of Multiplex PCR with 5 Pairs Specials Primer.
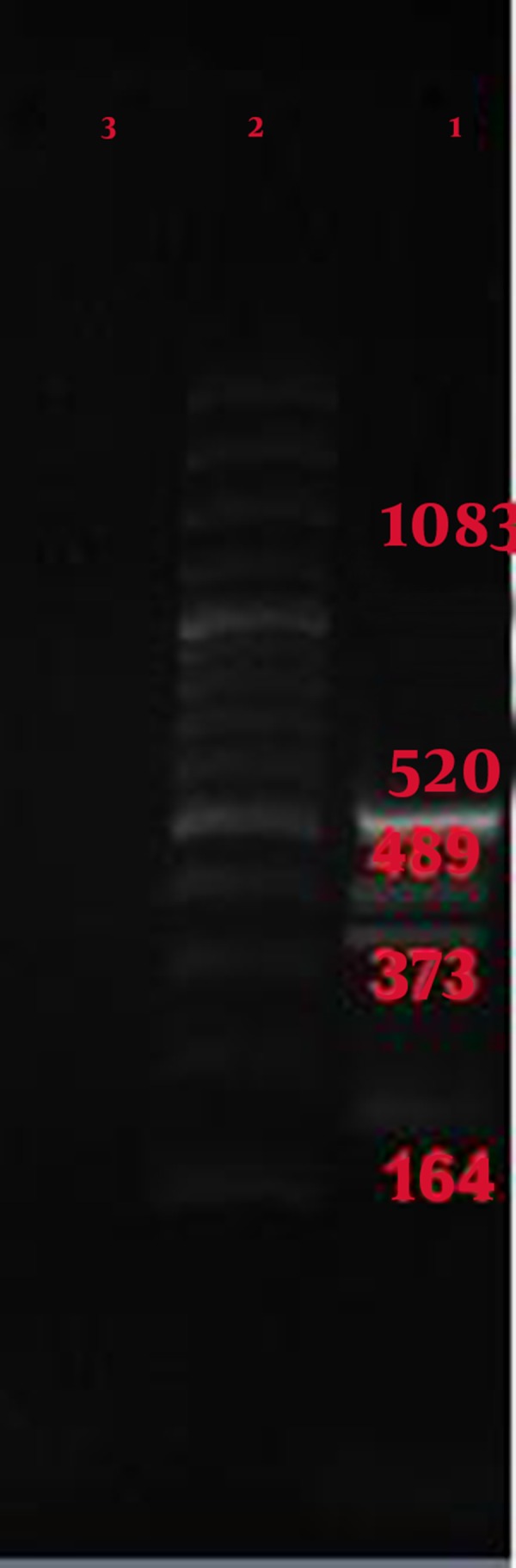
Lane 1: multiplex PCR, Bacillus anthracis with primer 129 (size 164 bp), Salmonella typhi with primer S12 (size 373 bp), Salmonella typhi with primer T (size 489 bp), Yersinia pestis with primer Yer (size 520 bp), and Bacillus anthracis with primer 125 (size 1083 bp); Lane 2: ladder; Lane 3: Negative control
4.2. The Specificity of PCR
Figures 1, 2, and 3 show the specificity of multiplex PCR and primers in that specific primers of Yersinia pestis, Bacillus anthracis , and Salmonella typhi did not create any bands with Staphylococcus aureus , Shigella sonnei , Escherichia coli , Enterococcus faecalis, Pseudomonas aeruginosa , Citrobacter freundii , Serratia marcescens, and Klebsiella pneumoniae .
4.3. Confirming the R Product of Bacillus anthracis Multiplex PCR With Primer 125
Results from enzyme cleavage product 1083 bp by the restriction enzyme cutting effect of HindIII, create 2 products with sizes of 696bp and 386bp, which confirm the accuracy of product generated by the primers (Figure 4).
Figure 4. HindIII Restriction Digestion of PCR Product.
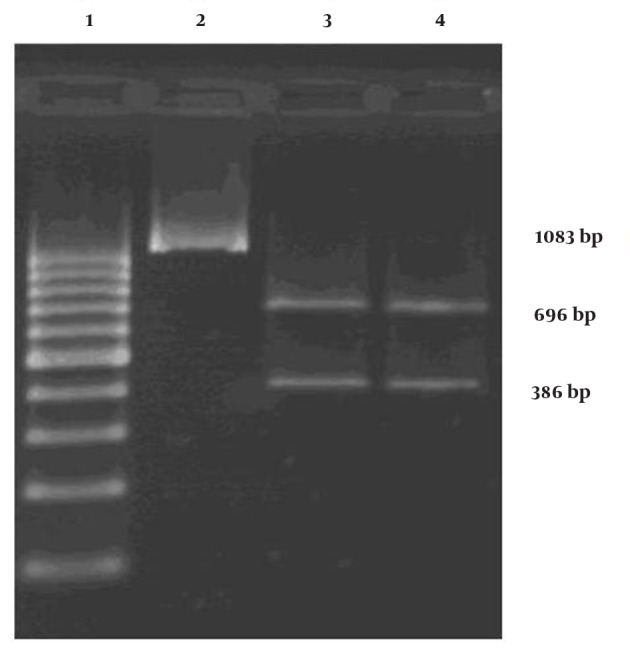
Lane1: Molecular marker; Lane 2: PCR fragment; Lane 3: Product digested by HindIII enzyme; Lane4: Product digested by HindIII enzyme
5. Discussion
Conventional methods to detect Salmonella typhi is a time consuming process without enough efficiency for rapid detection of bacteria (9). PCR has been used for detection and differentiation of typhoid from other enterobacteriaceae, as well as other salmonellas (17). To identify Salmonella typhi molecularly, researchers have designed and investigated several primers based on known gene sequence of tyv, flic-d, flic-a, viaB, prt, and spvC (18). In a study, researchers were able to detect Salmonella typhi on five pairs of primers based on genes viaB, fliC, tyv (rfbE), and prt (rfbS), by using multiplex method (19). The number of primers for this method is high, and yet the methods used is normal PCR at conventional speed. A variety of methods have been used for the detection of Bacillus anthracis in clinical animal and environmental specimens. Standard conventional detection methods are time consuming and inefficient for rapid detection of bacteria (20-22).
Different molecular methods have been used to detect Bacillus anthracis, and differentiating it from other bacilli (23-28). Hinbaj and Shwamm (1993) used gene primers pla to identify certain bacteria in their studies of bacterial genomes for Yersinia pestis under different species from Asia, Africa and America. Their research showed that performing PCR is reliable for control and diagnostic purposes, and epidemiological studies (29). Angeltaler et al. compared PCR method with inoculation to laboratory animals method to identify the bacteria causative for plague. Their research showed that not only PCR method eliminates false negative problem due to resistance of some laboratory animals against Yersinia pestis, but also is cost and time effective. Therefore, they concluded that this method is more sensitive and accurate compared to conventional diagnostic methods such as bacteriology cultivate and inoculation to laboratory animals, and can be a suitable method for detection, control and surveillance of plague (30). Multiplex method is not only less expensive and faster than uniplex, but also removes false negatives completely. Janse et al. performed multiplex real time PCR to detect Bacillus anthracis, Francisella tularensis, and Yersinia pestis. They used B. thuringiensis spores as internal controls. In another study by Skottman et al. (31) multiplex real time PCR was performed for simultaneous detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis . Also, they declared that simple multiplex PCR is much easier than real time PCR. In other study conducted by Skottman et al. (32) they applied multiplex PCR and RT-PCR enzyme hybridization assays for simultaneous detection of CDC category “A” bioterrorism agents is a complex and difficult method for rapid detection, and needs more simplification for filed application.
In this research performed based on invA and hp genes of Salmonella typhi, pA and chr of Bacillus anthracis, and pla of Yersinia pestis, all these genes were identified according to their specific primers. Due to the enormous progress in the field of molecular methods, such as multiplex PCR, they can be used to detect a variety of infectious agents, including detection of these three, i.e. Bacillus anthracis, Salmonella typhi, Yersinia pestis. The major factor in practical application of PCR is identification of specific primers. Primer designing needs studying different loci so that researchers used different existing parts of the sequences to design appropriate primers and probes. In this study, standard samples were used. Selected primers were able to identify the genomes of the virulent agents of these three bacteria specifically, and in direct samples. They neither respond to similar bacteria, nor to other bacteria that might exist in clinical samples. The size of PCR product of 164, 373, 489, 520, 1083 bp can be easily detected with usual agarose gel electrophoresis at concentration of 1% to 1.5%.
This approach allows rapid detection of genome, preparing reaction mixture, performing rapid PCR, electrophoresis cycles (containing ethidium bromide), and thus investigating result in the UV system in less than 90 min. Based on these results, the multiplex PCR can be used for rapid and simultaneous differentiation of three bacteria: Salmonella typhi, Bacillus anthracis, and Yersinia pestis from other similar bacteria. Given that these three factors can be named as biological and dangerous weapons, this study can provide a tool for rapid, accurate and low cost detection to detect them in case of bioterrorism operations and biological warfare.
Acknowledgments
Thanks to the Medical Research Deputy of Baqiyatallah Hospital, and the professors and colleagues from the Research Center for Molecular Biology and Institute of Military Medicine, whose sincere cooperation and assistance made this research possible.
Footnotes
Implication for health policy/practice/research/medical education:Raid Detection of Biological agent is crucial for Health workers and rapid response team like red crescent organization so in this research we have developed new technique for rapid and simultaneous detection of three important biological agents that could be used by rapid response team.
Authors’ Contribution:None declared.
Financial Disclosure:Dr. Ali Karami reported receiving research grants and writing proposal and funding the project.
Funding/Support:None declared.
References
- 1.Crump JohnA, Luby StephenP, Mintz EricD. The global burden of typhoid fever. Bulletin World Health Org. 2004;82(5):346–353. [PMC free article] [PubMed] [Google Scholar]
- 2.Purver RonaldG. Chemical and biological terrorism: the threat according to the open literature. Canadian Security Intelligence Service; 1995. [Google Scholar]
- 3.Okinaka R, Cloud K, Hampton O, Hoffmaster A, Hill K, Keim P, et al. Sequence, assembly and analysis of pX01 and pX02. J Appl Microbiol. 1999;87(2):261–2. doi: 10.1046/j.1365-2672.1999.00883.x. [DOI] [PubMed] [Google Scholar]
- 4.Franz DR, Jahrling PB, Friedlander AM, McClain DJ, Hoover DL, Bryne WR, et al. Clinical recognition and management of patients exposed to biological warfare agents. JAMA. 1997;278(5):399–411. doi: 10.1001/jama.278.5.399. [DOI] [PubMed] [Google Scholar]
- 5.Inglesby TV, Dennis DT, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, et al. Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 2000;283(17):2281–90. doi: 10.1001/jama.283.17.2281. [DOI] [PubMed] [Google Scholar]
- 6.Butler Thomas. Plague and other Yersinia infections. Plenum Medical Book Company; 1983. [Google Scholar]
- 7.Hinnebusch J, Cherepanov P, Du Y, Rudolph A, Dixon JD, Schwan T, et al. Murine toxin of Yersinia pestis shows phospholipase D activity but is not required for virulence in mice. Int J Med Microbiol. 2000;290(4-5):483–7. doi: 10.1016/S1438-4221(00)80070-3. [DOI] [PubMed] [Google Scholar]
- 8.Brubaker RR. Factors promoting acute and chronic diseases caused by yersiniae. Clin Microbiol Rev. 1991;4(3):309–24. doi: 10.1128/cmr.4.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivanoff Bernard. Typhoid fever: global situation and WHO recommendations. Southeast Asian J Tropical Med Pub Health. 1995;26:1–6. [Google Scholar]
- 10.Reeves MW, Evins GM, Heiba AA, Plikaytis BD, Farmer JJ, 3rd. Clonal nature of Salmonella typhi and its genetic relatedness to other salmonellae as shown by multilocus enzyme electrophoresis, and proposal of Salmonella bongori comb. nov. J Clin Microbiol. 1989;27(2):313–20. doi: 10.1128/jcm.27.2.313-320.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbull Peter, Böhm Reinhard, Hugh-Jones MartinE, Melling Jack. Guidelines for the surveillance and control of anthrax in humans and animals. 1998. [Google Scholar]
- 12.Leal NC, Almeida AM. Diagnosis of plague and identification of virulence markers in Yersinia pestis by multiplex-PCR. Rev Inst Med Trop Sao Paulo. 1999;41(6):339–42. doi: 10.1590/s0036-46651999000600002. [DOI] [PubMed] [Google Scholar]
- 13.Bahmanyar M, CAVANAUGH DanC. Plague manual. 1976. [Google Scholar]
- 14.Turnbull PCB, Hutson RA, Ward MandyJ, Jones MarieN, Quinn CP, Finnie NJ, et al. Bacillus anthracis but not always anthrax. J Appl Microbiol. 1992;72(1):21–28. doi: 10.1111/j.1365-2672.1992.tb04876.x. [DOI] [PubMed] [Google Scholar]
- 15.Joseph Sambrook, David WR. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. 2001;2. [Google Scholar]
- 16.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aabo S, Rasmussen OF, Roseen L, Sørensen PD, Olsen JE. Salmonella identification by the polymerase chain reaction. Molecular and cellular probes. 1993;7(3):171–178. doi: 10.1006/mcpr.1993.1026. [DOI] [PubMed] [Google Scholar]
- 18.Hirose K, Itoh K, Nakajima H, Kurazono T, Yamaguchi M, Moriya K, et al. Selective amplification of tyv (rfbE), prt (rfbS), viaB, and fliC genes by multiplex PCR for identification of Salmonella enterica serovars Typhi and Paratyphi A. J Clin Microbiol. 2002;40(2):633–6. doi: 10.1128/JCM.40.2.633-636.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrera-Leon S, McQuiston JR, Usera MA, Fields PI, Garaizar J, Echeita MA. Multiplex PCR for distinguishing the most common phase-1 flagellar antigens of Salmonella spp. J Clin Microbiol. 2004;42(6):2581–6. doi: 10.1128/JCM.42.6.2581-2586.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison LH, Ezzell JW, Abshire TG, Kidd S, Kaufmann AF. Evaluation of serologic tests for diagnosis of anthrax after an outbreak of cutaneous anthrax in Paraguay. J Infect Dis. 1989;160(4):706–10. doi: 10.1093/infdis/160.4.706. [DOI] [PubMed] [Google Scholar]
- 21.Johnson-Winegar A. Comparison of enzyme-linked immunosorbent and indirect hemagglutination assays for determining anthrax antibodies. J Clin Microbiol. 1984;20(3):357–61. doi: 10.1128/jcm.20.3.357-361.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logan NA, Carman JA, Melling J, Berkeley RC. Identification of Bacillus anthracis by API tests. J Med Microbiol. 1985;20(1):75–85. doi: 10.1099/00222615-20-1-75. [DOI] [PubMed] [Google Scholar]
- 23.Welkos SL, Lowe JR, Eden-McCutchan F, Vodkin M, Leppla SH, Schmidt JJ. Sequence and analysis of the DNA encoding protective antigen of Bacillus anthracis. Gene. 1988;69(2):287–300. doi: 10.1016/0378-1119(88)90439-8. [DOI] [PubMed] [Google Scholar]
- 24.Ramisse V, Patra G, Garrigue H, Guesdon JL, Mock M. Identification and characterization of Bacillus anthracis by multiplex PCR analysis of sequences on plasmids pXO1 and pXO2 and chromosomal DNA. FEMS Microbiol Lett. 1996;145(1):9–16. doi: 10.1111/j.1574-6968.1996.tb08548.x. [DOI] [PubMed] [Google Scholar]
- 25.Beyer W, Glockner P, Otto J, Bohm R. A nested PCR method for the detection of Bacillus anthracis in environmental samples collected from former tannery sites. Microbiol Res. 1995;150(2):179–86. doi: 10.1016/S0944-5013(11)80054-6. [DOI] [PubMed] [Google Scholar]
- 26.Henderson I, Duggleby CJ, Turnbull PC. Differentiation of Bacillus anthracis from other Bacillus cereus group bacteria with the PCR. Int J Syst Bacteriol. 1994;44(1):99–105. doi: 10.1099/00207713-44-1-99. [DOI] [PubMed] [Google Scholar]
- 27.Beyer W, Pocivalsek S, Bohm R. Polymerase chain reaction-ELISA to detect Bacillus anthracis from soil samples-limitations of present published primers. J Appl Microbiol. 1999;87(2):229–36. doi: 10.1046/j.1365-2672.1999.00875.x. [DOI] [PubMed] [Google Scholar]
- 28.Jackson PJ, Hugh-Jones ME, Adair DM, Green G, Hill KK, Kuske CR, et al. PCR analysis of tissue samples from the 1979 Sverdlovsk anthrax victims: the presence of multiple Bacillus anthracis strains in different victims. Proc Natl Acad Sci U S A. 1998;95(3):1224–9. doi: 10.1073/pnas.95.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinnebusch J, Schwan TG. New method for plague surveillance using polymerase chain reaction to detect Yersinia pestis in fleas. J Clin Microbiol. 1993;31(6):1511–4. doi: 10.1128/jcm.31.6.1511-1514.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janse I, Hamidjaja RA, Bok JM, van Rotterdam BJ. Reliable detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis by using multiplex qPCR including internal controls for nucleic acid extraction and amplification. BMC Microbiol. 2010;10:314. doi: 10.1186/1471-2180-10-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelthaler DM, Gage KL, Montenieri JA, Chu M, Carter LG. PCR detection of Yersinia pestis in fleas: comparison with mouse inoculation. J Clin Microbiol. 1999;37(6):1980–4. doi: 10.1128/jcm.37.6.1980-1984.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skottman T, Piiparinen H, Hyytiainen H, Myllys V, Skurnik M, Nikkari S. Simultaneous real-time PCR detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis. Eur J Clin Microbiol Infect Dis. 2007;26(3):207–11. doi: 10.1007/s10096-007-0262-z. [DOI] [PubMed] [Google Scholar]