

Graphical abstract
Abbreviations: SARS-CoV, Severe Acute Respiratory Syndrome coronavirus; 3CLpro, 3 Chymotrypsin-like cysteine protease; MD, molecular dynamics; VS, virtual screening; CSM, computational solvent mapping; ROC, receiver operating characteristic; RMSD, root mean square deviation; HTS, high-throughput screening; FRET, fluorescence resonance energy transfer; SPR, surface plasmon resonance; KD, equilibrium dissociation constant; Ki, inhibition constant; MLPCN, Molecular Libraries Probe Production Centers Network
Keywords: SARS 3CLpro, Virtual screening, Docking, Pharmacophore modeling, High-throughput screening, Surface plasmon resonance
Abstract
We have used a combination of virtual screening (VS) and high-throughput screening (HTS) techniques to identify novel, non-peptidic small molecule inhibitors against human SARS-CoV 3CLpro. A structure-based VS approach integrating docking and pharmacophore based methods was employed to computationally screen 621,000 compounds from the ZINC library. The screening protocol was validated using known 3CLpro inhibitors and was optimized for speed, improved selectivity, and for accommodating receptor flexibility. Subsequently, a fluorescence-based enzymatic HTS assay was developed and optimized to experimentally screen approximately 41,000 compounds from four structurally diverse libraries chosen mainly based on the VS results. False positives from initial HTS hits were eliminated by a secondary orthogonal binding analysis using surface plasmon resonance (SPR). The campaign identified a reversible small molecule inhibitor exhibiting mixed-type inhibition with a Ki value of 11.1 μM. Together, these results validate our protocols as suitable approaches to screen virtual and chemical libraries, and the newly identified compound reported in our study represents a promising structural scaffold to pursue for further SARS-CoV 3CLpro inhibitor development.
1. Introduction
A highly infectious respiratory illness, Severe Acute Respiratory Syndrome (SARS), emerged in Southern China and Hong Kong during late 2002.1 It rapidly spread to over 25 other countries, infecting nearly 8000 people worldwide with a mortality rate of over 10%.2 A novel human coronavirus (SARS-CoV) was identified as the causative agent of SARS. Although containment of the epidemic was successful through epidemiological and quarantine measures, the possibility of reemergence of the SARS-CoV or SARS-like diseases has been suggested based on the isolation of closely related strains from the horseshoe bat.3, 4 Two new human coronaviruses named HKU15, 6 and NL637, 8 have been identified since 2003, which turned out to be less lethal than SARS-CoV. However, a more recently identified human coronavirus, Middle East Respiratory Syndrome coronavirus (MERS-CoV), has infected 114 people since April 2012 with a nearly 50% mortality rate (54 deaths reported), and this number keeps rising daily.9, 10, 11 Two Asian bat coronaviruses, BtCoV-HKU4 and BtCoV-HKU5, have been shown to be the closest to MERS-CoV.12 This strongly suggests that there could be new SARS-like or new strains of SARS human coronaviruses that could cause another deadly outbreak in the future. There is still no effective therapy for SARS or other coronaviral infections.13
The SARS coronavirus gene encodes two overlapping polyproteins—pp1a and pp1ab.14 These polyproteins are processed by virally encoded peptidases to generate functional viral proteins. The SARS Chymotrypsin-like cysteine protease (3CLpro) is the primary enzyme responsible for proteolysis, cleaving the initial polyprotein synthesized by the virion at eleven of its fourteen cleavage sites. Due to its crucial role in maturation of SARS-CoV, 3CLpro is an important target for anti-SARS drug design. 3CLpro functions as a dimer,15 with one wall of the active site including a loop from the neighboring monomer. The N-terminus Ser1 of one monomer forms a hydrogen bond with Glu166 of the adjacent monomer in the S1 pocket. This interaction between the two monomers substantially stabilizes the protein, which then behaves as an active dimer where both monomers are found in the ‘active form’. However, the majority of the drug discovery efforts against SARS-CoV 3CLpro reported to date have employed ‘non-authentic’ protein, i.e. one which has either an N-terminal GST tag or a C-terminal His-tag, in the crystal structures used in computational studies or in the constructs used in enzyme assays. Since the modified N- and C-termini can influence the functionality and activity of the SARS-CoV 3CLpro,15, 16, 17 the inhibition data reported in literature should be interpreted with caution.
SARS-CoV 3CLpro has been the target of extensive inhibitor design, with drug design strategies ranging from substrate-based peptidomimetic design to identification of leads from direct high-throughput screening (HTS) and computational screening of large databases. Many peptidomimetic inhibitors of 3CLpro were initially developed, with these inhibitors including a ‘warhead’ moiety that formed a covalent interaction with the catalytic cysteine (Cys145) in 3CLpro.18, 19, 20, 21, 22 After the development of the first generation of covalently interacting peptidomimetic inhibitors, researchers moved on to identifying non-peptidic small molecule inhibitors.23, 24, 25, 26, 27, 28 These were also largely warhead-based covalently bonding inhibitors, which could cause unwanted problems such as toxicity and off-target side effects, especially for other cysteine proteases. Thus there is a strong need to identify novel, non-covalent inhibitors with enhanced selectivity and potency against 3CLpro.
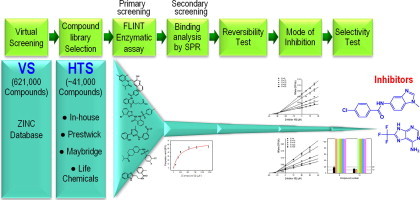
In this work, we describe structure-based virtual screening (VS) and subsequent high-throughput screening (HTS) methodology to identify novel non-peptidic, non-covalent small molecule inhibitors against 3CLpro of human SARS-CoV (Fig. 1 ). The lack of a rigid, well-defined deep pocket of SARS-CoV 3CLpro makes small molecule inhibitor design difficult using in silico approaches. We thus developed a screening protocol using tiered docking and dynamic pharmacophore model filtering to screen the ZINC database (Fig. 1A). Subsequently, we developed an experimental high-throughput screening assay to screen ∼40,000 diverse small molecules from four chemical libraries and optimized an orthogonal counter-screen binding assay to remove false positives (Fig. 1B). Through the use of virtual and high-throughput screens we present several novel chemical scaffolds, which serve as excellent lead candidates for optimization of biological activity for potential anti-SARS drug design.
Figure 1.
Schematic of (A) virtual and (B) high-throughput screening pipelines. The hit validation steps are shown within the HTS pipeline.
2. Results and discussion
2.1. Virtual screening challenges
2.1.1. Target flexibility
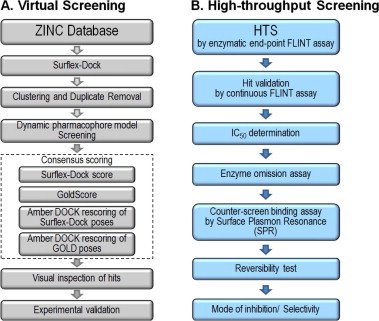
The SARS-CoV 3CLpro binding pocket has been shown to be quite flexible in previous studies based on crystal structures solved at various pH values and in molecular dynamics (MD) simulations.29 Our nanosecond-scale MD simulations of the authentic SARS-CoV 3CLpro dimer structure show that the four loops surrounding the active site region can sample alternative conformations (Fig. 2 A). The plot in Figure 2B displays the average per-residue RMSD for the N-terminal monomer relative to the crystal structure. Significant conformational flexibility can be observed in the active site, particularly in loops L1, L2 and L4 that constitute the walls of the binding pocket. Such flexible loops are expected to play an important role in the correct positioning of amino acid residues during substrate binding and may adversely affect the virtual screening performance for a single rigid-receptor docking approach, as is often undertaken.
Figure 2.
Conformational flexibility of 3CLpro studied using MD simulations. (A) Superposition of MD-generated structurally diverse snapshots showing the flexibility of the loops L1–L4 (L1: residues 185–193, L2: residues 165–172, L3: residues 139–150, and L4: residues 40–53). The snapshots were selected based on heavy atom RMSD of the binding site residues. (B) Average per-residue RMSD (in angstroms) of 3CLpro monomer from the MD simulation of dimer structure. Grey panels correspond to the flexible loops around the active site.
2.1.2. Quantitative characterization of docking efficiency
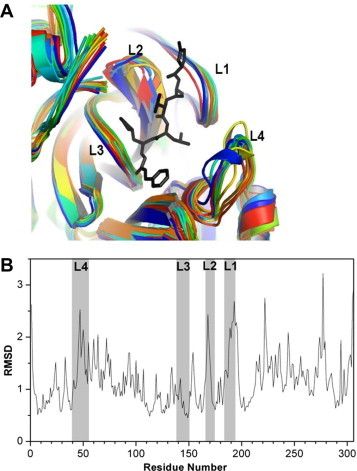
The available apo and ligand-bound crystal structures of SARS-CoV 3CLpro elucidate the structural features of the enzyme’s substrate binding pocket and the key interactions important for molecular recognition, allowing for structure-based screens of large compound libraries. The success of docking approaches relies on the overall classification accuracy of the scoring functions as well as their early enrichment, which is particularly important in high-throughput virtual screening. We examined the overall predictive power of scoring functions on the compounds in the validation dataset using rank order correlation between the computed docking scores and the experimentally reported IC50 values, and the area under the receiver operating characteristic (ROC) curve (AUC) values (see Methods). The correlation coefficients and the AUC values for all the assessed scoring functions are listed in Figure 3 A. We found that both Surflex-Dock and GOLD yielded poor prediction accuracy. Rescoring with Amber-DOCK improved the binding free energy estimation of the docked poses from GOLD, but reduced the classification accuracy of the Surflex-Dock poses. The initial slope of the ROC curve quantifies the early enrichment of the scoring function. We found that Amber-DOCK rescoring of GOLD binding poses and the GoldScore function show superior early enrichments (Fig. 3B). These results show that Amber-DOCK rescoring of GOLD binding poses displays the best prediction accuracy in terms of early and overall enrichment for 3CLpro inhibitors. However, there remains an issue in using the Amber-DOCK rescoring method for screening large compound libraries, primarily because pose refinement employs energy minimization and molecular dynamics of the binding-site residues, making it computationally slow.
Figure 3.
Comparison of different docking/scoring methods on the 3CLpro validation dataset. (A) The rank-order correlation (r) between the docking scores and the experimental IC50 values and area under the curve (AUC) values for ROC plots of the assessed docking methods. (B) ROC plots for Surflex-Dock, GoldScore, Amber-DOCK rescoring of GOLD and Surflex docked conformations.
2.2. Virtual screening workflow
We examined tiered docking and consensus scoring along with dynamic pharmacophore based screening approaches to address some of the limitations of molecular docking mentioned above. The tiered docking approach aims at increasing the computational efficiency by making use of fast docking methods at an early stage to quickly remove non-binders, followed by using more rigorous and accurate algorithms on the promising subsets to rank potential binders. The consensus scoring method combines estimates from different scoring functions to capitalize on strengths and to compensate for errors from individual functions, thereby improving the hit rates. The dynamic pharmacophore model accommodates 3CLpro active site loop flexibility into virtual screening.
2.2.1. Tiered docking
Surflex-Dock provides a good compromise between the overall predictive power (as characterized by its AUC value on the validation dataset) and high computational speed, and was used for the first round of elimination to rapidly discard compounds that did not fit in the binding pocket of the 3CLpro enzyme. This enriched the dataset with the compounds that could form favorable steric and electrostatic interactions with the enzyme’s active site, reducing the Vernalislead dataset from 97 K compounds to 50 K compounds and the Cleanlead dataset from 524 K compounds to 106 K compounds. We then clustered the hits from Surflex-Dock to generate a representative set of structurally diverse compounds, still achieving maximum coverage of the chemical diversity space. A reciprocal nearest neighbor (RNN) packing algorithm in Sybyl8.0 was used to generate this representative set. A compound was not selected when 6 of its closest 30 neighbors were selected and those 6 neighbors were at least 85% similar. For a more exhaustive search, the representative sets were docked with GOLD followed by rescoring of the docked conformations from GOLD and Surflex-Dock using Amber-DOCK.
2.2.2. Consensus scoring
We evaluated several methods for combining ranks from individual scoring functions to improve enrichment. Combining predictions from different models to generate a consensus has been widely employed in other areas and has been shown to be more robust than the individual component models.30, 31, 32 The rationale behind combining ranks is that different scoring functions may have different biases and may sometimes wrongly predict the binding affinity of a compound, but consensus estimates from multiple scoring functions can potentially approximate the true activity of a compound.33, 34 The evaluated consensus scoring methods and their performances are described in the Supplementary data. The best accuracy and enrichment was obtained from the rank-sum approach by summing ranks from GoldScore, Surflex-Dock and Amber-DOCK rescoring of GOLD and Surflex-Dock conformations. The method not only retrieved maximum actives at the beginning of the ordered list, but also showed a significantly higher AUC (= 0.8) than other scoring functions (Figs. S1 and S2). The entire representative set from tiered docking was ranked using the rank-sum consensus method.
2.2.3. Pharmacophore screening
The Computational Solvent Mapping (CSM) algorithm determines the energetically favorable binding positions for small probes in the active site, conserved over multiple conformers of the protein, thus making the generated features independent of the binding site conformation. Five different probes were selected such that they covered the common functional groups found in inhibitor molecules as well as offered a good coverage of the substrate-recognition pocket of 3CLpro. The S1 subsite includes His163 and Glu166 residues, which could potentially make hydrogen bonding interactions with inhibitors. The hydrophobic S2 subsite is lined with Met, Pro, Asp, His and Tyr residues. Methanol, acetate, and methylammonium probes were used to identify hydrogen bonding interactions; benzene probes were used for mapping of aromatic interactions, and ethane probes were used to distinguish hydrophobic interactions from aromatic. The method identified five hot spots (Fig. 4 ), an aromatic feature interacting with the S1’ subsite, two aromatic/hydrophobic features in the S1 and S2 subsites, an acceptor site near the catalytic cysteine, and a donor site interacting with residues His163 and His164 in the S1 subsite. The validity of the generated pharmacophore model was ascertained using two techniques. The water molecule was used as a probe and the positions of consensus clusters were compared with the crystallographic waters in the SARS-CoV 3CLpro active site. The method reproduced the binding sites of all the three binding pocket crystallographic waters. We also evaluated the performance of the generated pharmacophore model on the validation dataset. Although the non-inhibitors closely resembled the active compounds in chemical structure and molecular properties, the model differentiated well between the two classes. The model correctly predicted 18 out of 34 active compounds and had an overall accuracy of around 73%.
Figure 4.
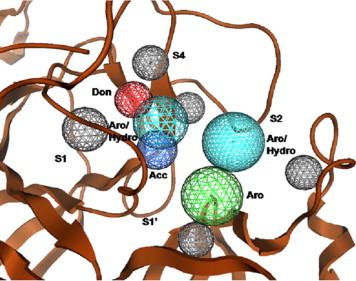
Pharmacophore model generated using computational solvent mapping. Pharmacophore features shown within 3CLpro active site. Aromatic (Aro) affinity sites are in green, H-bond donor (Don) sites are in red, H-bond acceptors (Acc) sites are in blue, aromatic/hydrophobic (Aro/Hydro) sites are in turquoise, and excluded volume regions are in grey.
The final pharmacophore query was used to search the pre-generated ligand conformers of the representative set obtained from tiered docking. Alignment with the pharmacophore coordinates gave ∼3400 hits. The ranks from the consensus scoring method were used to pick out top scoring commercially available compounds from the pharmacophore hits. The prioritized compounds were visually inspected to eliminate reactive compounds and those that did not efficiently span the binding site. Finally, 68 compounds from the Vernalislead and Cleanlead set of Zinc v8.0 were ordered for biological assays.
2.3. Assay development and optimization for HTS
The SARS-CoV main protease 3CLpro exists as a functional dimer. It has been known that any affinity tags or extra amino acid residues at either the N-terminus or C-terminus decrease 3CLpro enzyme activity significantly via disturbing the formation of dimmers.35, 36 Hence, we over-expressed and purified the authentic 3CLpro without any additions on either termini. Currently, Dabcyl-KTSAVLQSGFRKME-Edans,37 is the only commercially available fluorescence resonance energy transfer (FRET)-based substrate to monitor the 3CLpro enzyme activity. The fluorescence due to cleavage of the Edans/Dabcyl substrate can be monitored at excitation and emission wavelengths of 355 nm and 538 nm. In addition to this substrate, the long-wavelength fluorophore/quencher pair Alexa Fluor/QSY has been developed for compound screening, which can be monitored at excitation/emission of 490 nm/535 nm or 595 nm/620 nm depending on the types of Alexa Fluor and QSY.38 Typically, substrates with higher wavelength fluorescence signals work better for HTS because of less interference from intrinsically fluorescent compounds. For this reason, AlexaFluor/QSY would be a better substrate than Edans/Dabcyl. However, it was not used in this study primarily due to its much greater expense. Instead, we developed a new 3CLpro FRET substrate peptide labeled with fluorophore, 5-FAM and a quencher, QXL, for our studies. The FAM/QXL pair is a cost-effective alternative with a higher excitation/emission (492 nm/520 nm) wavelength than that of Edans/Dabcyl. The new 3CLpro substrate, 5-FAM-TSATLQSGFRK (QXL520)-NH2, has the same peptide cleavage sequence as the Edans/Dabcyl-containing substrate. We investigated our new FAM/QXL substrate by comparing its kinetic parameters with Edans/Dabcyl, which are summarized in Table 1 . The Michaelis constant (K M) value of the FAM/QXL substrate (16.1 μM) is similar to that of Edans/Dabcyl (23.8 μM), however, the FAM/QXL substrate exhibited a ∼22-fold greater k cat value over the Edans/Dabcyl substrate. This resulted in ∼37-fold higher k cat/K M of our FAM/QXL substrate than the Edans/Dabcyl substrate. Furthermore, there was no inner filter effect up to almost 100 μM for our developed substrate. Thus, we have successfully developed a new 3CLpro substrate with good enzyme efficiency, a higher wavelength and reduced inner filter effect.
Table 1.
Comparison of kinetic parameters with two FRET-based substrates
| FRET-peptide substrate | KM (μM) | kcat (s−1) | kcat/KM (M−1 s−1) |
|---|---|---|---|
| Edans/Dabcyl | 23.8 ± 1.2 | 0.48 ± 0.3 | 17,547 |
| 5-FAM/QXL520 | 16.1 ± 0.8 | 10.9 | 649,010 |
3CLpro enzyme was incubated with the substrates in 50 mM HEPES, pH 7.5, 2 mM GSH, 0.01% Triton X-100. The KM values were determined from three to four independent assays.
Before HTS was done, a thorough assay optimization was done to determine the optimal substrate and enzyme concentrations. In addition, the reducing agent effect, DMSO tolerance, and enzyme stability were also studied. In order to determine the correct substrate concentration for the HTS assay, the Michaelis constant (K M) was determined in the presence and absence of reducing agents as described.39 A series of DMSO concentrations ranging from 0% to 10% were tested for enzyme activity and substrate stability. Less than 10% inhibition of enzyme activity was observed with up to 10% of DMSO, and the substrate of 3CLpro showed sensitivity to DMSO starting at 10%. Therefore, a final DMSO concentration of less than 4% was used for all assays.
2.4. Enzymatic characterization of hits from virtual screening
The compounds selected from VS were experimentally evaluated for inhibitory activity against SARS-CoV 3CLpro using our new FAM/QXL FRET-based substrate. A total of 68 virtual screening hits were purchased, and initial experimental screening was conducted at a single concentration of 100 μM, which identified eight compounds with over 50% inhibition. IC50 values for the active compounds were determined by dose-response curves. We were able to obtain IC50 values for six compounds, all of which exhibited micromolar activities, with the most potent one (Compound 8) showing an IC50 value slightly less than 1 μM (Fig. 5 ). Seven analogs of compound 8 were identified from ZINC v8.0 and were evaluated. However, none of the analogs were found to inhibit the enzyme by more than 50% at 100 μM concentration. Due to the presence of a catalytic cysteine in the 3CLpro active site, the hits from primary screening were evaluated in follow-up assays containing reduced glutathione (GSH) as the reducing agent39 to eliminate any covalent binders and to maintain the protein in its active state. Out of the eight hits identified in the preliminary assays, four compounds (1, 2, 3 and 6) retained IC50 values in the micromolar range (Fig. 5), with the rest losing their inhibitory activity in the presence of GSH. The IC50 values of these four hits ranged from 40.9 to 76.7 μM in the presence of GSH. None of the analogs of compound 8 were found to inhibit the enzymatic activity of SARS-CoV 3CLpro by more than 50% at 100 μM concentrations when GSH was present in the assay buffer.
Figure 5.
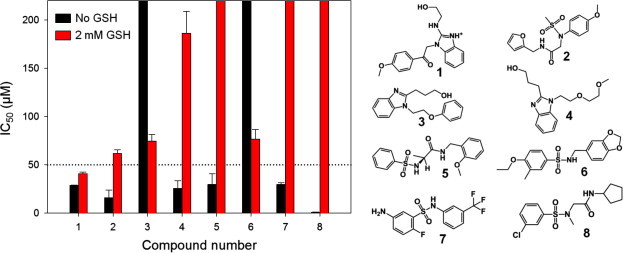
IC50 value comparison of hits from virtual screening. IC50 determination was done in triplicate and each plate contained a total of 32 positive and 32 negative controls. IC50 values were calculated by fitting the data to the three parameter Hill equation with OriginPro 8.5 (OriginLab, Inc.). Bars that reach the top of the graph represent IC50 values of over 200 μM (no inhibitory effect.)
2.5. High-throughput screening
Based on the VS results, three structurally diverse libraries (an in-house collection, a Maybridge diversity set, and a specially constructed antimicrobial/antiviral focused set from Life Chemicals) were selected for HTS. The compounds in the selected libraries exhibit molecular weight distributions and the basic scaffolds similar to those of the VS hits. In addition, the Prestwick FDA-approved drug library was also screened to identify the activity of any known drugs against 3CLpro. Together, a total of 41,022 compounds were screened against 3CLpro by enzymatic end-point fluorescence intensity assays. The in-house library included ten control compounds reported in PubChem (AID1890), from high-throughput screening assay of the MLPCN compound library against SARS-CoV 3CLpro. The primary screens were done in duplicate and the Z’-factors varied between 0.57 and 0.75, indicating high quality of the screening campaign. The replicate plot of percent inhibition shown in Figure 6 A also illustrates the good quality of the screens. The primary hits with percent inhibition greater than 50% at 50 μM compound concentration were cherry-picked and retested for their inhibitory activities by continuous enzymatic assays by hand. The compounds exhibiting IC50 values less than 50 μM were reordered from their commercial vendors and activity confirmed. The primary screening of the in-house library identified three hits, but none of the compounds showed an IC50 of less than 50 μM. All ten compounds from PubChem exhibited inhibitory activities similar to their reported values (PubChem AID1890). The IC50 values determined for one of the PubChem compounds (9) is shown in Figure 6B. The primary screening of the Prestwick FDA-approved drug library resulted in one hit (Alexidine dihydrochloride, compound 10 in Fig. 6B) with an IC50 value of 25.1 μM. Alexidine dihydrochloride is a tyrosine phosphatase inhibitor that displays anticancer activities in some cells.40, 41 It also kills harmful germs and bacteria and is often used in cosmetics, antiseptic, and healthcare products.42 The primary screening of the Maybridge library resulted in 14 hits and thirteen compounds that showed inhibition with IC50 values below 50 μM. We reordered nine of these and confirmed inhibitory activities of five compounds with IC50 values below 50 μM. The primary screening of the focused Life Chemicals library resulted in 13 hits, out of which eight compounds showed IC50 values below 50 μM. We reordered six of these, and the IC50 values of the three compounds (12–14) varied between 13.2 and 28.3 μM (Fig. 6B).
Figure 6.
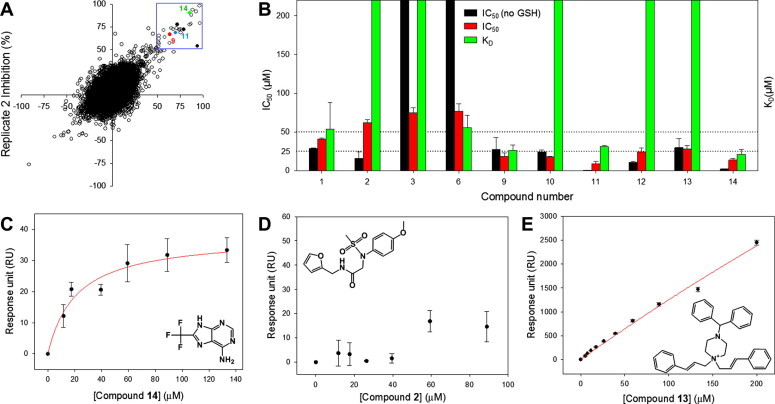
Primary hits from HTS and hit validation using SPR binding assay. (A) Replicate plot from screening 41,022 compounds from structurally diverse in-house, Prestwick FDA-approved drugs, Maybridge and Life Chemicals libraries. The blue box indicates hit compounds with over 50% inhibition at 50 μM compound concentration. The initial percent inhibitions of the three confirmed hits from HTS are shown in red (9), blue (11) and green (14). (B) IC50 values and the dissociation equilibrium constants (KD) of four virtual screening and six HTS hits. All data were normalized for immobilization levels of target and reference proteins. Bars that reach the top of the graph represent KD values of over 200 μM (no binding). (C) The binding data of compound 14 fitted to a single rectangular hyperbolic equation (See Methods). The determined KD of compound 14 was 21.3 ± 6.3 μM, similar to its IC50 value (13.9 ± 2.2 μM). (D) Response units of compound 2 at a series of increasing concentrations (0–90 μM), showing lack of binding to 3CLpro. (E) Response units of compound 13 at a series of increasing concentrations (0–200 μM), showing non-specific binding to 3CLpro.
2.6. Validation of VS and HTS hits
The HTS hits were tested in enzyme omission assays to eliminate any false positives caused by interference mediated by compound fluorescence rather than inhibition of 3CLpro enzyme activity. Only compound 10 showed minor interference, while the other hit compounds showed no effect on the intensity of fluorescence signal. An orthogonal binding assay, surface plasmon resonance (SPR), was used for further validation of the hits from virtual and high-throughput screening. In addition, we investigated the inhibition by compound 9 deposited in the PubChem bioassay database (AID 1890), since to our knowledge, it has not been well characterized. Of the four VS hits and the six HTS hits, the dissociation equilibration constants (K D) of five compounds (1, 6, 9, 11, and 14) were successfully determined. The calculated K D values of the five hits ranged between 26.4 and 55.3 μM, indicating direct binding to the 3CLpro enzyme. Figure 6C shows the K D curve fit for one of the identified hits (14). The remaining compounds either non-specifically bound to the enzyme or did not bind at all. Specifically, as shown in Figure 6D, compound 2 did not bind to 3CLpro, and four compounds (3, 10, 12, and 13) exhibited non-specific binding patterns (Fig. 6E). Unfortunately the only drug hit, Alexidine dihydrochloride from the Prestwick FDA-approved drug library, also turned out to be a false positive based on SPR because it did not bind to the 3CLpro enzyme. Therefore, we considered five compounds (1, 6, 9, 11, and 14) worth pursuing for further validation.
The reversibility of the five confirmed hit compounds was tested by incubating 3CLpro with saturating concentrations of the inhibitors (20-fold concentration of their respective IC50 values), which inhibited 3CLpro activity by more than 95%. Each compound was removed by desalting column and 3CLpro activity was monitored to determine the recovery rate. The observed recovery rates of 3CLpro were over 50% except for compound 1, which showed only 14.3%. Consequently, four compounds (6, 9, 11, and 14) are potentially non-covalent reversible inhibitors. Compound 6 is a new scaffold that can be an excellent lead compound for further optimization to improve potency. Although the remaining three hits show similar inhibitory activities against 3CLpro, compounds 9 and 14 were better candidates than 11 due to the unattractive structure of compound 11. The sequential elimination of primary hits in the follow-up assays is summarized in Table 2 .
Table 2.
Summary statistics for experimental screening of VS and HTS hits
| Library | Number of compounds | Primary hits ⩾ 50% inh or IC50 ⩽ 50 μM | Reordered | Confirmed hits IC50 ⩽ 50 μM | Enzyme omission assay | Binding confirmed by SPR | Reversible hits | Final hit rate (%) |
|---|---|---|---|---|---|---|---|---|
| ZINC library | 650,000 (75)a | 8/5b | NA | NA | NA | 2 | 1 | 1.3c |
| In-house | 422 | 3/0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Prestwick | 1200 | 5/5 | 1 | 1 | 1 | 0 | 0 | 0 |
| Maybridge | 14,400 | 14/13 | 9 | 5 | 5 | 1 | 1 | 0.007 |
| Life Chemicals | 25,000 | 13/8 | 6 | 3 | 3 | 1 | 1 | 0.004 |
Number of virtual hit compounds ordered for experimental testing. NA—not applicable.
This number was counted from IC50 values in the absence of a reducing agent.
Final hit rate was calculated with the number of compounds ordered and experimentally tested ones only.
2.7. Binding mode of inhibition and selectivity
The majority of currently reported 3CLpro inhibitors are competitive inhibitors with respect to the substrate. We investigated the mechanism of inhibition for compounds 9 and 14. The kinetics of inhibition by compound 9 have not been determined (PubChem AID1890). Our kinetic analysis was done with varying concentrations of substrate and enzyme–inhibitor complexes for each compound. The different models of enzyme inhibition (competitive, non-competitive, uncompetitive, and mixed-type) were fit to the kinetic data and were assessed using Michaelis–Menten, Lineweaver–Burke, and Dixon plots, using the SigmaPlot Enzyme kinetics Module. The best fit equations had the lowest Akaike Information Criterion corrections (AICc) value,43 which should have a minimum of 2 AICc units difference from the next lowest to be significant. Compound 9 was determined to be a competitive inhibitor with respect to the substrate, with a K i value of 27.5 μM. Compound 14 showed a mixed-type inhibition with different affinities for free 3CLpro enzyme and the enzyme–substrate complex, with a K i value of 11.1 μM (Fig. 7 ). Their determined K i values were similar to each of their IC50 values (Table 3 ). Since compound 9 is a competitive inhibitor while 14 is a mixed-type inhibitor, 9 apparently binds to the active site of 3CLpro, while 14 apparently binds at a site distinct from the active site. We further studied the binding of compound 9 to SARS-CoV 3CLpro by molecular docking. The predicted binding pose and the key interactions are shown in Fig. S3. The inhibitor makes good van der Waals contacts with the active site and spans mainly the S1–S2 subsites blocking access to the catalytic cysteine. Specifically, the inhibitor makes hydrogen bond with the side chain hydroxyl group of Ser144 in the S1 subsite and the p-chlorophenyl moiety of the inhibitor makes hydrophobic contacts with the residues in the S2 subsite.
Figure 7.
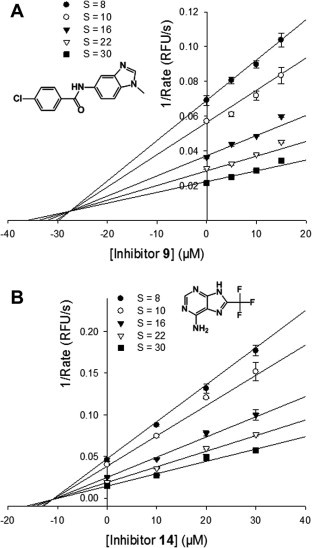
Kinetics of inhibition of 3CLpro activity by compounds 9 and 14. Dixon plot for competitive inhibition of (A) Compound 9 and mixed inhibition of (B) compound 14 with respect to the substrate 5-FAM-TSATLQSGFRK (QXL520)-NH2. Determined Ki values of 9 and 14 were 27.5 μM and 11.1 μM, respectively. Four equations (see Methods) in SigmaPlot Enzyme Kinetics Module 1.3 were used to fit the experimental data. The competitive inhibition model was the best fit for 9 and mixed inhibition model for 14.
Table 3.
Inhibition modes of of two compounds
| Compound | SARS-CoV 3CLpro |
|
|---|---|---|
| IC50a (μM) | Kinetic mode (Kib, μM) | |
| 9 | 18.2 ± 4.9 | Competitive inhibition (27.5) |
| 14 | 13.9 ± 2.2 | Mixed inhibition (11.1) |
Kinetic modes were determined from three independent assays.
IC50 value is a half maximum inhibitory concentration.
Ki is inhibition constant value.
Because our newly identified compound 14 is a small compound, there could be some concern about non-specificity. Thus, we examined the selectivity of compounds 9 and 14 against SARS-CoV 3CLpro. The compounds were tested against three other proteases, namely SARS-CoV PLpro (a cysteine protease), human UCH-L1 (a cysteine protease) and Hepatitis C Virus NS3/4A (a serine protease); and two non-proteolytic enzymes, Bacillus anthracis dihydroorotase and Streptococcus pneumoniae PurC. Compound 9 was found to be selective for SARS-CoV 3CL cysteine protease and did not show inhibitory activity against other tested enzymes (Fig. 8 ). Compound 14 displayed selectivity against the two SARS cysteine proteases, 3CLpro and PLpro over other enzymes. Since low molecular weight compounds typically lack high specificity, no inhibition of compound 14 for other enzymes, especially the UCH-L1 cysteine protease, is particularly noteworthy.
Figure 8.
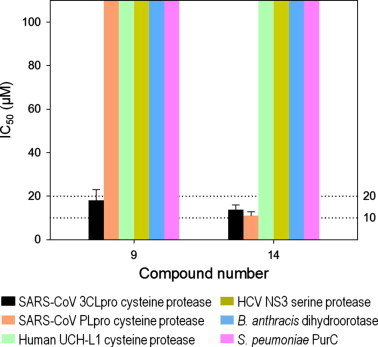
Selectivity of two confirmed hit compounds. IC50 determination was done in triplicate with a total of 32 positive and 32 negative controls in a plate. IC50 values were calculated by fitting the data to the three parameter Hill equation with OriginPro 8.5 (see Methods). Compound 9 had inhibitory activity against only 3CLpro, while compound 14 inhibited both 3CLpro and PLpro. Bars that reach the top represent IC50 values of over 200 μM (no inhibitory effect).
3. Conclusion
In this work, we present a high-throughput experimental screening approach guided by structure-based computational methods to discover new chemical scaffolds to be developed as non-covalent inhibitors of SARS-CoV 3CLpro. We have identified two compounds that exhibit IC50 values in the low micromolar range. Unlike the majority of 3CLpro inhibitor leads reported to date, which either contain reactive warheads that may potentially engage in non-specific interactions with other cysteine-containing proteins, or which have not been tested for their inhibitory activities in the presence of reducing agents, the identified inhibitors act by binding reversibly to 3CLpro and are functional in the presence of the physiological reducing agent, GSH.
This study places particular emphasis on the quality of the results at each step of the screening pipeline and the rigorous experimental validation of the hits. The virtual screening protocol was designed for low computational complexity, improved enrichment, and its ability to accommodate protein flexibility into computational screening. The VS hits provided insights for choosing appropriate compound libraries for HTS instead of screening random compound libraries. The favorable binding positions of small probes in the CSM studies and the molecular docking studies of the VS hits suggest the preference of the S2 subsite for hydrophobic or aromatic groups and hydrogen bonding interactions with the residues in the S1 subsite. Accordingly, the libraries were selected which were enriched in aromatic groups and hydrogen bond donors and acceptors while exhibiting a molecular size distribution similar to that of the VS hits. The enzyme assays and the HTS screens employed the untagged construct of the 3CLpro enzyme and a new FRET substrate designed for high k cat/K M value and longer excitation/emission wavelength for minimal interference. The primary hits identified from VS and HTS were examined for binding in an independent SPR assay, followed by kinetic characterization of the mode of compound inhibition. The identified hits were further tested against a number of enzymes and were found to be selective for SARS-CoV proteases over other enzymes. Together, the results indicate the well-characterized hit compound 14 identified in this study to be sufficiently potent to provide a new scaffold for further development.
4. Materials and methods
4.1. Dataset used for virtual screening
The starting protein conformation for all computations was taken from the dimeric crystal structure PDB code: 2HOB. Residues His163 and His172 were protonated to match the low pH endosomal environment where proteolytic processing is believed to occur.44 The validation of the screening protocol was carried out using a set of 42 compounds identified from the literature that have been tested experimentally against 3CLpro by various research groups.45, 46, 47, 48, 49, 50, 51 After tagging the non-covalent inhibitors with IC50 values below 100 μM as actives, the dataset had 34 active and 8 inactive compounds. We used two subsets from the ZINC v7 library52, Vernalislead (comprising about 97 K compounds) and Cleanlead (comprising about 524 K compounds) for virtual screening.
4.2. Molecular dynamics simulations
We performed molecular dynamics (MD) simulations using the Molecular Dynamics Package AMBER1053 with the AMBER03 force field.54 The protein structure was solvated in a 10 Å cubic water box (TIP3P). Chloride ions were added to neutralize the system. Periodic boundary conditions were applied with a 10 Å non-bonded cutoff. The system was first subjected through a steepest decent energy minimization followed by a conjugate gradient minimization. The system was then heated from 0 K to 300 K using the canonical (NVT) ensemble for 30 ps. Finally, a 20 ns MD was performed at 300 K. The trajectories were analyzed using the ptraj module of Amber10.
4.3. Docking calculations
The protein structure preparation for docking calculations was done using Sybyl8.0 (Tripos, Inc., St. Louis, MO). After comparing the performances of several docking programs, we selected Surflex-Dock55 and GOLD56 for our study. The docked poses from Surflex-Dock and GOLD were then used for input into the DOCK6 Amber rescoring protocol57 wherein the protein residues within 10 Å of the ligand were kept flexible during the calculations. The rank-sum and the voting techniques of consensus scoring were investigated to improve the enrichment compared to single scoring functions (see Supplementary data). The performance of docking programs was assessed on the validation dataset using two metrics: (1) rank-order correlation between the computed docking scores and the experimental IC50 values, and (2) AUC. The ROC curve describes sensitivity or the true positive rate as a function of (1-specificity) or the false positive rate for varying values of thresholds for compound selection. The early enrichment was assessed from the plot by comparing true and false positive rates at the low thresholds. The AUC indicates the overall performance of the classifier compared to a random classification. An AUC value of 0.5 would correspond to a random classification and an AUC value of 1.0 would correspond to a perfect classification.
4.4. Pharmacophore model generation
A dynamic, structure-based pharmacophore model was generated through a computational solvent mapping (CSM) protocol as described.58, 59, 60, 61, 62, 63 Briefly, fifteen snapshots from the last 6 ns MD simulation of the bound form of the protein with the ligand removed were extracted based on their active site pocket heavy atom RMSD diversity compared to the original crystal structure. The RMSD for all frames was calculated using the ptraj module in Amber10 and the snapshots were clustered using the kclust program of the MMTools.64 Five small organic molecule probes, benzene, propane, methanol, acetate, and methylammonium were moved on the surface of the protein active site using 10,000 operations of the Monte Carlo simulated annealing algorithm as implemented in Autodock v4.0. 100 docking trials for each probe type were carried out followed by the conformational clustering of the final docked poses. The best scoring molecule of each Autodock determined cluster for each probe type was retained as the parent molecule for that cluster. On aligning all parent molecules for a particular probe type, consensus clusters (containing at least 3 parent probes) were formed. The center point of each cluster was computed from the center averages of the parent molecules of each cluster. The radius of each cluster was computed from the average deviation of parent probe centers forming a cluster from the cluster center. The pharmacophore model was built using the Molecular Operating Environment (MOE) software package (v 2008.10, Chemical Computing Group, www.chemcomp.com) from the center points and the radius of each consensus cluster for each probe type. Residues Leu27, Met49, Phe150, Met165 and Glu166 were used to define the center points of the excluded volumes with a radius of 1.5 Å to define the boundaries of the pharmacophore search space. TIP3P water molecule probes were also docked using the same protocol to evaluate the reproduction of crystallographic water sites. The stringency of the pharmacophore model was examined by varying the required number of pharmacophore elements that must be matched by enabling partial match and varying the radii of elements. Rule-based torsion driving in OMEGA (OpenEye Scientific Software) was used to produce multiple conformations of each molecule of the representative set, using an energy cut-off of 14 kcal/mol calculated with the MMFF force field and a heavy-atom RMSD criterion of 1 Å. These pre-generated conformations were compared to the pharmacophore model.
4.5. Plasmid construction and purification of SARS-CoV 3CLpro
The native 3CLpro gene (SARS-CoV polyprotein residues 3241-3544) was prepared by codon-optimized gene synthesis (BioBasic Inc.) and cloned into pGEX6p-1 vector between BamHI and EcoRI. 3 L of Rosetta2(DE3) cells were grown to an OD600 of 0.6 at 37 °C in LB medium. The cells were then induced with 0.5 mM IPTG for 16 hours at 25 °C. Cells were harvested and lysed by sonication in lysis buffer (1 mg/mL lysozyme and protease inhibitor cocktail in buffer A: 20 mM Tris–HCl (pH 8.0), 500 mM NaCl, 10 mM Imidazole, and 5 mM β-MCE). The original construct has a GST-tag at the N-terminus and a His-tag at the C-terminus. The GST-tag was auto-cleaved by 3CLpro itself upon expression, and the His-tag fused 3CLpro at the C-terminus was purified by HisTrap HP column with gradient of buffer B (20 mM Tris–HCl (pH 8.0), 500 mM NaCl, 500 mM Imidazole, and 5 mM β-MCE). The pooled 3CLpro was then dialyzed with buffer A and the His-tag was cleaved by 2 units/mL of HRV 3C protease (Novagen) overnight at 4 °C, producing a 3CLpro with an authentic C terminus. Finally, 3CLpro was again loaded on to the HisTrap column to clean up HRV protease, cleaved His-tags, and uncleaved His-tagged 3CLpro. The native SARS-CoV 3CLpro was then further purified by S-200 size exclusion column chromatography with a buffer containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 1 mM DTT, 1 mM EDTA, and 20% glycerol for storage at −80 °C.
4.6. Assay development and optimization
A new FRET substrate, 5-FAM-TSATLQSGFRK (QXL520)-NH2, was designed and synthesized through Anaspec. The 3CLpro activity was measured by continuous kinetic assay with this substrate and with a commercially available substrate, Dabcyl-KTSAVLQSGFRKME-Edans. The Michaelis constant (K M) values were determined with both substrates with 20 μL final assay volume in assay buffer (50 mM HEPES, pH 7.5, 0.01% Triton X-100, 0.1 mg/mL BSA, and 2 mM GSH) in 384-well plate (Corning Inc.). A series of substrate concentrations (0 to 100 μM) was prepared, and the enzyme reaction was initiated by adding 3CLpro (100 nM final concentration). The same series of substrate concentrations without any enzyme was also measured as a control. Fluorescence intensity (492/520 nm for FAM/QXL and 355/538 nm for Edans/Dabcyl, excitation/emission) was monitored continuously for 10 minutes with a POLARstar OPTIMA microplate reader (BMG LABTECH). The Michaelis constant (K M) and maximal activity (V max) were calculated by fitting the data with the hyperbolic Eq. 1 where y is initial velocity, and x is the concentration of substrate:
| (1) |
For testing virtual screening hits, 10 mM solutions of 75 compounds (Chembridge, Asinex, Bioscreen, ChemDiv, Life Chemicals, and Enamine) were prepared in 100% DMSO as stock solutions, diluted to 100 μM final concentrations with assay buffer containing 2% DMSO and incubated with 50 nM of 3CLpro for 20 min. The reaction was initiated by adding 15 μM final concentration of FAM/QXL substrate, and fluorescence intensity was measured by the same way as K M determination.
4.7. Primary high-throughput screening
We used an in-house library and three commercially available compound libraries for HTS. The in-house collection included 563 compounds. Three libraries were purchased from Prestwick, Maybridge, and Life Chemicals, which consisted of 1200, 14,400 and 25,000 compounds, respectively. All compounds were stored as 10 mM stock solutions dissolved in 100% DMSO in desiccated condition at −30 °C. The primary HTS assay was performed by a Tecan Freedom EVO 200 robot equipped with a Te-Mo 3 × 3 96-channel Liquid Handler dispenser and a 384-pin stainless steel pin tool (V&P Scientific) with a 200 nL capillary capacity. All assays were done in duplicate in black 384-well plates (Matrix Technologies) at room temperature. The 3CLpro enzyme (100 nM final concentration) was prepared in assay buffer (50 mM HEPES, pH 7.5, 0.01% Triton X-100, 0.1 mg/mL BSA, and 2 mM GSH). 30 μL of enzyme solution was dispensed into wells, and then 200 nL of 10 mM compound (50 μM final concentrations) were added and incubated for 5 min. Enzyme reactions were initiated with 10 μL of substrate (15 μM final concentration) dissolved in assay buffer and incubated for 6 min followed by adding 10 μL of 10% SDS as a stop solution. Fluorescence intensity was monitored with a Tecan Genios Pro microplate reader. Each plate contained a total of 32 positive and 32 negative controls.
4.8. Confirmation assay and IC50 value determination by dose response curve
All hit compounds from the HTS were cherry-picked and reanalyzed by continuous kinetic assay by hand for confirmation. For those that showed over 50% inhibition in the confirmation assay, IC50 values were measured using the same assay conditions as the primary screen by hand in triplicate. A series of compound concentrations (0–200 μM final concentration at 2-fold serial dilution) in 100% DMSO were prepared in a 384-well plate. 20 μL of enzyme solution was distributed into wells, and 0.5 μL of varying concentration of compounds were added and incubated for 5 min. The enzyme reaction was initiated by adding 5 μL of the substrate, and its activity was continuously monitored for 6 min. The IC50 values were calculated by fitting with the Hill Eq. 2, using OriginPro 8.5 (OriginLab, Inc.) where y is percent inhibition, x is inhibitor concentration, n is the slope of the concentration–response curve (Hill slope), and V max is maximal inhibition from two to four independent assays:
| (2) |
The enzyme omission assay was done by exactly the same method as IC50 determination, but without the 3CLpro enzyme in order to test for fluorescence signal interference by tested compounds.
4.9. Determination of dissociation equilibrium constant (KD) by SPR
Native 3CLpro enzyme was prepared in PBS (10 mM phosphate, pH 7.4, 2.7 mM KCl, 137 mM NaCl) and immobilized on a CM5 sensor chip using standard amine-coupling at 20 °C with running buffer HBS-P (10 mM HEPES, 150 mM NaCl, 0.05% surfactant P-20, pH 7.4) using a Biacore T100 instrument. Flow channel 1 was activated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrocholoride (EDC)/N-hydroxy succinimide (NHS) mixture, and the activated surface was blocked by ethanolamine (pH 8.5) as a control. 3CLpro enzyme was diluted in 10 mM sodium acetate (pH 5.0) and immobilized to flow channels 2 and 3 after sensor surface activation with EDC/NHS followed by ethanolamine blocking on unoccupied surface area. 3CLpro immobilization levels of flow channels 2 and 3 were ∼11,500 RU and ∼16,500 RU, respectively. An unrelated reference protein (∼25 kDa) was also immobilized to flow channel 4 as another control to be compared with 3CLpro (33.8 kDa as a monomer). Six initial HTS hits and one VS hit were prepared as 10 mM DMSO stock solutions. Compound solutions with a series of increasing concentrations (0–200 μM at 1.5-fold dilution) were applied to all four channels at a 10 μL/min flow rate at 20 °C. Sensorgrams were analyzed using BIAevaluation software 2.0.3, and response unit difference (ΔRU) values at each concentration were measured during the equilibration phase. Data were either single referenced with a blank (enthanolamine) or double referenced with both blank and reference protein RU values. SigmaPlot 12.0 was used to fit the data to a single rectangular hyperbolic curve to determine K D values. The hyperbolic function, y = y max·x/(K D + x), was used to plot response units and corresponding concentration, where y is the response, y max is the maximum response and x is the compound concentration.
4.10. Reversibility of inhibition
100 nM 3CLpro was incubated with screened compounds at 20× the concentration of the IC50 for each compound for 1 h at room temperature in assay buffer containing 50 mM HEPES (pH 7.5), 2 mM GSH, 0.1 mg/ml BSA, 0.01% Triton X-100 and 1% DMSO in a final volume of 500 μL. Control 3CLpro without any compound was also prepared in the same way. Then, each sample including positive control without any compound was buffer exchanged with assay buffer with desalting column (Pierce) to remove compounds. 3CLpro activity was measured in the same way as IC50 measurements before and after buffer exchange.
4.11. Mechanism of inhibition
3CLpro activity was monitored in the same way as the primary screening with varying concentration of inhibitor compounds and the substrate (0–200 μM). The concentration of compounds was varied from 0 to at least 10× the IC50 value of each fragment. The data were fit to the following equations using SigmaPlot Enzyme Kinetics Module 1.3 in order to determine the best fit inhibition mechanism and kinetic parameters for each compound:
| (3) |
| (4) |
| (5) |
| (6) |
where ν is the reaction rate, V max is the maximum rate of the reaction, K m is the Michaelis–Menten constant for the substrate, [S] is the substrate concentration, [I] is the inhibitor concentration, K i is the dissociation constant of the inhibitor I to the free enzyme and αK i is the dissociation constant for the inhibitor I to the ES complex.
Acknowledgments
We thank Dr. Pavel Petukhov for providing access to MOE software package. This work was supported by National Institutes of Health Grants R56 AI089535 and P01AI60915. We thank Dr. Kiira Ratia for performing HTS and primary screening data analysis. We thank ChemAxon for a free academic license of their cheminformatics suite including JChem and JChem for excel for HTS data analysis. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575.
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bmc.2013.11.041.
Supplementary data
This document file contains supplementary data.
References and notes
- 1.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2004. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. Communicable Disease Surveillance & Response (CSR). http://www.who.int/csr/sars/country/table2004_04_21/en.
- 3.Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A.D., Baker S.C. J. Virol. 2005;79:15189. doi: 10.1128/JVI.79.24.15189-15198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lau S.K., Woo P.C., Li K.S., Huang Y., Tsoi H.W., Wong B.H., Wong S.S., Leung S.Y., Chan K.H., Yuen K.Y. Proc. Natl. Acad. Sci. U.S.A. 2005;102:14040. doi: 10.1073/pnas.0506735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woo P.C., Lau S.K., Chu C.M., Chan K.H., Tsoi H.W., Huang Y., Wong B.H., Poon R.W., Cai J.J., Luk W.K., Poon L.L., Wong S.S., Guan Y., Peiris J.S., Yuen K.Y. J. Virol. 2005;79:884. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pyrc K., Berkhout B., van der Hoek L. J. Virol. 2007;81:3051. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fouchier R.A., Hartwig N.G., Bestebroer T.M., Niemeyer B., de Jong J.C., Simon J.H., Osterhaus A.D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:6212. doi: 10.1073/pnas.0400762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van der Hoek L., Pyrc K., Jebbink M.F., Vermeulen-Oost W., Berkhout R.J., Wolthers K.C., Wertheim-van Dillen P.M., Kaandorp J., Spaargaren J., Berkhout B. Nat. Med. 2004;10:368. doi: 10.1038/nm1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. N. Engl. J. Med. 2012;367:1814. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 10.Bermingham A., Chand M.A., Brown C.S., Aarons E., Tong C., Langrish C., Hoschler K., Brown K., Galiano M., Myers R., Pebody R.G., Green H.K., Boddington N.L., Gopal R., Price N., Newsholme W., Drosten C., Fouchier R.A., Zambon M. Euro. Surveill. 2012;17:20290. [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention, 2013.
- 12.Van Boheemen S., de Graaf M., Lauber C., Bestebroer T.M., Raj V.S., Zaki A.M., Osterhaus A.D., Haagmans B.L., Gorbalenya A.E., Snijder E.J., Fouchier R.A. MBio. 2012:3. doi: 10.1128/mBio.00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skowronski D.M., Astell C., Brunham R.C., Low D.E., Petric M., Roper R.L., Talbot P.J., Tam T., Babiuk L. Annu. Rev. Med. 2005;56:357. doi: 10.1146/annurev.med.56.091103.134135. [DOI] [PubMed] [Google Scholar]
- 14.Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 15.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. J. Mol. Biol. 2007;366:965. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G.F., Anand K., Bartlam M., Hilgenfeld R., Rao Z. Proc. Natl. Acad. Sci. U.S.A. 2003;100:13190. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 20.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H., Hsu M.C. J. Med. Chem. 2006;49:4971. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 22.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. J. Virol. 2008;82:2515. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh A.K., Xi K., Johnson M.E., Baker S.C., Mesecar A.D. Annu. Rep. Med. Chem. 2006;41:183. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh A.K., Gong G., Grum-Tokars V., Mulhearn D.C., Baker S.C., Coughlin M., Prabhakar B.S., Sleeman K., Johnson M.E., Mesecar A.D. Bioorg. Med. Chem. Lett. 2008;18:5684. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. Bioorg. Chem. 2008;36:229. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee P., Desai P., Ross L., White E.L., Avery M.A. Bioorg. Med. Chem. 2008;16:4138. doi: 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen T.T., Ryu H.J., Lee S.H., Hwang S., Breton V., Rhee J.H., Kim D. Bioorg. Med. Chem. Lett. 2011;21:3088. doi: 10.1016/j.bmcl.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regnier T., Sarma D., Hidaka K., Bacha U., Freire E., Hayashi Y., Kiso Y. Bioorg. Med. Chem. Lett. 2009;19:2722. doi: 10.1016/j.bmcl.2009.03.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan J., Verschueren K.H.G., Anand K., Shen J., Yang M., Xu Y., Rao Z., Bigalke J., Heisen B., Mesters J.R., Chen K., Shen X., Jiang H., Hilgenfeld R. J. Mol. Biol. 2005;354:25. doi: 10.1016/j.jmb.2005.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han-Yu, C.; Hongfang, L.; Brown, S.; McMunn-Coffran, C.; Cheng-Yan, K.; Hsu, D. F. Identifying significant genes from microarray data, 2004.
- 31.Dwork, C.; Kumar, R.; Naor, M.; Sivakumar, D. In Proceedings of the 10th international conference on World Wide Web; ACM: Hong Kong, Hong Kong, 2001, 613.
- 32.Lyons D.M., Hsu D.F. Inf. Fusion. 2009;10:124. [Google Scholar]
- 33.Charifson P.S., Corkery J.J., Murcko M.A., Walters W.P. J. Med. Chem. 1999;42:5100. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 34.Wang R., Wang S. J. Chem. Inf. Comput. Sci. 2001;41:1422. doi: 10.1021/ci010025x. [DOI] [PubMed] [Google Scholar]
- 35.Xue X., Yang H., Shen W., Zhao Q., Li J., Yang K., Chen C., Jin Y., Bartlam M., Rao Z. J. Mol. Biol. 2007;366:965. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S., Jonas F., Shen C., Hilgenfeld R. Protein Cell. 2010;1:59. doi: 10.1007/s13238-010-0011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuo C.J., Chi Y.H., Hsu J.T., Liang P.H. Biochem. Biophys. Res. Commun. 2004;318:862. doi: 10.1016/j.bbrc.2004.04.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee H., Torres J., Truong L., Chaudhuri R., Mittal A., Johnson M.E. Anal. Biochem. 2012;423:46. doi: 10.1016/j.ab.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yip K.W., Ito E., Mao X., Au P.Y., Hedley D.W., Mocanu J.D., Bastianutto C., Schimmer A., Liu F.F. Mol. Cancer Ther. 2006;5:2234. doi: 10.1158/1535-7163.MCT-06-0134. [DOI] [PubMed] [Google Scholar]
- 41.Doughty-Shenton D., Joseph J.D., Zhang J., Pagliarini D.J., Kim Y., Lu D., Dixon J.E., Casey P.J. J. Pharmacol. Exp. Ther. 2010;333:584. doi: 10.1124/jpet.109.163329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coburn R.A., Baker P.J., Evans R.T., Genco R.J., Fischman S.L. J. Med. Chem. 1978;21:828. doi: 10.1021/jm00206a024. [DOI] [PubMed] [Google Scholar]
- 43.Burnham K.P.A., Anderson D.R. Sociol. Methods Res. 2004;33:261. [Google Scholar]
- 44.Hilgenfeld R., Anand K., Mesters J., Rao Z., Shen X., Jiang H., Tan J., Verschueren K.G. In: Perlman S., Holmes K., editors. Vol. 581. Springer; US: 2006. pp. 585–590. (The Nidoviruses). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L.-R., Wang Y.-C., Lin Y.W., Chou S.-Y., Chen S.-F., Liu L.T., Wu Y.-T., Kuo C.-J., Chen T.S.-S., Juang S.-H. Bioorg. Med. Chem. Lett. 2005;15:3058. doi: 10.1016/j.bmcl.2005.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kao R.Y., To A.P.C., Ng L.W.Y., Tsui W.H.W., Lee T.S.W., Tsoi H.-W., Yuen K.-Y. FEBS Lett. 2004;576:325. doi: 10.1016/j.febslet.2004.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kao R.Y., Tsui W.H.W., Lee T.S.W., Tanner J.A., Watt R.M., Huang J.-D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K.-H., Tse H., To A.P.C., Ng L.W.Y., Wong B.C.W., Tsoi H.-W., Yang D., Ho D.D., Yuen K.-Y. Chem. Biol. 2004;11:1293. doi: 10.1016/j.chembiol.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mukherjee P., Desai P., Ross L., White E.L., Avery M.A. Bioorg. Med. Chem. 2008;16:4138. doi: 10.1016/j.bmc.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang J., Huitema C., Niu C., Yin J., James M.N.G., Eltis L.D., Vederas J.C. Bioorg. Chem. 2008;36:229. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. J. Med. Chem. 2006;49:3440. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 52.Irwin J.J., Shoichet B.K. J. Chem. Inf. Model. 2005;45:177. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Case, D.; Darden, T.; Cheatham, T.E.; Simmerling, C. L.; Wang, J.; Duke, R. E.; Luo, R.; Crowley, M.; Ross C. Walker; W. Zhang; Merz, K. M.; Wang, B.; Hayik, S.; Roitberg; G. Seabra, A.; I. Kolossváry; K.F.Wong; F. Paesani; J. Vanicek; X.Wu; S.R. Brozell; T. Steinbrecher; H. Gohlke; L. Yang; C. Tan; J. Mongan; V. Hornak; G. Cui; D.H. Mathews; M.G. Seetin; C. Sagui; Babin, V.; Kollman, P. A. AMBER 10. University of California, San Francisco 2008
- 54.Duan Y., Wu C., Chowdhury S., Lee M.C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P. J. Comput. Chem. 2003;24:1999. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 55.Jain A.N. J. Med. Chem. 2003;46:499. doi: 10.1021/jm020406h. [DOI] [PubMed] [Google Scholar]
- 56.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. J. Mol. Biol. 1997;267:727. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 57.Graves A.P., Shivakumar D.M., Boyce S.E., Jacobson M.P., Case D.A., Shoichet B.K. J. Mol. Biol. 2008;377:914. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feig M., Karanicolas J., Brooks C.L., 3rd. J. Mol. Graph. Model. 2004;22:377. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 59.Carlson H.A., Masukawa K.M., Rubins K., Bushman F.D., Jorgensen W.L., Lins R.D., Briggs J.M., McCammon J.A. J. Med. Chem. 2000;43:2100. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 60.Allen K.N., Bellamacina C.R., Ding X., Jeffery C.J., Mattos C., Petsko G.A., Ringe D. J. Phys. Chem. 1996;100:2605. [Google Scholar]
- 61.Mattos C., Ringe D. Nat. Biotechnol. 1996;14:595. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]
- 62.Kortvelyesi T., Dennis S., Silberstein M., Brown L., 3rd., Vajda S. Proteins. 2003;51:340. doi: 10.1002/prot.10287. [DOI] [PubMed] [Google Scholar]
- 63.Kortvelyesi T., Dennis S., Silberstein M., Brown L., Vajda S. Proteins: Struct., Funct., Bioinf. 2003;51:340. doi: 10.1002/prot.10287. [DOI] [PubMed] [Google Scholar]
- 64.Feig M., Karanicolas J., Brooks Iii C.L. J. Mol. Graph. Model. 2004;22:377. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This document file contains supplementary data.