

Abstract
Background and Purpose
Prohibitin (PHB) is a multi-functional protein involved in numerous cellular activities. PHB overexpression protects neurons from injury in vitro, but it is unclear whether PHB can protect selectively vulnerable hippocampal CA1 neurons in a clinically relevant injury model in vivo and, if so, whether the salvaged neurons remain functional.
Methods
A mouse model of transient forebrain ischemia that mimics the brain damage produced by cardiac arrest in humans was used to test whether PHB expression protects CA1 neurons from injury. PHB-expressing viral vector was microinjected in mouse hippocampus to upregulate PHB.
Results
PHB overexpression protected CA1 neurons from transient forebrain ischemia. The protection was associated with dampened post-ischemic ROS generation, reduced mitochondrial cytochrome c release and decreased caspase-3 activation. Importantly, the improvement in CA1 neuronal viability translated into an improvement in hippocampal function: PHB expression ameliorated the spatial memory deficit induced by ischemia, assessed by the Y-maze test, and restored post-ischemic synaptic plasticity assessed by long-term potentiation, indicating that the neurons spared form ischemic damage were functionally competent.
Conclusions
These data demonstrate that PHB overexpression protects highly vulnerable CA1 neurons from ischemic injury in vivo, and suggest that the effect is mediated by reduction of post-ischemic ROS generation and preservation of mitochondrial outer membrane integrity that prevents activation of apoptosis. Measures to enhance PHB expression could have translational value in ischemic brain injury and, possibly, other forms of brain injury associated with mitochondrial dysfunction.
Keywords: prohibitin, Mitochondria, neuroprotection, transient forebrain ischemia
Introduction
Prohibitin (PHB) is a highly conserved protein that participates in a wide variety of cellular processes 1, 2 in multiple cellular compartments 3. In mitochondria, PHB acts as a scaffolding protein and, as such, is essential for the formation and maintenance of mitochondrial structure, and for the stability of their nucleoids 4, 5. Deletion of PHB is lethal, highlighting its essential biological role, and its upregulation is protective for intestinal epithelial cells, cardiomyocytes, and pancreatic-beta cells 6–8.
PHB is also emerging as a key regulator of neuronal survival. It is upregulated in models of ischemic tolerance and in exercise-induced neuroplasticity 9, 10, suggesting a cytoprotective role in brain. Accordingly, PHB expression in neuronal cultures is protective against excitotoxicity, reactive oxygen species (ROS) and apoptosis inducers 10. Conversely, ablation of PHB expression in mouse forebrain results in neuronal death 11. A major mechanism of PHB-mediated cytoprotection involves suppression of mitochondrial ROS production. Whereas upregulation of PHB expression in neurons dampens complex I-dependent ROS generation, down-regulation of PHB expression increases mitochondrial ROS formation in response to glutamate 10. Therefore, PHB is an important modulator of neuronal survival through mitochondrial mechanisms. However, due to the lack of pharmacological approaches to specifically upregulate PHB in vivo, it has not been possible to determine whether PHB is neuroprotective in a clinically relevant animal model of neuronal injury. Considering the central role that mitochondria and oxidative stress play in cerebral ischemia-reperfusion injury 12–14, it is conceivable that PHB could confer neuroprotection in this highly prevalent and devastating form of brain injury.
In the present study, we tested this possibility using adeno-associated viral (AAV) gene transfer to express human PHB in the hippocampal CA1, a region selectively vulnerable to the deleterious effects of ischemia-reperfusion. We found that PHB expression in CA1 neurons protects them from the delayed cell death induced by transient forebrain ischemia, a model that mimics the brain damage produced by cardiac arrest in humans 15–17. The effect was associated with suppression of the early phase of post-ischemic ROS generation, as well as cytochrome (cyt) c release and caspase-3 activation. Notably, the protection of CA1 neurons resulted in a marked improvement in post-ischemic hippocampal long-term potentiation (LTP) and in Y-maze performance. The findings provide the first evidence to date that the powerful neuroprotective capacity of PHB can be harnessed in a clinically relevant model of delayed neuronal death in vivo, and raise the possibility that PHB upregulation could be a novel preventive strategy in ischemic injury and neurodegeneration.
Materials and methods
AAV production and injection in vivo
PHB is a nuclear encoded protein. It is translated in the cytosol and imported into mitochondria through its N-terminal mitochondrial targeting sequence (MTS, amino acids 1–18) 18. The AAV transduced PHB likely follows the same mitochondrial import pathway as the endogenous PHB. Recombinant AAV vectors were constructed in a di-cistronic vector containing GFP and PHB expressed under the control of a CMV promoter, as previously described10. The same AAV vector, expressing GFP but not PHB, was used as control. The PHB expressing AAV is designated as AAV-PHB-GFP. The AAV without PHB is designated as vector control. Serotype 2 (AAV-2) was used in all experiments. We chose AAV-2 because of its low host immune response and minimal inflammatory stimulation 19, 20, and its high propensity for neuronal expression 21. Furthermore, our initial screening showed that AAV-2 achieved high neuron/glia transduction ratio in mixed primary cultures (data not shown). The AAV stock was produced by the University of Iowa Vector Core Facility.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Stereotaxic AAV injections into hippocampal CA1 sector were performed in 4-week-old C57BL/6J mice as previously described 22 and detailed in the supplements. AAV injection did not cause animal fatality.
Transient forebrain ischemia by bilateral common carotid artery occlusion (BCCAO)
BCCAO was performed 3 weeks after AAV injection according to a procedure described previously23 and detailed in the supplemental methods. Briefly, after anesthesia, both common carotid arteries were exposed through a midline incision of the neck and occluded for 22 min. Sham-operated mice underwent the same procedures, except that their arteries were not occluded. The physiological parameters of mice before and after BCCAO are shown in supplemental material (Supplemental Fig. I). Of the total 210 mice that were subjected to the BCCAO procedure, 24 died before various experiments were performed, resulting in 11% attrition rate.
Immunohistochemistry
Mice were perfused transcardially with PBS followed by 4% paraformaldehyde in PBS. The brains were removed and coronal sections (thickness 10μm) were cut in a cryostat and collected at 100 μm intervals for immunofluorescence staining with antibodies (MAP2 from Sigma; NeuN and GFAP from Millipore). Nuclear staining was performed with TO-PRO-3 iodide (Invitrogen).
Cell counts in CA1
Cell counts were performed by an operator blinded to the treatment groups. Hippocampal cell death was assessed according to a standard protocol described previously 23. Briefly, brain sections were stained with hematoxylin and eosin (HE). One 10 μm-thick section was collected every 100μM. Injury to the CA1 subfield was evaluated in the medial and intermediate portion of the CA1 under a light microscope at 100× magnification by manually counting the viable neurons using NIH ImageJ. Viable neurons were defined as those with a blue hue, an intact plasma membrane, and a hypochromatic oval or round nucleus. Eosinophilic cells with a shrunken nucleus were defined as dead cells. Cell counting was performed in 5 sections per hippocampus starting at 1.4 mm posterior to bregma encompassing the core of the lesioned area (rostro-caudal levels -1.4, -1.5, -1.6, -1.7, -1.8 mm from bregma).
Determination of ROS production
ROS production was assessed in vivo using hydroethidine microfluorography, as previously described 24. Dihydroethidine (DHE; 10 mg/kg) was injected into the jugular vein 2.5 hrs before brains were removed, frozen, and cut in a cryostat (thickness 20 μm). Images were acquired by a computer-controlled digital monochrome camera. After subtracting the camera dark current, the pixel intensity of the fluorescent oxidized hydroethidine signal was assessed in the hippocampal CA1 region, 5 sections per animal. The sum of the fluorescence intensities for each region was divided by the total number of pixels analyzed and expressed as relative fluorescence units 24. Fluorescence intensity was assessed 0, 1, 3, 6, 8, 12, 24, 48, and 72 hours after BCCAO/reperfusion.
Western blotting and cytochrome c release
The CA1 subregion of hippocampus was dissected as described25 and cyt c released in the cytosol was assessed by a subcellular fractionation procedure described previously 26. For PHB western blotting, AAV-PHB or vector control injected hippocampus were homogenized in RIPA buffer. Equal amounts of proteins were gel separated, transferred to membranes and incubated with anti-cytochrome c antibody (Cell Signaling) or with anti-PHB antibody (NeoMarker) in appropriate dilutions. Protein bands were detected and intensity quantified using a Kodak Digital Imaging Station.
Caspase-3 activity assay
Caspase-3 activity in hippocampal CA1 tissues was assayed as described previously 26 and detailed in the supplemental methods.
Hippocampal slice preparation and electrophysiology
Electrophysiology experiments were performed as described previously 27 and are described in supplemental methods.
Y-maze test
Spatial working memory in mice was evaluated by the Y-maze test, as previously described 28, 29. We chose this test because it is less stressful and more consistent with the natural behavior of mice than other spatial memory tests, such as the Morris water maze 30. Furthermore, the Y maze test does not involve learning new rules and takes advantage of the natural tendency of rodents to explore new environments 31. The operator was blinded to the treatment that the mice had received. Before and 7 days after BCCAO, each mouse was placed at the end of one arm and allowed to freely explore the apparatus for 8 minutes. The sequence and number of all arm entries were recorded for each animal throughout the period. Alternation rate was defined as entries into all three arms on consecutive occasions using the following formula: . Trials in which the number of total arm entries was less than 10 were not included in the analysis.
Statistical analysis
Data are expressed as Mean±SEM. Differences between multiple groups were statistically evaluated by the one-way ANOVA Analysis of Variance followed by the appropriate post hoc tests.
Results
AAV-mediated PHB gene transfer in mouse hippocampus
First, we examined the regional pattern and cellular localization of gene expression in the hippocampus injected with AAV-PHB-GFP. Maximal levels of gene expression of PHB-GFP di-cistronic construct were observed 3 weeks after AAV-PHB-GFP injection. PHB-GFP expression was mostly restricted to the injected hippocampus (Fig. 1A) and observed mostly in neurons, although sparse astrocytic processes were also positive (Supplemental Fig. II). PHB expression assessed by western blot was also increased in the hippocampus injected with AAV-PHB-GFP, but not vector (Fig. 1B, C).
Figure 1.
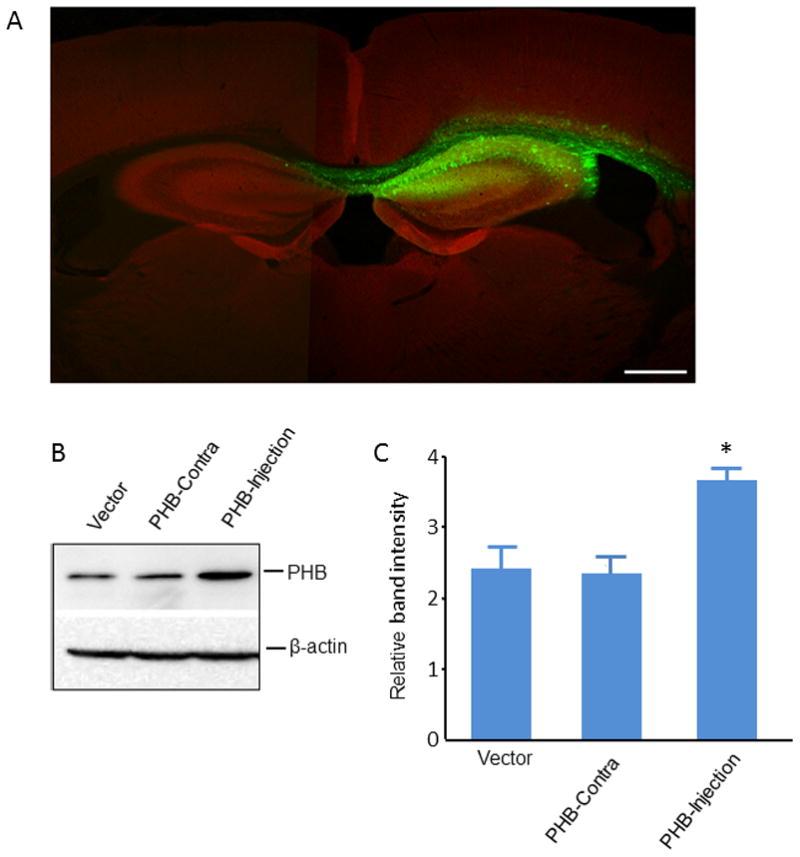
AAV-mediated PHB expression in the mouse hippocampus. Three weeks after AAV injection, mouse brains were collected either for immunostaining or for western blotting. (A) Robust AAV mediated GFP expression in hippocampus. Coronal sections were immunostained with the neuronal marker Map-2 (red) to facilitate the identification of GFP expression in neurons. Scale bar is 500 μm. (B) AAV-mediated PHB expression was higher in the injected hippocampus (right side) compared to the contralateral hippocampus (left side) and naïve control hippocampus (non-AAV injected). Total tissue lysate (20μg/lane) was used for the experiments. β-actin was used as a loading control. A representative blot from 4 independent experiments is shown. (C) Protein band intensity quantification. *p<0.05, n=4/group.
PHB viral gene transfer in CA1 protects against BCCAO-induced cell death
Next, we tested the effect of PHB viral gene transfer on hippocampal CA1 injury produced by transient forebrain ischemia. In mice injected with the control vector, BCCAO caused extensive cell death in CA1 assessed by morphological criteria. However, in mice that received AAV-PHB-GFP injections, post-ischemic cell death was markedly reduced (Fig. 2A). In post-ischemic mice treated with control vector, cell nuclei in CA1 appeared smaller and GFP expression was not observed in cells with neuronal morphology (Fig. 2B upper panels). In contrast, in mice treated with AAV-PHB-GFP most nuclei had a normal appearance and several PHB-GFP positive neurons were observed in CA1 (Fig. 2B lower panels). Because the magnitude of cell death induced by BCCAO in CA1 differs rostrocaudally, we examined the degree of protection conferred by AAV-PHB-GFP gene transfer at different rostrocaudal levels of the hippocampus (Fig. 2C). 3D reconstruction from serial sections demonstrated that the ischemic lesion was located entirely within the volume of tissue expressing PHB (Fig. 2C). A reduction in cell death was observed at all rostrocaudal levels, although the protection tended to be more pronounced caudally, where the injury was less severe (Fig. 2D). Collectively, these results demonstrate that AAV-PHB-GFP gene transfer protects CA1 cells from the deleterious effects of transient forebrain ischemia.
Figure 2.
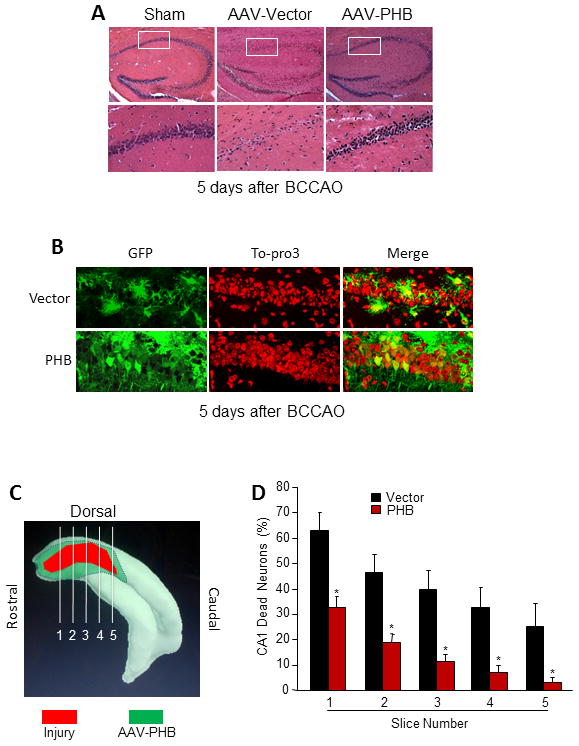
PHB expression protected CA1 neurons from cell death after BCCAO. (A) H & E staining of hippocampal sections 5 days after BCCAO. Vector injected hippocampus suffered extensive cell death in CA1 (middle panels), an effect reduced by PHB expression (right panels). (B) Cell morphology in hippocampus 5 days after BCCAO. Hippocampal sections were stained with Alexa 488 conjugated anti-GFP antibody and counterstained with the nuclear dye TO-PRO-3. Top panels, vector injected. Lower panels, PHB injected. Note the astrocytic labeling due to weak GFP expression in glia. (C) Diagram illustrating the location of cell injury after BCCAO in a 3D reconstruction of the hippocampus (red). The green area marks the extent of PHB gene transfer. Note that the area of injury is comprised within the area of PHB gene transfer. Lines indicate the 5 levels at which cell counts were obtained. (D) PHB gene transfer reduced cell death at all levels of the lesion indicated in C. *p<0.05 in each section, n=15 for vector and 20 for PHB.
PHB viral gene transfer in CA1 attenuates post-ischemic ROS production
Next, we sought to investigate the mechanisms of the protective effect of PHB gene transfer in CA1. Oxidative stress is a key factor in ischemic brain damage 13, 32. Therefore, we examined whether the protective effect of PHB could be attributed to reduced post-ischemic ROS production. ROS production in mice not injected with AAV, assessed by DHE fluoromicrography, exhibited a biphasic time course with peaks at 3 and 72 hrs after reperfusion (Fig. 3A). As shown in Fig. 3B and Fig. 3C, AAV-PHB-GFP injection significantly decreased the DHE signal in hippocampus at both time points (3 hrs and 72 hrs) compared to samples from vector injected mice. Therefore, PHB viral gene transfer suppresses the post-ischemic increase in damaging ROS in CA1.
Figure 3.
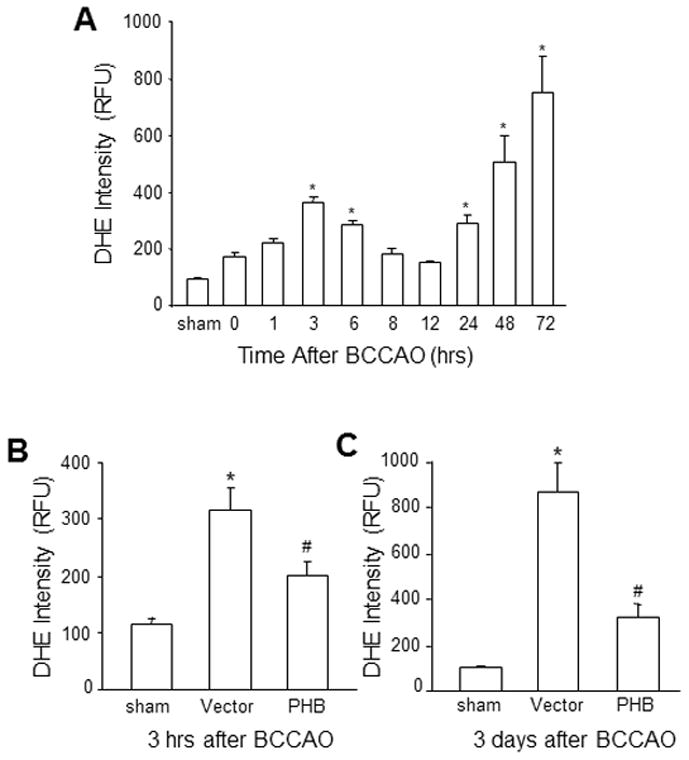
PHB expression attenuated ROS production in the hippocampus after BCCAO. (A) Biphasic temporal profile of ROS generation after BCCAO in mouse hippocampus detected by DHE fluoromicrography. (B) PHB expression reduced the ROS signal in hippocampus at 3 hrs and (C) at 3 days. *p<0.05 compared to sham group; #p<0.05, compared to vector group by one way ANOVA analysis followed by Dunnett’s test, n=5/group.
PHB viral gene transfer attenuates post-ischemic cyt c release from mitochondria and caspase-3 activation
Having established that PHB viral gene transfer attenuated post-ischemic ROS production, we examined whether cyt c release and caspase activation, critical steps downstream of ROS in delayed post-ischemic hippocampal damage 33, were also attenuated by PHB expression. Released hippocampal cyt c was assayed in the cytosolic fraction, following subcellular fractionation. As previously reported 34, cytosolic cyt c increased at 6 hrs, returned to baseline at 12 hrs, rose again at 24 hrs and reached a second peak at 48 hr, after BCCAO (Supplemental Fig. IIIA, B). PHB viral gene transfer attenuated cyt c release at 48hrs after BCCAO (Fig. 4A, B). Since cyt c release has been linked to post-ischemic caspase activation and apoptotic cell death in CA1 35, we next sought to determine if PHB viral gene transfer reduces also post-ischemic caspase 3 activation. Hippocampal caspase-3 activation exhibited a biphasic pattern, which was delayed relative to that of cyt c release, with peaks at 8 and 72 hrs after BCCAO (Fig. 4C). Because the 72 hrs peak in caspase activation is thought to contribute to neuronal death after forebrain ischemia 36, we assessed the effect of PHB viral gene transfer at this time point. As illustrated in Fig. 4D, injection of AAV-PHB-GFP, but not vector, attenuated caspase 3 activity. Therefore, PHB viral gene transfer attenuated post-ischemic cyt c release from mitochondria and the ensuing caspase 3 activation in CA1.
Figure 4.

PHB expression attenuated cyt c release and post-ischemic caspase-3 activity in the hippocampus after BCCAO. (A) PHB expression attenuated the cyt c release induced by BCCAO. Cytosolic fractions (10 μg/lane) from sham, vector and PHB injected hippocampal CA1 48 hrs after BCCAO were used. The blot shown was from one of 6 experiments. (B) Protein band intensity quantitation from data in (A). *p<0.05, compared to sham group, #p<0.05 compared to vector group, one-way ANOVA analysis followed by Dunnett’s test; n=6/group. β-actin was used as a loading control to normalize the cyt c band intensity. (C)Time course of caspase-3 activity following BCCAO in hippocampal CA1 of non-injected mice. *p<0.05 compared to sham group, one-way ANOVA analysis followed by Tukey’s test, n=4/group. (D) PHB expression reduced caspase-3 activity in hippocampal CA1 measured at 3 days after BCCAO. *p<0.05 compared to sham group; #p<0.05 compared to vector group, n=6/group.
PHB viral gene transfer ameliorates post-ischemic alterations in hippocampal LTP and Y maze performance
Finally, we sought to determine if the neuroprotection exerted by PHB viral gene transfer in CA1 is associated with improvement in post-ischemic hippocampal function. Hippocampal LTP is impaired by transient forebrain ischemia 37, 38. Therefore we examined whether PHB viral gene transfer could counteract the LTP alterations induced by BCCAO. Consistent with hippocampal injury, LTP was suppressed in hippocampal slices 7 days after BCCAO in mice injected with vector (Fig. 5A, B, C). In contrast, hippocampal LTP was well developed in mice that received AAV-PHB-GFP (Fig. 5A, B, C). In addition, PHB viral gene transfer did not alter LTP in hippocampal slices from vector injected mice (Fig. 5D). To provide evidence of cognitive preservation by PHB viral gene transfer we examined the performance of the mice at the Y-maze (arm alternation) test, a sensitive and widely used approach to assess hippocampus-dependent spatial memory39–41. Mice injected with vector or PHB-AAV were tested before and 7 days after sham operation or BCCAO. Before BCCAO, there were no differences in spontaneous arm alternations among groups (Fig. 6B). Seven days after BCCAO, the vector group showed reduced spontaneous alternations (Fig 6B), suggestive of hippocampal dysfunction42. However, AAV-PHB-GFP injection eliminated such dysfunction, and the performance was not different from that before BCCAO or sham treatment (Fig. 6B, p>0.05). Of note, no motor deficits were observed in the mice after BCCAO; rather, there was a tendency for BCCAO to increase the total number of arm entries, suggesting hyperactivity, but this difference did not reach statistical significance (Fig. 6A). Thus, hippocampal PHB viral gene transfer improves the post-ischemic impairments of LTP and Y maze performance.
Figure 5.

PHB expression ameliorated the long-term potentiation (LTP) impairments induced by BCCAO in mice. (A) High-frequency stimulation (HFS) induced LTP was inhibited in hippocampal slices from mice injected with vector 7 days after BCCAO (dark gray circles, n=10). In contrast, hippocampal LTP was well maintained in slice from mice injected with PHB (gray triangles, n=8), comparable to levels of LTP elicited in control mice (open squares, n=7). (B) Representative fEPSP traces before and 60 minutes after HFS for LTP experiments shown in panel (A). Scale: 1 mv/10ms. (C) Cumulative data showing mean fEPSP slopes 60 minutes post-HFS based from the LTP experiments in panel (A). (D) Baseline fEPSPs were not affected by vector or PHB AAV injection. HFS-induced hippocampal LTP was expressed similarly in mice injected with either vector (open square, n=6) or PHB (grey circles, n=6) but not subjected to BCCAO.
Figure 6.
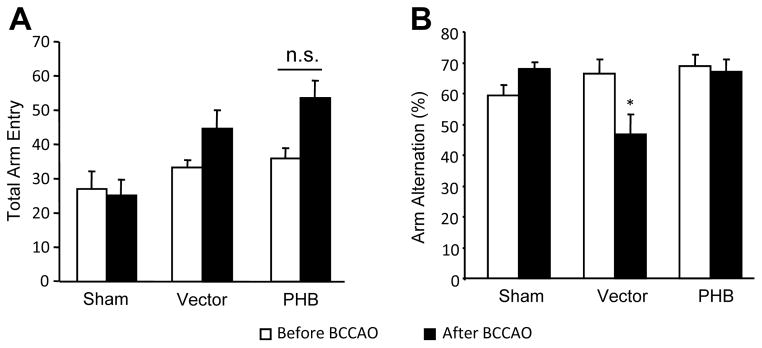
PHB expression restored spatial working memory loss following BCCAO in mice. (A) Total number of arm entry in 8 min Y-maze tests before and 7 day after BCCAO. An increase in motor activity after BCCAO was observed, but the difference between vector and PHB injected mice was non-significant (n.s.). (B) Y-maze arm entry alteration test for PHB and vector injected mice before and 7 days after BCCAO. PHB expression prevented the decrease in arm entry alternations. *p<0.05, n=8 /group.
Discussion
PHB viral gene transfer protects CA1 neurons from ischemic cell death
We used a mouse model of transient forebrain ischemia that recapitulates some of the neuropathological features of the brain damage suffered during global cerebral hypoperfusion, as it occurs in systemic hypotension or cardiac arrest16. The CA1 hippocampal damage produced by transient forebrain ischemia is spatially and temporally defined16, 17, 43–45 and, as such, is amenable to viral gene transfer using local microinjection22. We found that PHB viral gene transfer in the hippocampus increases PHB expression and ameliorates the CA1 damage induced by transient forebrain ischemia. The effect is observed throughout the damaged regions of the hippocampus and is associated with suppression of post-ischemic ROS production, cyt c release from mitochondria, and caspase-3 activation. Remarkably, the protective effect of PHB is associated with improved hippocampal LTP and Y-maze performance, suggesting that PHB upregulation protects hippocampal function.
Body temperature is a critical variable in experiments of cerebral ischemia 46. However, the findings of this study cannot be attributed to differences in body temperature because temperature was carefully controlled during the critical period up to 24 hrs after ischemic insult, according to our established protocol 23. In addition, the protective effect of PHB expression observed at multiple rostrocaudal levels of the damaged hippocampus rules out the possibility that the protection was observed only in areas of minimal damage. However, it could not be established whether only the neurons expressing PHB were spared from damage or whether there was a protective “bystander effect” on nearby neurons, as suggested by other studies47, 48.
PHB viral gene transfer suppresses post-ischemic ROS production in CA1
The major contributing factors causing delayed neuronal death in the vulnerable hippocampal CA1 subregion in the transient forebrain ischemia model include oxidative stress and activation of the apoptotic cell death pathway45. In agreement with previous reports49, we observed a biphasic increase in ROS in CA1 3 hrs and 3 days after BCCAO. The initial ROS peak is thought to trigger downstream events leading to delayed cell death49. Therefore, the observation that PHB viral gene transfer dampens the early ROS peak suggests that this effect is involved in the attendant protection from delayed neuronal death.
The mechanisms by which PHB suppresses ROS production remain to be elucidated. PHB reduces complex I generated ROS due to rotenone inhibition in neuronal cultures10. Accordingly, one possibility is that PHB directly protects complex I and reduces ROS generation from complex I, a major source of ROS after cerebral ischemia-reperfusion injury50, 51. In addition, PHB expression may contribute to maintaining mitochondrial inner membrane structural integrity52, possibly due to its role in the assembly of respiratory chain components53. Consequently, more stress could be sustained before the respiratory chain complexes succumb to oxidative damage, giving rise to a vicious cycle of ROS production. Irrespective of the mitochondrial mechanisms of the effect, the attenuation of post-ischemic ROS production by PHB is partial, suggesting that either mitochondrial radical sources are not completely stabilized or other, non respiratory chain dependent, ROS generating pathways are also involved. An alternative source of ROS after transient global ischemia is the superoxide producing enzyme NADPH oxidase54, but it remains to be established whether PHB overexpression dampens this ROS source as well. Given the proximity of NADPH oxidase subunits to neuronal mitochondria55, it is likely that NADPH oxidase generated ROS may trigger additional ROS production from neuronal mitochondria and vice-versa56, 57. Therefore, inhibition of either source may lower total ROS production.
Inflammatory response is another factor contributing to post-ischemic brain injury58. However, recent data indicate that inflammation is likely to be downstream of the early ROS production observed in transient forebrain ischemia 59. Therefore, it would be of interest to investigate whether PHB also suppresses post-ischemic inflammation in this model.
PHB viral gene transfer reduces post-ischemic cyt c release and caspase-3 activation
Since mitochondria play a key role in apoptotic cell death in CA114, 60, we examined the effect of PHB-viral gene transfer on the mitochondrial release of cyt c and on the attendant caspase-3 activation. PHB viral gene transfer markedly attenuated cyt c release and caspase-3 activation in the post-ischemic hippocampus, implicating inhibition of the mitochondrial apoptotic pathway in the mechanisms of protection. Considering that oxidative stress leads to mitochondrial permeability transition (MPT), cyt c release, and apoptosis61, 62, it is conceivable that PHB suppresses the ROS increase at 3 hrs after ischemia, thereby preventing MPT, the resulting reduction in cyt c release and caspase-3 activation.
PHB viral gene transfer improves post-ischemic hippocampal function and the Y-maze performance
The damage to CA1 induced by forebrain ischemia is associated with deficits in hippocampal function and behavioral performance 41. The decrease in the post-ischemic suppression of LTP indicates that the CA1 neurons salvaged by PHB gene transfer are able to express synaptic strengthening in response to tetanic stimulation of their inputs, a physiological characteristic of these neurons thought to play a role in learning and memory 63. The improvement in LTP involved the maintenance phase and not the induction phase of the potentiation. The detailed neurophysiological and molecular mechanisms associated with the phenomenon together with the effect of PHB expression on the alterations in LTP induced by forebrain ischemia remain to be elucidated.
In agreement with the LTP improvement, we also observed an improvement in the cognitive performance assessed by the Y-maze test, indicating preservation of hippocampal circuits involved in spatial working memory 39. However, we cannot rule out that the behavioral improvement resulted from an enhancement of recovery of function induced by PHB expression. We used the Y-maze to explore cognitive function because this method relies on the natural inclination of mice to explore and, as such, does not require training, is not stressful and is a sensitive indicator of post-ischemic hippocampal dysfunction 42. Collectively, these observations provide evidence that the CA1 neurons spared from cell death by PHB viral gene transfer not only appear morphologically intact, but are also functionally competent. Another issue concerns whether PHB expression improves the baseline performance of intact CA1 neurons. Our data suggest that this is not the case, because PHB viral gene transfer did not enhance LTP or Y-maze performance in naïve mice. Therefore, the improvement of hippocampal function is likely the direct consequence of the protection from post-ischemic degeneration of CA1 neurons due to viral mediated PHB expression.
In conclusion, we have shown for the first time that PHB expression mediated by viral gene transfer protects hippocampal CA1 neurons from injury and preserves neuronal function after transient forebrain ischemia. Although the full translational potential of PHB remains to be assessed, the present results provide proof of principle that upregulation of PHB is beneficial to the post-ischemic brain in vivo and, as such, could be a novel approach to protect the brain in neurological diseases associated with acute or chronic neuronal degeneration due to mitochondrial dysfunction.
Supplementary Material
Acknowledgments
Funding Sources
Supported by NIH grants NS67078 (PZ), NS34179 (CI).
Footnotes
Disclosures
No
References
- 1.Thuaud F, Ribeiro N, Nebigil CG, Desaubry L. Prohibitin ligands in cell death and survival: Mode of action and therapeutic potential. Chem Biol. 2013;20:316–331. doi: 10.1016/j.chembiol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta. 2011;1813:1137–1143. doi: 10.1016/j.bbamcr.2011.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mishra S, Ande SR, Nyomba BL. The role of prohibitin in cell signaling. FEBS J. 2010;277:3937–3946. doi: 10.1111/j.1742-4658.2010.07809.x. [DOI] [PubMed] [Google Scholar]
- 4.Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res. 2008;314:988–996. doi: 10.1016/j.yexcr.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Lower B, Wunderlich FT, et al. Prohibitins control cell proliferation and apoptosis by regulating opa1-dependent cristae morphogenesis in mitochondria. Genes Dev. 2008;22:476–488. doi: 10.1101/gad.460708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa b nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. 2009;20:4412–4423. doi: 10.1091/mbc.E09-05-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu X, Ren Z, Zhan R, Wang X, Wang X, Zhang Z, et al. Prohibitin protects against oxidative stress-induced cell injury in cultured neonatal cardiomyocyte. Cell stress & chaperones. 2009;14:311–319. doi: 10.1007/s12192-008-0086-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Nguyen KH, Mishra S, Nyomba BL. Prohibitin is expressed in pancreatic beta-cells and protects against oxidative and proapoptotic effects of ethanol. Febs J. 2010;277:488–500. doi: 10.1111/j.1742-4658.2009.07505.x. [DOI] [PubMed] [Google Scholar]
- 9.Ding Q, Vaynman S, Souda P, Whitelegge JP, Gomez-Pinilla F. Exercise affects energy metabolism and neural plasticity-related proteins in the hippocampus as revealed by proteomic analysis. Eur J Neurosci. 2006;24:1265–1276. doi: 10.1111/j.1460-9568.2006.05026.x. [DOI] [PubMed] [Google Scholar]
- 10.Zhou P, Qian L, D’Aurelio M, Cho S, Wang G, Manfredi G, et al. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J Neurosci. 2012;32:583–592. doi: 10.1523/JNEUROSCI.2849-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merkwirth C, Martinelli P, Korwitz A, Morbin M, Bronneke HS, Jordan SD, et al. Loss of prohibitin membrane scaffolds impairs mitochondrial architecture and leads to tau hyperphosphorylation and neurodegeneration. PLoS genetics. 2012;8:e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sims NR, Muyderman H. Mitochondria, oxidative metabolism and cell death in stroke. Biochim Biophys Acta. 2010;1802:80–91. doi: 10.1016/j.bbadis.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Chan PH. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem Res. 2004;29:1943–1949. doi: 10.1007/s11064-004-6869-x. [DOI] [PubMed] [Google Scholar]
- 15.Kirino T. Delayed neuronal death. Neuropathology: official journal of the Japanese Society of Neuropathology. 2000;20 (Suppl):S95–97. doi: 10.1046/j.1440-1789.2000.00306.x. [DOI] [PubMed] [Google Scholar]
- 16.Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- 17.Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Annals of neurology. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- 18.Ikonen E, Fiedler K, Parton RG, Simons K. Prohibitin, an antiproliferative protein, is localized to mitochondria. FEBS Lett. 1995;358:273–277. doi: 10.1016/0014-5793(94)01444-6. [DOI] [PubMed] [Google Scholar]
- 19.Feng X, Eide FF, Jiang H, Reder AT. Adeno-associated viral vector-mediated apoe expression in alzheimer’s disease mice: Low cns immune response, long-term expression, and astrocyte specificity. Front Biosci. 2004;9:1540–1546. doi: 10.2741/1323. [DOI] [PubMed] [Google Scholar]
- 20.McPhee SW, Janson CG, Li C, Samulski RJ, Camp AS, Francis J, et al. Immune responses to aav in a phase i study for canavan disease. J Gene Med. 2006;8:577–588. doi: 10.1002/jgm.885. [DOI] [PubMed] [Google Scholar]
- 21.Taymans JM, Vandenberghe LH, Haute CV, Thiry I, Deroose CM, Mortelmans L, et al. Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther. 2007;18:195–206. doi: 10.1089/hum.2006.178. [DOI] [PubMed] [Google Scholar]
- 22.Sun HS, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal trpm7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12:1300–1307. doi: 10.1038/nn.2395. [DOI] [PubMed] [Google Scholar]
- 23.Shimamura M, Zhou P, Casolla B, Qian L, Capone C, Kurinami H, et al. Prostaglandin e type 1 receptors contribute to neuronal apoptosis after transient forebrain ischemia. J Cereb Blood Flow Metab. 2013;33:1207–1214. doi: 10.1038/jcbfm.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- 25.Lein ES, Zhao X, Gage FH. Defining a molecular atlas of the hippocampus using DNA microarrays and high-throughput in situ hybridization. J Neurosci. 2004;24:3879–3889. doi: 10.1523/JNEUROSCI.4710-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou P, Qian L, Glickstein SB, Golanov EV, Pickel VM, Reis DJ. Electrical stimulation of cerebellar fastigial nucleus protects rat brain, in vitro, from staurosporine-induced apoptosis. J Neurochem. 2001;79:328–338. doi: 10.1046/j.1471-4159.2001.00585.x. [DOI] [PubMed] [Google Scholar]
- 27.Ma T, Hoeffer CA, Wong H, Massaad CA, Zhou P, Iadecola C, et al. Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J Neurosci. 2011;31:5589–5595. doi: 10.1523/JNEUROSCI.6566-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, et al. Innate immunity receptor cd36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013;110:3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarnyai Z, Sibille EL, Pavlides C, Fenster RJ, McEwen BS, Toth M. Impaired hippocampal-dependent learning and functional abnormalities in the hippocampus in mice lacking serotonin(1a) receptors. Proc Natl Acad Sci U S A. 2000;97:14731–14736. doi: 10.1073/pnas.97.26.14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dellu F, Contarino A, Simon H, Koob GF, Gold LH. Genetic differences in response to novelty and spatial memory using a two-trial recognition task in mice. Neurobiology of learning and memory. 2000;73:31–48. doi: 10.1006/nlme.1999.3919. [DOI] [PubMed] [Google Scholar]
- 32.Moskowitz MA, Lo EH, Iadecola C. The science of stroke: Mechanisms in search of treatments. Neuron. 2010;67:181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke; a journal of cerebral circulation. 2009;40:e331–339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- 34.Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK. Biphasic cytochrome c release after transient global ischemia and its inhibition by hypothermia. J Cereb Blood Flow Metab. 2005;25:1119–1129. doi: 10.1038/sj.jcbfm.9600111. [DOI] [PubMed] [Google Scholar]
- 35.Ouyang YB, Tan Y, Comb M, Liu CL, Martone ME, Siesjo BK, et al. Survival- and death-promoting events after transient cerebral ischemia: Phosphorylation of akt, release of cytochrome c and activation of caspase-like proteases. J Cereb Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, et al. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyazaki S, Katayama Y, Furuichi M, Kinoshita K, Kawamata T, Tsubokawa T. Impairment of hippocampal long-term potentiation following transient cerebral ischaemia in rat: Effects of bifemelane, a potent inhibitor of ischaemia-induced acetylcholine release. Neurological research. 1993;15:249–252. doi: 10.1080/01616412.1993.11740144. [DOI] [PubMed] [Google Scholar]
- 38.Kiprianova I, Sandkuhler J, Schwab S, Hoyer S, Spranger M. Brain-derived neurotrophic factor improves long-term potentiation and cognitive functions after transient forebrain ischemia in the rat. Experimental neurology. 1999;159:511–519. doi: 10.1006/exnr.1999.7109. [DOI] [PubMed] [Google Scholar]
- 39.Ellen P, Deloache J. Hippocampal lesions and spontaneous alternation behavior in rat. Physiology & behavior. 1968;3:857–860. [Google Scholar]
- 40.Douglas RJ. Sab as a tool: Advice from a veteran. In: Dember WNRC, editor. Spontaneous alternation behavior. New York: Springer; 1989. pp. 145–159. [Google Scholar]
- 41.Volpe BT, Pulsinelli WA, Tribuna J, Davis HP. Behavioral performance of rats following transient forebrain ischemia. Stroke; a journal of cerebral circulation. 1984;15:558–562. doi: 10.1161/01.str.15.3.558. [DOI] [PubMed] [Google Scholar]
- 42.Kiyota Y, Miyamoto M, Nagaoka A. Relationship between brain damage and memory impairment in rats exposed to transient forebrain ischemia. Brain Res. 1991;538:295–302. doi: 10.1016/0006-8993(91)90443-y. [DOI] [PubMed] [Google Scholar]
- 43.Petito CK, Pulsinelli WA. Sequential development of reversible and irreversible neuronal damage following cerebral ischemia. Journal of neuropathology and experimental neurology. 1984;43:141–153. doi: 10.1097/00005072-198403000-00004. [DOI] [PubMed] [Google Scholar]
- 44.Petito CK, Pulsinelli WA. Delayed neuronal recovery and neuronal death in rat hippocampus following severe cerebral ischemia: Possible relationship to abnormalities in neuronal processes. J Cereb Blood Flow Metab. 1984;4:194–205. doi: 10.1038/jcbfm.1984.28. [DOI] [PubMed] [Google Scholar]
- 45.Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurologic clinics. 2006;24:1–21. doi: 10.1016/j.ncl.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 46.Corbett D, Thornhill J. Temperature modulation (hypothermic and hyperthermic conditions) and its influence on histological and behavioral outcomes following cerebral ischemia. Brain pathology. 2000;10:145–152. doi: 10.1111/j.1750-3639.2000.tb00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman SM, Abboud CN, Whartenby KA, Packman CH, Koeplin DS, Moolten FL, et al. The “bystander effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–5283. [PubMed] [Google Scholar]
- 48.Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, et al. Repressor element-1 silencing transcription factor (rest)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Natl Acad Sci U S A. 2012;109:E962–971. doi: 10.1073/pnas.1121568109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, et al. Overexpression of sod1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke; a journal of cerebral circulation. 1996;27:327–331. doi: 10.1161/01.str.27.2.327. [DOI] [PubMed] [Google Scholar]
- 51.Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, et al. The oxygen free radicals originating from mitochondrial complex i contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Merkwirth C, Langer T. Prohibitin function within mitochondria: Essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 53.Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial phb complex: Roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. Nadph oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42:341–348. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, et al. Nmda receptor activation increases free radical production through nitric oxide and nox2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, et al. Role of nad(p)h oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin ii. Hypertension. 2005;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- 57.Wenzel P, Mollnau H, Oelze M, Schulz E, Wickramanayake JM, Muller J, et al. First evidence for a crosstalk between mitochondrial and nadph oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid Redox Signal. 2008;10:1435–1447. doi: 10.1089/ars.2007.1969. [DOI] [PubMed] [Google Scholar]
- 58.Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang HK, Park UJ, Kim SY, Lee JH, Kim SU, Gwag BJ, et al. Free radical production in ca1 neurons induces mip-1alpha expression, microglia recruitment, and delayed neuronal death after transient forebrain ischemia. J Neurosci. 2008;28:1721–1727. doi: 10.1523/JNEUROSCI.4973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal ca1 neurons in rats after transient global cerebral ischemia. J Neurosci. 1999;19:RC39. doi: 10.1523/JNEUROSCI.19-22-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chernyak BV, Bernardi P. The mitochondrial permeability transition pore is modulated by oxidative agents through both pyridine nucleotides and glutathione at two separate sites. European journal of biochemistry / FEBS. 1996;238:623–630. doi: 10.1111/j.1432-1033.1996.0623w.x. [DOI] [PubMed] [Google Scholar]
- 62.Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- 63.Malenka RC. The long-term potential of ltp. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.