

Abstract
In the photocycle of bacteriorhodopsin at pH 7, proton release from the proton releasing group (PRG) to the extracellular medium occurs during formation of the M intermediate. This proton release is inhibited at acidic pH, below the pKa of the PRG, ∼6 in M, and instead occurs later in the cycle as the initial state is restored from the O intermediate. Here, structural changes related to deprotonation of the PRG have been investigated by time-resolved FTIR spectroscopy at 25°C. The vibrational features at 2100-1790 cm-1, 1730-1685 cm-1, 1661 cm-1, and 1130-1045 cm-1 have greater negative intensity in the pure M-minus-BR spectrum and even in the M-minus-BR spectrum, that is present earlier together with the L-minus-BR spectrum, at pH 7, than in the corresponding M-minus-BR spectra at pH 5 or pH 4. The D212N mutation abolishes the decreases in the intensities of the broad feature between 1730 and 1685 cm-1 and the band at 1661 cm-1. The 1730-1685 cm-1 feature may arise from transition dipole coupling of the backbone carbonyl groups of Glu204, Phe208, Asp212 and Lys216 interacting with Tyr57 and C15-H of the chromophore. The 1661 cm-1 band, which is insensitive to D2O substitution, may arise by interaction of the backbone carbonyl of Asp212 with C15-H. The 2100-1790 cm-1 feature with a trough at 1885 cm-1 could be due to a water cluster. Depletion of these bands upon deprotonation of the PRG is attributable to disruption of a coordinated structure, held in place by interactions of Asp212. Deprotonation of the PRG is accompanied also by disruption of the interaction of the water molecule near Arg82. The liberated Asp212 may stabilize the protonated state of Asp85, and thus confer uni-directionality to the transport.
The extremely halophilic Archaeon, Halobacterium salinarum, harvests energy from sunlight by means of the light-driven proton pump, bacteriorhodopsin. This protein is concentrated into patches in the cell membrane known as “purple membrane” which can be isolated as essentially pure bacteriorhodopsin and lipids (1). Light is absorbed by the retinal chromophore of bacteriorhodopsin, which is linked to the amino group of Lys216 through a protonated Schiff base. Absorption of a photon by the state that contains the all-trans isomer of the retinal (BR1) causes isomerization of the C13═C14 bond and thus initiates a catalytic cycle involving intermediates L, M, N, and O (2, 3), and the result is the net translocation of a proton from the cytoplasmic to the extracellular medium against an electrochemical potential (4).
Bacteriorhodopsin is one of a very few enzymes whose precise reaction scheme has been elucidated. All of the stepwise protonation reactions, which together result in translocation of a proton across the membrane in this light dependent proton pumping protein, have been clarified (reviewed in 5-11). The first proton transfer occurs in the L-to-M transition, from the Schiff base to Asp85 (12), the counterion on the extracellular side of the Schiff base. The release of a proton to the extracellular medium is from the proton-release group (PRG) during formation of M (13, 14). Reprotonation of the Schiff base takes place in the M-to-N transition, when it receives a proton from As96 on the cytoplasmic side of the Schiff base. The N-to-O transition is accompanied by the back-isomerization of the retinal to the all-trans form (15). After a short delay a proton is taken up from the cytoplasm by Asp96 (16). The O intermediate prevails at acidic pH in the N-O equilibrium in which the pKa value of Asp96 is ∼7.2 (17). The PRG is refilled by a proton from Asp85 in the final O-to-BR step. On the basis of these known facts, we can now address the problem of elucidating the sub-molecular events that assure unidirectional proton transport across the protein.
The proton release in M is suppressed at alkaline and acidic pH, and two pKa values, one at ∼9 in BR (18, 19) and another at ∼6 in M (14) are responsible for the inhibition: The pKa of the PRG drops from ∼9 to ∼6 upon protonation of Asp85 in M, causing release of a proton from the PRG in M at neutral pH. It has been proposed that the unidirectionality of the re-protonation of the Schiff base in the M-to-N transition is created by increasing the proton affinity of Asp85 in M through coupling of the proton transfer to deprotonation of the PRG (18, 19). Such coupling was shown in spectroscopic titration studies of BR, where deprotonation of a group, postulated to be the PRG, increased the pKa of Asp85 from ∼2.5 to ∼7 (19, 20). The M intermediate with protonated Asp85 is present even at pH 10.5. This has been attributed to elevation of the pKa of Asp85 above 10.5 (21). Additional structural changes are expected to occur in M to attain such a high pKa at Asp85 and thereby prevent the backflow of the proton to the Schiff base.
Extensive studies have been conducted to define and characterize the PRG. Mutations of several different residues on the extracellular side of the Schiff base have been shown to affect the normal proton release mechanism. These residues are Glu204, Glu194, Tyr57, and Arg82 (20, 22-25). The search for the site of deprotonation has been carried out through studies of the effects of mutating these residues on light dependent difference FTIR spectra, and in particular on the continuum absorbance changes at frequencies between 1800 and 1900 cm-1 (26) and between 2600 and 2900 cm-1 (27) and on the broad band between 1580 and 1550 cm-1 (28, 29). The authors of the former references (26, 27) proposed that the proton is released from a protonated water cluster surrounded by Arg82, Glu194 and Glu204, while those of the latter reference proposed that Arg82 is the site of the release (28, 29). Neither of the two carboxylic acids, Glu204 and Glu194, which are unprotonated in the initial state (26), can be sites for the release.
Infrared absorption is sensitive to polarity changes in chemical bonds. Hence, FTIR spectroscopy is a powerful tool to detect chemical changes related to deprotonation of the PRG and the process that increases the proton affinity of Asp85. Since the pKa of the PRG in BR is ∼9 (20), the PRG will have already largely deprotonated in BR at pH 10, as in M. In comparing FTIR spectra at pH 7 and pH 10 (30), the contribution from the deprotonation of the PRG in M appeared with larger negative amplitudes in the spectrum at pH 7 than pH 10 in the 1730-1685 cm-1 region and the 1130-1045 cm-1 region, in addition to the 2120-1790 cm-1 region.
Below pH ∼6, the pKa of PRG, release of the proton is not observed in M (14, 31). At this lower pH, the PRG remains protonated in the M intermediate, as it is in BR, and thus differences between the FTIR spectra of the M intermediate at pH 7 and pH 5 can be ascribed to deprotonation of the PRG. There are no known pH dependent changes in the initial state between pH 7 and pH 4 (6), meaning that the photocycles at pH 7 and pH 5 start from the same initial state. Structural changes which are accompanied by deprotonation of the PRG could be revealed in a comparison of the spectral changes in M at pH 7 with those at pH 5 as the reference.
The spectral change in the region between 1725 and 1685 cm-1 has been proposed to be due to release of Asp212 from the constraints exerted by helix G, Arg82, and C15—H of the chromophore (30). Asp212 is located in a unique position: it interacts with Arg82 through a water molecule in the initial BR state. In D212N/R82Q, the proton affinity of Asp85 is lowered (32). The R82Q mutation increases the pKa of Asp85 in BR up to ∼7 and accelerates M formation (19, 20), suggesting that the role of Asp212 is to stabilize the protonated state of Asp85. In support of this notion, the photocycle of the D212N mutant does not include M (33, 34). M formation is partially restored in the presence of chloride (35).
In this article, FTIR spectral changes in the M intermediate at pH 7 are compared with those at pH 5 and pH 4 in order to reveal structural changes which may cause stabilization of the protonated Asp85 in M. The effects of the D212N mutation and D2O substitution on vibrational bands were investigated to reveal features near Asp85 specific to this issue.
Materials and Methods
Sample Preparation
Purple membrane containing wild-type bacteriorhodopsin was isolated according to a standard procedure (36). The D212N mutant was described previously (33). The sample of wild-type bacteriorhodopsin at pH 5 for FTIR measurements was prepared as follows: purple membranes were suspended in 1.4 mL of 0.1 M phosphate buffer (pH 5.0), centrifuged, resuspended in water and centrifuged again. The same cycle was repeated once more. To a 100 μL aliquot of the washed sample, the same buffer at pH 5 (2 μL) was added. About 60 μL of the suspension was applied to the center of a 25 mm diameter BaF2 window, and dried under gentle aspiration. The remaining part of the suspension (∼40 μL) was then overlaid on the dried film and dried again. The resulting film was hydrated with ∼0.5 μL of water (or D2O). The sample cell was sealed and installed in the spectrophotometer as described previously (37). The samples at pH 4 and pH 6.5 were made in the same way except for the use of 0.1 M citrate buffer at pH 4, and 0.1 M phosphate buffer at pH 6.5, respectively. In the case of D212N, the sample was supplemented with 0.1 M NaCl (10 μL) before it was applied to the window. The ratio of the absorbance at 1658 cm-1 to that at 1548 cm-1 in the absolute spectra is 1.3-1.5. The absorbance at 3350 cm-1 is >1.4. These figures correspond to a water content of 65-70% as defined in (38).
Data Acquisition and Processing
Details of the laser and data acquisition are described in Morgan et al. (37). Spectra were measured using a Varian FTS-6000 spectrophotometer in step-scan mode at 25°C. A data file was typically obtained by summing 96 scans (about 3 hours). Samples were replaced after being used to acquire two files. A global multi-exponential fit was applied to the data in the 2500-900 cm-1 region of the complete data set using SplMod (39: http://s-provencher.com/). The rate constants thus obtained were used to calculate the consecutive spectra di using a Gaussian elimination method. The resulting rate constants and spectra were processed by the method of Chizhov et al. (40) to calculate a difference spectrum with respect to the initial unphotolyzed state (BR) for each intermediate in terms of a kinetic model consisting of a linear irreversible sequence of intermediates. In this scheme, di denotes the spectrum of the kinetic intermediate that is formed at the rate of ki and decays at the rate of ki+1. The spectrum d0 corresponds to the component immediately after the flash within in the time resolution (5 μs), unless otherwise stated.
Data were acquired for 20 ms (4000 data points) of which 2 ms was prior to the laser flash, unless otherwise noted. The data set (24 files) from a time-resolved study on wild type at pH 5 was fitted by six exponentials with the rate constants (in ms-1) of 22 (k1), 3.4 (k2), 1.3 (k3), 0.38 (k4), 0.16 (k5), and 0.01 (k6). The data set (14 files) obtained from the photocycle at pH 4 was fitted by 5 exponentials with the rate constants (in ms-1) of 20 (k1), 3.4 (k2), 0.39 (k3), 0.37 (k4), and 0.10 (k5). The stored processed data set of wild type in H2O at pH 7 (37) was used in the current studies. A data set of 24 files, with 18 ms of post-trigger recording, was acquired at pH 7, but we have chosen to show data from an earlier set consisting of 44 files, but with only 8 ms of post-trigger data, which had less noise. However, the results in both data sets are consistent with the conclusions in this paper. The fitted results for pH 7 included a fast component (rate of formation 416 ms-1) that was not detected in our study at pH 5, consistent with a previous paper (41). In the current paper, to simplify comparison of the pH 7 results to those at other pH values, this fastest step has been omitted from the description of the reaction, which is thus presented as a five-step process. The spectrum of the product of the 416 ms-1 phase is thus designated d0, and time constants at pH 7 (in ms-1) are 23 (k1), 8.1 (k2), 1.9 (k3), 0.67 (k4), and 0.16 (k5). The spectrum d3 in our earlier paper (37), which was assigned to the M-minus-BR transition, is thus d2 in the description used here.
The results for wild-type bacteriorhodopsin hydrated with D2O (14 files), which included 8 ms of post-trigger data, was fitted by five-exponentials. As in the case of the H2O data above, the first step (k = 416 ms-1) of this five-step process has been omitted. The rate constants (in ms-1) in this four-step description of the reaction are: 7.8 (k1), 1.3 (k2), 0.39 (k3), and 0.025 (k4). The ratios of these rates to those in H2O are nearly coincident with those recorded in solution in a previous visible spectroscopic study (16).
The data set of D212N in the presence of chloride at pH 6.5 was fitted by 6 exponentials. As in the case of wild type at pH 7, to simplify comparison with other data in this paper, the first step (k=309 ms-1) has been omitted. The rate constants (in ms-1) in this five-step description of the reaction are: 18 (k1), 7.0 (k2), 1.5 (k3), 0.43 (k4), and 0.017 (k5). All the calculated M-minus-BR spectra were adjusted on the basis of the intensity at 1168 cm-1 to match the M-minus-BR spectrum of wild type at pH 7.
Results
M-minus-BR spectrum at pH 7 vs. at pH 4 or 5
The residence times of the kinetic intermediates d1 (time constant for the decay of 0.29 ms) and d2 (0.77 ms) at pH 5 largely overlap with those of d1 (0.12 ms) and d2 (0.53 ms) at pH 7. The spectra of d1 and d2 are almost the same in shape. In Figure 1 the spectrum of d2 at pH 5 (green line) is superimposed on the spectrum of d2 at pH 7 (blue line), which has been shown to be the M-minus-BR spectrum (37). The presence of the hydrogen out-of-plane band at 987 cm-1 and the larger positive intensity of the 1188 cm-1 band in the spectrum of d2 at pH 5 (green line) than in d2 at pH 7 (blue line) indicate the presence of a considerable fraction of long-lived L intermediate at the lower pH, as was previously suggested (41, 42).
Figure 1.

The chromophore region (1700-900 cm-1) of the M-minus-BR spectrum (d2) of wild type at pH 7 (37, blue line), the spectrum of d2 of wild type at pH 5 (green line), and the calculated spectrum of the M-minus-BR component of wild type at pH 5 (red line) obtained by subtracting 40% of the spectrum of d0 (mainly due to the L-minus-BR spectrum) from the spectrum of d2. The M-minus-BR spectra at pH 7 and pH 5 were normalized on the basis of the negative band at 1168 cm-1. One division of the ordinate corresponds to 0.002 absorbance unit for bacteriorhodopsin at pH 7. The level of zero absorbance is shown by a horizontal broken line.
Subtraction of 40% of the spectrum of d0, which is largely due to the L-minus-BR spectrum, from the spectrum of d2 at pH 5 (green line) reduces the amplitude at 1188 cm-1 to the level of the M-minus-BR spectrum at pH 7 (blue line), resulting in the M-minus-BR spectrum at pH 5 (red line). A broad band between 1130 and 1045 cm-1 in the M-minus-BR spectrum at pH 7 is absent at pH 5. This band, which is also absent from the spectrum at pH 10, has been attributed to the vibrations of the side chain of Lys216 coupled with the vibration of C15—H of the retinal (30). Also, the M-minus-BR spectrum of wild type at pH 4, obtained by subtracting 35% of the spectrum of d0 from the spectrum of d2 at pH 4 (not shown), is identical in shape in the 1650-900 cm-1 region to the M-minus-BR spectrum at pH 5 (red line).
Spectral Changes Associated with Deprotonation of the PRG in M
In Figure 2a, vibrational bands in the 1800-1650 cm-1 region, mostly due to the protein, in the spectrum of the calculated M-minus-BR component from d2 at pH 5 (solid line), are compared with those in the M-minus-BR spectrum at pH 7 (dotted line). The calculated spectrum at pH 4 (not shown) is almost same as that at pH 5. A similar frequency shift of the C=O stretching band of Asp85 at 1761 cm-1 upon lowering the pH has previously been noted (29, 43). Throughout the region between 1730 and 1685 cm-1, the spectrum at pH 5 appears above the spectrum at pH 7. The decrease in negative intensity at pH 5 in this frequency region shows that a broad feature, present in the initial BR state, disappears upon deprotonation of the PRG, presumably because the PRG deprotonates at pH 7 but not at pH 5 and pH 4, which are below the pKa of PRG, ∼6 in M (14). Decreases observed in the M-minus-BR spectrum at pH 10 (broken line in Figure 2a, adapted from 30), where the PRG had already deprotonated in the unphotolyzed state, since this is above the pKa value of the PRG, ∼9 in BR (20), are similar to those at pH 5 in this frequency region, except for the presence of an overlapping positive broad band between 1732 and 1706 cm-1. This is probably due to protonation of Glu194/Glu204 by a proton released from the PRG, as claimed by Wolf et al. (44). This band, however, is much smaller in intensity than another C=O stretching vibrational band at 1761 cm-1 due to Asp85 (Figure 2a), suggesting that the release in M at pH 5 occurs in only a minor population. The 1694 cm-1 band itself does not decrease at pH 5. The sharp negative band at 1661 cm-1 at pH 7 loses its intensity at pH 5, leaving a twin band between 1675 and 1660 cm-1. The same differences had been noted in a previous time-resolved attenuated total reflectance FTIR spectroscopic study (43), in which films immersed in solution at pH 7 and pH 4 were compared, though this was not described explicitly in the text.
Figure 2.

The protein region (1800-1650 cm-1): (a) the M-minus-BR spectra of wild type of d2 at pH 7 (dotted line), and at pH 5 (solid line). (b) the M-minus-BR spectra of wild type of d1 at pH 7 (dotted line), and at pH 5 (solid line). (c) the spectra of d0 (mainly due to the L-minus-BR spectra) of wild type at pH 7 (dotted line) and at pH 5 (solid line). The M-minus-BR spectrum at pH 10 (30) was overlaid as dashed lines in (a) and (b). One division of the ordinate corresponds to 0.0005 absorbance unit for bacteriorhodopsin at pH 7.
The 1761 cm-1 band of M is undetectable in the spectrum of d0 (Figure 2c), but appears in the M-minus-BR spectrum of d1 at pH 7 (dotted line in Figure 2b) that was obtained by subtraction of 40% of the spectrum of d0 at pH 7. The shape in the 1650-900 cm-1 region, including the band at 1130-1045 cm-1 (not shown), is completely coincident with the M-minus-BR spectrum at pH 7 of the spectrum d2 (blue line in Figure 1). The spectrum of the M-minus-BR component from d1 at pH 5 was obtained by subtracting the contribution of the L-minus-BR spectrum of d0 in the same way. Decreases in negative intensity of the broad feature at 1730-1685 cm-1 and of the band at 1661 cm-1 at pH 5 (solid line in Figure 2b) relative to those at pH 7 (dotted line) can also be observed in the parallel pH dependent differences in the calculated spectra of the M-minus-BR components from d1, as they are in the M-minus-BR spectra of d2. However, the broad positive band between 1732 and 1706 cm-1, which could have been revealed by comparison with the M-minus-BR spectrum at pH 10 (broken line), was not detected, in contrast to the case of the spectra of d2. The coincidence of the spectra of d0 at pH 7 (solid line in Figure 2c) and pH 5 (dotted line), both of which show the same typical 1740 cm-1 band of the L-minus-BR spectrum, is consistent with the fact that deprotonation of the PRG does not occur in L. The results show that deprotonation of the PRG causes the depletion of the broad feature at 1730-1685 cm-1 and the band at 1661 cm-1 in M formation, but not at all in L.
Besides these pH dependent changes, the positive broad feature between 1580 and 1550 cm-1 in the M-minus-BR spectrum at pH 7 (blue line in Figure 1) has been shown to be less intense in the early M spectrum (41). This band decreases intensity in the M-minus-BR spectra from spectra d2 (red line) and d1 (not shown) at pH 5, and at pH 4 (data not shown, also 29).
Absorbance Change with a Trough at 1885 cm-1 Associated with M Formation
The broad negative feature in the M-minus-BR spectrum above 1800 cm-1 has been called the continuum absorbance change. It was explained as arising from deprotonation of the PRG, through studies comparing the spectrum of the wild-type protein with that of E204Q and other mutants, as well as the wild type at low pH (26, 44-46). The analysis of this feature is biased by the overlapping broad positive band, which had been attributed to heating (46). This latter feature appears in the spectrum of d0 at pH 7 with a maximum at ∼1960 cm-1 (cyan solid line in Figure 3a). The amplitude of this band decreases significantly in the ensuing spectrum of d1 (brown solid line). This putative heating effect has disappeared entirely in the spectra of d2 (red solid line) and d3 (blue solid line) at pH 7, as judged from their coincident shapes, at least in the 2000-1790 cm-1 region. This is consistent with the reported dissipation time of the heating effect of 0.3 ms (46). In a similar way, the spectrum of d2 at pH 5 (red dotted line), which is coincident with the spectrum of d3 (blue dotted line), can be regarded as being free from the heating effect.
Figure 3.

(a) The continuum absorbance changes of wild type at pH 7 (solid lines) and at pH 5 (dotted lines). The first spectra (d0) are shown in cyan, the second spectra (d1) in brown, the third spectra (d2) in red, and fourth spectra (d3) in blue. (b) The difference between the spectra at pH 7 and pH 5 of d1 (brown), d2 (red), and d3 (blue). One division of the ordinate corresponds to 0.00005 and 0.00002 absorbance unit for bacteriorhodopsin at pH 7 in (a) and (b), respectively.
Comparison of these spectra, whose main contribution is the M-minus-BR spectrum, indicates that the negative amplitudes at pH 5 are smaller than at pH 7 in the broad region between 2100 and 1790 cm-1 for the spectra of d2, d3 and d4. A similar pH effect on the absorbance changes in the frequency region of 1900-1800 cm-1 was recently reported (44). The calculated difference between the spectra of d3 at pH 7 and pH 5 (blue line in Figure 3b), each of which is composed of 80% of the M-minus-BR spectrum and 20% of the N-minus-BR spectrum, shows a broad feature ranging from 2200 to 1800 cm-1 with a trough at ∼1885 cm-1. A similar spectral shape was observed in the difference between the spectra of d2 at pH 7 and pH 5 (red line), and even in the spectra of d1 (brown line), which still retain the heating effect. The amplitude at 1885 cm-1 of this broad band in the difference spectrum is about -0.05 milli-absorbance unit (Figure 3b). This value is comparable to the approximately -0.08 milli-absorbance unit change of the sharp negative band at 3645 cm-1 (26, 37), which is due to the stretching vibration of non-hydrogen bonding water O—H, suggesting that the changes originate from a stoichiometric quantity of water molecules.
Even though each of these spectra at pH 7 and at pH 5 (Figure 3a) increases in negative amplitude progressively with decreasing frequency toward 1790 cm-1, as has been shown previously for the spectrum at pH 7 (26, 45, 46), the difference between pH 7 and pH 5 (Figure 3b) becomes smaller beyond 1885 cm-1. Hence, the broad band induced by deprotonation of the PRG is distinct in shape from the residual band at pH 5, which must be due to something other than pH dependent deprotonation of the PRG.
Water O—H Stretching Vibrations at pH 5
The water vibration band at 3657 cm-1 in the M-minus-BR spectrum at pH 7 (Figure 4, dotted line), which persists in the N-minus-BR spectrum, was shown to be less intense at pH 10 (30). The calculated M-minus-BR spectrum at pH 5 (solid line, see also Figure 2) shows a recognizable negative band at 3645 cm-1 superimposed on the tilted baseline produced by subtraction of an intense feature in the spectrum of d0. However, it does not exhibit any trace of the peak at 3657 cm-1 that is present at pH 7. Thus, the 3657 cm-1 band is due to a water O—H vibration that arises in M at pH 7 by loss of hydrogen bonding upon deprotonation of the PRG.
Figure 4.

The O—H stretching vibrations of water in the calculated M-minus-BR spectrum at pH 5 (solid line) compared with the M-minus-BR spectrum at pH 7 (dotted line). One division of the ordinate corresponds to 0.0001 absorbance unit for bacteriorhodopsin at pH 7.
Effects of D2O-Substitution
No pure M-minus-BR spectrum for bacteriorhodopsin hydrated by D2O was separated in the global fit. The spectrum of d2, which has the largest fraction of the M-minus-BR spectrum, has the same place in the progression of kinetic model intermediates as the spectrum d2 of bacteriorhodopsin in H2O at pH 7, though it arises much more slowly (1.3 vs. 8.1 ms-1 in k2). Subtraction of 30% of the spectrum of d3, which is mainly due to the N-minus-BR spectrum, from the spectrum of d2 yielded a pure M-minus-BR spectrum, as judged by the complete elimination of the 1670 cm-1 band due to the N-minus-BR spectrum (47, 48). This calculated spectrum of the M-minus-BR component for bacteriorhodopsin hydrated with D2O (Figure 5a, solid line) is compared with the corresponding spectrum from a sample hydrated with H2O (dotted line) by normalizing at the 1168 cm-1 band, which is due to the chromophore and is not affected by either solvent D2O or by C15-deuteration (49).
Figure 5.

The effect of D2O substitution on the M-minus-BR spectrum of wild type at pH 7. (a) The calculated spectrum of the M-minus-BR component of wild type in D2O at pD 7 (red line) was compared with the M-minus-BR spectrum in H2O at pH 7 (blue line), which is duplicated from Figure 2 for the sake of comparison. The labels are shown in blue and red for the bands in H2O and D2O, respectively. One division of the ordinate corresponds to 0.0005 absorbance unit for bacteriorhodopsin at pH 7. (b) Comparison of the time courses of the intensity of the averages between 1663 and 1658 cm-1 (1661 cm-1) in H2O (cyan circles) and D2O (blue circles) with those between 1189 and 1188 cm-1 (1188 cm-1, brown circles for D2O and pink circles for H2O). See the details in the text. Horizontal broken line is zero absorbance for the plots at 1661 cm-1. One division of the ordinate corresponds to 0.0002 absorbance unit for the 1661 cm-1 band of bacteriorhodopsin in H2O at pH 7. An arrow indicates the level of the zero line for the traces at 1188 cm-1.
The 1761 cm-1 band of Asp85 has shifted to 1751 cm-1 as expected for D2O substitution. The C=N stretching band of the Schiff base in BR at 1640 cm-1 has shifted to 1633 cm-1. Also the N—H bending vibration bands at 1348 and 1254 cm-1 undergo the same changes in D2O as those observed in the M-minus-BR spectra at 230 K (50). A positive band of M at 1556 cm-1 splits into two bands at 1569 and 1555 cm-1. Strikingly, the negative band at 1661 cm-1 is retained in D2O.
In assessing the D2O sensitivity of the 1661 cm-1 band it is important to take into account the possibility that the larger absorbance at this frequency in the absolute spectrum in H2O (∼1 absorbance unit), due to the O—H bending vibration of water and the correspondingly smaller transmitted intensity, could potentially lead to a less linear spectrophotometric response, so that the intensity of the peak at 1661 cm-1 could be artificially decreased in the H2O data compared to the D2O data. However, it is unlikely that our data are significantly distorted by this effect. If the 1661 cm-1 band were sensitive to D2O substitution, its intensity in D2O would be expected to decrease to the level seen in the pH 5 sample (see Figure 2a). This is not observed. This conclusion is supported also by the data shown in Figure 5b, in which the time course of absorbance changes at 1661 cm-1 in H2O (in pink) and D2O (in red) are compared with data at 1188 cm-1 (in cyan for H2O and in blue for D2O), which show the time courses of the L-to-M transition in H2O and D2O. The 1661 cm-1 data have not been re-scaled; the two 1188 cm-1 curves have been shifted so that the initial amplitudes match the corresponding 1661 cm-1 data, and then scaled by the same multiplication factor (0.7). In both cases, this brings the maximum absorbance change (initial amplitude to point of maximum depletion) into line with the 1661 cm-1 data. The fact that the initial amplitudes of the 1661 cm-1 traces in H2O and D2O are almost identical, shows that there is no significant spectrophotometric artifact due to the underlying H2O absorbance. The L-minus-BR contribution at 1661 cm-1 does not include any contribution from deprotonation of the PRG (Figure 2c), and therefore is expected to be the same in H2O and D2O. Given the time-resolution of these measurements, extrapolation to zero time point should reflect the sample in the L state, and the amplitudes should correspond to the L-minus-BR spectrum. The extrapolated initial amplitudes at 1661 cm-1 in H2O and D2O are essentially identical. Furthermore, for both H2O and D2O, the subsequent changes at 1661 cm-1 follow almost exactly the same time course as those at 1188 cm-1 which are due to the L-to-M transition and are insensitive to D2O substitution. Both of these observations show that there is no significant artificial lowering of the intensity at 1661 cm-1 in H2O, and confirm our conclusion that the 1661 cm-1 feature is retained in D2O. The differences in amplitude between the H2O and D2O data at 1661 cm-1 (Figure 5b) presumably reflect differences in the kinetics of the reaction consistent with the fact that the d3 spectrum is not pure M (above). The traces at 1188 cm-1 (pink and brown) both exhibit a plateau in the millisecond time range corresponding to the formation of the N and O intermediates where the negative intensity of the 1661 cm-1 band (cyan and blue) decreases. The structural changes associated with the depletion of the 1661 cm-1 band are restored in the N and O intermediates.
Effects of the D212N Mutation on the M-minus-BR Spectrum
D212N undergoes a blue-to-purple transition upon lowering of the pH (with a pKa ∼7) (33), but no M is observed in the photocycle at pH values either above or below the pKa. When chloride is added, in the pH range of 3.8 to 7.3 about one-third of the population of D212N undergoes an active photocycle which, as in the wild-type protein, includes the M intermediate (35). The hierarchy of time constants in the kinetics of the photocycle of D212N in the presence of chloride is similar to that of wild type (see Materials and Methods). In the spectrum of d2, the Asp85 band at 1761 cm-1 has about 30% of the intensity it exhibits in the M-minus-BR spectrum of wild type at pH 7. Subtraction of 70% of the spectrum of d0, mainly due to the L-minus-BR spectrum, from the spectrum of d2 revealed a spectrum in which the 1188 cm-1 band is below the baseline (not shown) typical for M. The calculated spectrum of the M-minus-BR component from d2 of D212N (solid line in Figure 6) shows smaller intensities in the band at 2100-1790 cm-1 and the feature at 1730-1685 cm-1 than those in wild type (dotted line), while the 1761 cm-1 band of Asp85 appeared with identical shape. Long-range effects of the D212N mutation seem to be suppressed by chloride that replaces the negative charge of Asp212. The sharp negative band at 1661 cm-1 of the M-minus-BR spectrum of wild type (dotted line) is missing in D212N (solid line), and is replaced by a large negative band at 1673 cm-1. This band makes comparison around 1685 cm-1 ambiguous, but it is certain that the broad band at 1725-1685 cm-1 loses intensity, at least above 1692 cm-1.
Figure 6.

Comparison of the calculated spectrum of the M-minus-BR component from d2 of D212N at pH 6.5 in the presence of chloride (solid line) with that of wild type at pH 7 (dotted line), which is duplicated from Figure 2 for the sake of comparison. The one division of the ordinate corresponds to 0.001 absorbance unit for wild type at pH 7.
Discussion
Depletion of Spectral Features Which Do Not Occur in M at pH 5
Comparison of the M-minus-BR spectrum at pH 7 with that at pH 5 in the sub-millisecond time range reveals that the negative amplitudes (depletion) for the features at 1130-1045 cm-1 (Figure 1), 1661 cm-1, 1730-1685 cm-1 (Figure 2), and 2100-1790 cm-1 (Figure 3) and the positive amplitude (appearance) for the feature at 1580-1550 cm-1 (Figure 1) are larger at pH 7 than pH 5. The decrease in intensity of these features at pH 5 is correlated with deprotonation of the PRG in M (pKa ∼6) (14). This is consistent with the observation that these negative bands were also abolished in the M-minus-BR spectrum at pH 10 (30), where the PRG is in a deprotonated state in both BR and M (20). Comparison of the spectra at neutral and acidic pH is based on idea that the reactions start from the same initial BR state, because the PRG is protonated at both pH 7 and 5.
These pH dependent spectral changes are detected in the M-minus-BR spectrum of d1 at pH 7, which is present together with L, but not in the spectrum d0, which contains only L. In other words, the depletion induced by deprotonation of the PRG can be observed even in an early phase of M, where a sub-population of L still remains. This means that, in the wild-type protein, deprotonation of the PRG occurs as soon as M is formed, and the delay in the externally observed proton release is due to barriers and diffusion within the protein (13). This conclusion is qualified by the possibility that a small delay of the depletion in the early phase of the spectrum of d1 might not be detected by a failure to isolate such a component in the global fit, even if present. Earlier results (51) have shown that the depletion of the 1661 cm-1 band in M does not take place earlier than 0.05 ms after the initiation of the photocycle. The rise time constant of our spectrum of d1 is ∼0.04 ms.
The component of d2 at pH 7 is almost exclusively composed of M, and is virtually free from L. By contrast, the component of d2 at pH 5 or at pH 4 arises with a slower rate (k2=1.8 ms-1 vs. 3.4 ms-1 at pH 7) and retains the nearly same fraction of L as the component of d1. The equilibration of the proton between Asp85 and the Schiff base has completely shifted toward Asp85 in d2 at pH 7 but not at acidic pH values below the pKa of ∼6 of the PRG in M, indicating that the protonated Asp85 in M is stabilized by deprotonation of the PRG. This is consistent with the proposed coupling between the deprotonation of Asp85 and the PRG (6, 14, 19, 20).
Changes in Water Molecules and Arg82
The broad continuum band between 1900 and 1800 cm-1 has been proposed to arise from the water molecules responsible for proton release (26, 45). The current study separates this band into two parts. One feature that is apparent in the spectrum at pH 5 shows progressively larger negative amplitude toward lower frequency (dotted lines in Figure 3a). This is not related to deprotonation of the PRG. The other feature, which is related to deprotonation of the PRG, spans between 2100 and 1790 cm-1 with a trough at 1885 cm-1 (Figure 3b). The trough is present in a frequency region where protonated water clusters would be expected to appear: specifically the protonated water dimer has a peak at 1770 cm-1, the trimer at 1880 cm-1, and the tetramer at 2665 cm-1 (52) along with a combination of the bending and librational modes of water at ∼2140 cm-1 (46), although these frequencies and the band widths could be altered by the protein environment, which might stabilize the possible protonated clusters. Although a discernible shift in H218O could not be detected in our data, the possibility that the broad band at 2100-1790 cm-1 is due to depletion of the bending vibration of a protonated water cluster (52, 53) cannot be excluded.
The broad feature between 1580 and 1550 cm-1 has been previously referred to as the 1556 cm-1 band, and attributed to deprotonation of Arg82 by use of a bacteriorhodopsin sample in which the two η-nitrogens of arginine were labeled with 15N (29). The highly asymmetric character of the two η-nitrogen atoms of Arg82 in M in solid-state NMR spectra (54) suggests that one of the guanidyl C–N bonds of Arg82 acquires a high degree of double bond character with a localized positive charge in M, but seems not to be in a completely deprotonated state when compared with the authentic deprotonated arginine. However, a subsequent NMR result with a model system suggests that deprotonation of Arg82 is likely when it is present in the kind of apolar environment expected to be in M (55).
A crystallographic structure of the initial unphotolyzed state (see Figure 7, based on protein databank entry, 1c3w from Ref. 56) shows that Arg82 interacts with two water molecules, each of which further interacts with one of the oxygen atoms of the carboxyl group of Asp212 and the phenolic oxygen atom of Tyr57. Liberation of a water molecule interacting with Arg82 was shown by the appearance of the non-hydrogen bonding water O—H at 3657 cm-1 (Figure 4), which decreases in intensity in mutants of Arg82 (58). These lines of evidence are compatible with the idea that deprotonation of the PRG is accompanied by disruption of the interaction of the water molecule near Arg82. This disruption could further affect Asp212.
Figure 7.
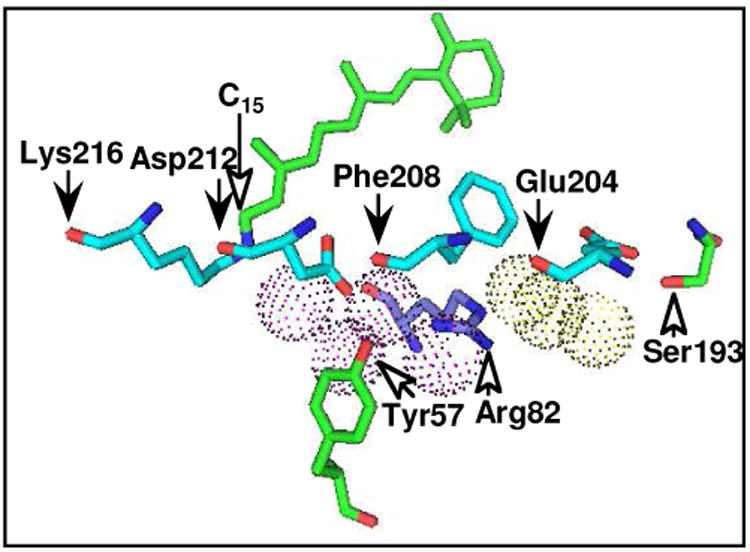
Structural elements involved in structural changes upon deprotonation of the PRG on the basis of the crystallographic structure of unphotolyzed bacteriorhodopsin in protein data bank entry 1c3w (56). All the residues are shown by sticks. Water molecules are dotted spheres in magenta (above Arg82) or yellow (below Arg82). The other oxygen, and nitrogen atoms are shown in red, and blue, respectively. Carbonyl oxygen atoms are indicated by black arrows, and side chains by white arrows. The molecular graphic representation was generated with PYMOL (57).
Disruption of the Interactions of Asp212
The unstructured broad feature at 1730-1685 cm-1 was suggested to be due to backbone carbonyl groups interacting with C15-H of the retinal (30) and with the ring of Tyr57 (discussed in 30 based on Ref. 59). The crystallographic structure of BR (Figure 7) shows that the nearest backbone carbonyl groups are those of Asp212 and Phe208. Involvement of the vibrations due to the backbone carbonyl of Lys216 and Glu204 has also been suggested (discussed in 30). The S193A mutation abolishes this feature in M at pH 7 (44). The side chain O—H group of Ser193 forms a hydrogen bond with one of the carboxyl oxygen atoms of Glu204 (Figure 7). The backbone carbonyl groups of Lys216, Asp212, Phe208, and Glu204 align in parallel along the distorted helix G, which is further disrupted at one end by the π-bulge at Ala215 and at the other by the extended structure at Val199 (56). The broad feature between 1730 and 1685 cm-1 probably arises from transition dipole coupling between these vibrational modes in the short helical segment (60, 61). Interactions with Tyr57 and the C15-H of the chromophore may be further included, as suggested in (30). The current study has shown that in D212N this broad feature is absent in the BR state or remains unchanged upon M formation (Figure 6), suggesting the disruption of interactions of Asp212 in the formation of M at neutral pH.
Previous study has shown that 15N-arginine substitution substantially reduces the intensity of the 1661 cm-1 band, though a fraction of the intensity is retained (29). The 1661 cm-1 band in the M-minus-BR spectrum is composed of two contributions; one of which is dependent on the deprotonated PRG, while the other remains at pH 5 (Figure 2). The C=N stretching vibration of Arg82, that should be coupled with the N—H bending vibrations, is expected to be sensitive to D2O substitution, because the N—D stretching vibration bands of Arg82 were recorded using a similar procedure for hydration by D2O (62). However, both of the bands contributing to the 1661 cm-1 trough in the M-minus-BR spectrum are retained in D2O (Figure 5). The intensity at 1661 cm-1, which was sensitive to 15N-substitution in the previous spectrum obtained at 0°C (29), is nearly two times larger (∼40% relative to the 1528 cm-1 band) than those in the current spectrum (∼25%). In other published spectra, the size of this band is also ∼25% of the 1528 cm-1 band (41, 45, 63). It should be noted that the photocycle in our measurement system at 25°C is very similar to that previously obtained at the same temperature by visible spectroscopy in solution (40) with respect to the time constants and composition of the kinetic intermediates, as discussed previously (37). Together, this suggests that the contribution to negative intensity at 1661 cm-1, which was reported in (29) to be lost in 15N-arginine bacteriorhodopsin, is not present in the spectra in our current study, and is thus distinct from either of the D2O insensitive contributions described above. Our results show that the 1661 cm-1 band, which is located in the frequency region for backbone carbonyl groups, is completely absent in D212N (Figure 6) and decreases in intensity in C15-D bacteriorhodopsin (30). Its depletion in M could be interpreted to be the result of loss of interaction between the C15-H of the retinal and the nearby backbone carbonyl group of Asp212. A detailed mechanism of the complex coupling will require further work beyond the scope of this paper.
Possible Structure Changes Associated with Deprotonation of PRG
It is interesting that the oxygen atoms (Figure 7, red sticks) of Tyr57, Phe208 and Asp212 form a cluster over a layer of four water molecules (spheres, in magenta). The guanidyl group of Arg82 is embedded in this water cluster. Another water cluster is present further below (spheres in yellow). Depletion of the features at 1730-1685 cm-1, the band at 1661 cm-1 and also the feature at 2100-1790 cm-1 together with the appearance of the feature at 1580-1550 cm-1 may be due to deprotonation and the ensuing disruption in this coordinated structure.
Effects of Asp212 on the Protonated Asp85
The M state is not formed in the photocycle of D212N (33) and D212N/R82Q (32), suggesting that the role of the anionic Asp212 is to stabilize the protonated form of Asp85. Formation of M in D212N can be partially restored by supplying chloride as a replacement of the missing negative charge (35), and in D212N/R82Q by decreasing the pKa of the Schiff base through the replacement with of C14-fluoro retinal (32). This could be interpreted to mean that Asp212 is involved in stabilizing the protonated form of Asp85 in M. The rationale would be to avoid reverse flow of the proton from Asp85 to the Schiff base, ensuring unidirectional proton transfer from the Schiff base to Asp85. Asp212 would take part in this stabilization when released from its constraints in the coordinated structure.
A Possible Mechanism for Stabilization of the Protonated Asp85 by Asp212
In the initial state, BR, two water molecules (Water401 and Water406) are sandwiched between Asp85 and Asp212 (56). Interaction between Asp85 and one water molecule in M (Water401) has been suggested from spectral changes of Asp85, which occurred in parallel with those of Water401 throughout the photoreactions of M at 80 K, 100 K and 133 K (64, discussed in 9). The same water molecule also forms hydrogen bonding with Trp86 in M (65; discussed in 11). Liberation of Asp212 from the constraints exerted by the complex coordinated structure (see above) would place Asp212 in a favorable orientation to interact with Asp85 through this water molecule. Interaction of water molecules with Asp85 in a proper geometry, which is supported by Asp212 and Trp86, would bring about an extremely high pKa state, similarly to Asp115 and Asp96 (66, 67), which interact with the O—H groups of Thr90 and Thr46, respectively (see 1c3w of Ref. 56). Asp115 also interacts with the carbonyl oxygen atom of Leu87 through a water molecule (Water511).
Footnotes
This work was supported by NIH grants HL 16101 (to R.B.G.) and 5R37GM029498 (to J.K.L.), and by a DOE grant DEFG03-86ER13525 (to J.K.L.).
Abbreviations: BR, initial unphotolyzed state of bacteriorhodopsin with all-trans retinal chromophore; PRG, proton release group.
References
- 1.Oesterhelt D, Stoeckenius W. Rhodopsin-like protein from the purple membrane of Halobactrium halobium. Nature New Biol. 1971;233:149–152. doi: 10.1038/newbio233149a0. [DOI] [PubMed] [Google Scholar]
- 2.Lozier RH, Bogomolni RA, Stoeckenius W. Bacteriorhodopsin: A light-driven proton pumping in Halobacterium halobium. Biophys J. 1975;15:955–962. doi: 10.1016/S0006-3495(75)85875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie A, Nagle JF, Lozier RH. Flash spectroscopy of purple membrane. Biophys J. 1987;51:627–635. doi: 10.1016/S0006-3495(87)83387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oesterhelt D, Stoeckenius W. Function of a new photoreceptor membrane. Proc Natl Acad Sci USA. 1973;70:2853–2857. doi: 10.1073/pnas.70.10.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haupt U, Tittor J, Oesterhelt D. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu Rev Biophys Biomol Struct. 1999;28:367–399. doi: 10.1146/annurev.biophys.28.1.367. [DOI] [PubMed] [Google Scholar]
- 6.Balashov SP. Protonation reactions and their coupling in bacteriorhodopsin. Biochim Biophys Acta. 2000;1460:75–94. doi: 10.1016/s0005-2728(00)00131-6. [DOI] [PubMed] [Google Scholar]
- 7.Herzfeld J, Lansing JC. Magnetic resonance studies of the bacteriorhodopsin pump cycle. Annu Rev Biophys Biomol Struct. 2002;31:73–95. doi: 10.1146/annurev.biophys.31.082901.134233. [DOI] [PubMed] [Google Scholar]
- 8.Lanyi JK, Schobert B. Local—global conformational coupling in a heptahelical membrane protein: transport mechanism from crystal structures of the nine states in the bacteriorhodopsin photocycle. Biochemistry. 2004;43:3–8. doi: 10.1021/bi035843s. [DOI] [PubMed] [Google Scholar]
- 9.Maeda A, Morgan JE, Gennis RB, Ebrey TG. Water as a cofactor in the unidirectional light-driven proton transfer steps in bacteriorhodopsin. Photochem Photobiol. 2006;82:1398–1405. doi: 10.1562/2006-01-16-IR-779. [DOI] [PubMed] [Google Scholar]
- 10.Lanyi JK. Proton transfers in the bacteriorhodopsin photocycle. Biochim Biophys Acta. 2006;1757:1012–1018. doi: 10.1016/j.bbabio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Morgan JE, Gennis RB, Maeda A. A role for internal water molecules in proton affinity changes in the Schiff base and Asp85 for one-way proton transfer in bacteriorhodopsin. Photochem Photobiol. 2008;84:1038–1045. doi: 10.1111/j.1751-1097.2008.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Braiman MS, Mogi T, Marti T, Stern LJ, Khorana HG, Rothschild KJ. Vibrational spectroscopy of bacteriorhodopsin mutants. Light-driven proton transport involves protonation changes of aspartic acid residues 85, 96, and 212. Biochemistry. 1988;27:8516–8520. doi: 10.1021/bi00423a002. [DOI] [PubMed] [Google Scholar]
- 13.Heberle J, Dencher NA. Surface-bound optical probes monitor proton translocation and surface potential changes during the bacteriorhodopsin photocycle. Proc Natl Acad Sci USA. 1992;89:5996–6000. doi: 10.1073/pnas.89.13.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimányi L, Váró G, Chang M, Ni B, Needleman R, Lanyi JK. Pathway of proton release in the bacteriorhodopsin photocycle. Biochemistry. 1992;31:8535–8543. doi: 10.1021/bi00151a022. [DOI] [PubMed] [Google Scholar]
- 15.Smith SO, Pardoen JA, Mulder PPJ, Curry B, Lugtenburg J, Mathies R. Chromophore structure in bacteriorhodopsin's O640 photointermediate. Biochemistry. 1983;22:6141–6148. [Google Scholar]
- 16.Cao Y, Brown LS, Sasaki J, Maeda A, Needleman R, Lanyi JK. Relationship of proton release at the extracellular surface to deprotonation of the Schiff base in the bacteriorhodopsin photocycle. Biophys J. 1995;68:1518–1530. doi: 10.1016/S0006-3495(95)80324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balashov SP, Lu M, Imasheva ES, Govindjee R, Ebrey TG, Othersen B, III, Chen Y, Crouch RK, Menick DR. The proton release group of bacteriorhodopsin controls the rate of the final step of its photocycle at low pH. Biochemistry. 1999;38:2026–2039. doi: 10.1021/bi981926a. [DOI] [PubMed] [Google Scholar]
- 18.Balashov SP, Govindjee R, Imasheva ES, Misra S, Ebrey TG, Feng Y, Crouch RK, Menick DR. The two pKa's of aspartate-85 and control of thermal isomerization and proton release in the arginine-82 to lysine mutant of bacteriorhodopsin. Biochemistry. 1995;34:8820–8834. doi: 10.1021/bi00027a034. [DOI] [PubMed] [Google Scholar]
- 19.Richter HT, Brown LS, Needleman R, Lanyi JK. A linkage of the pKa's of asp-85 and glu-204 forms part of the reprotonation switch of bacteriorhodopsin. Biochemistry. 1996;35:4054–4062. doi: 10.1021/bi952883q. [DOI] [PubMed] [Google Scholar]
- 20.Balashov SP, Govindjee R, Kono M, Imasheva ES, Lukashev E, Ebrey TG, Crouch RK, Menick DR, Feng Y. Effect of arginine-82 to alanine mutation in bacteriorhodopsin on dark adaptation, proton release, and the photochemical cycle. Biochemistry. 1993;32:10331–10343. doi: 10.1021/bi00090a008. [DOI] [PubMed] [Google Scholar]
- 21.Braiman MS, Dioumaev AK, Lewis JR. A large photolysis-induced pKa increase of the chromophore counterion in bacteriorhodopsin: Implications for ion transport mechanism of retinal proteins. Biophys J. 1996;70:939–947. doi: 10.1016/S0006-3495(96)79637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown LS, Sasaki J, Kandori H, Maeda A, Needleman R, Lanyi JK. Glutamic acid 204 in the terminal proton release group at the extracellular surface of bacteriorhodopsin. J Biol Chem. 1995;270:27122–27126. doi: 10.1074/jbc.270.45.27122. [DOI] [PubMed] [Google Scholar]
- 23.Govindjee R, Kono M, Balashov SP, Imasheva E, Sheves M, Ebrey TG. Effects of substitution of tyrosine 57 with asparagine and phenylalanine on the properties of bacteriorhodopsin. Biochemistry. 1995;34:4828–4838. doi: 10.1021/bi00014a040. [DOI] [PubMed] [Google Scholar]
- 24.Govindjee R, Misra S, Balashov SP, Ebrey TG, Crouch RK, Menick DR. Arginine-82 regulates the pKa of the group responsible for the light-driven proton release in bacteriorhodopsin. Biophys J. 1996;71:1011–1023. doi: 10.1016/S0006-3495(96)79302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balashov SP, Imasheva ES, Ebrey TG, Chen N, Menick DR, Crouch RK. Glutamate-194 to cysteine mutation inhibits fast light-induced proton release in bacteriorhodopsin. Biochemistry. 1997;36:8671–8676. doi: 10.1021/bi970744y. [DOI] [PubMed] [Google Scholar]
- 26.Garczarek F, Brown LS, Lanyi JK, Gerwert K. Proton binding within a membrane protein by a protonated water cluster. Proc Natl Acad Sci USA. 2005;102:3633–3638. doi: 10.1073/pnas.0500421102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439:105–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- 28.Hutson MS, Alexiev U, Shilov S, Wise KJ, Braiman MS. Evidence for a perturbation of arginine-82 in the bacteriorhodopsin photocycle from time-resolved infrared spectra. Biochemistry. 2000;39:13189–13200. doi: 10.1021/bi000426q. [DOI] [PubMed] [Google Scholar]
- 29.Xiao U, Hutson MS, Belenky M, Herzfeld J, Braiman MS. Role of arginine-82 in fast proton release during the bacteriorhodopsin photocyle: a time-resolved FT-IR study of purple membranes containing 15N-labeled arginine. Biochemistry. 2004;43:12809–12818. doi: 10.1021/bi049238g. [DOI] [PubMed] [Google Scholar]
- 30.Morgan JE, Vakkasoglu AS, Lugtenburg J, Gennis RB, Maeda A. Structural changes due to the deprotonation of the proton release group in the M-photointermediate of bacteriorhodopsin as revealed by time-resolved FTIR spectroscopy. Biochemistry. 2008;47:11598–11605. doi: 10.1021/bi801405v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanogami J, Kikukawa T, Miyauchi S, Muneyuki E, Kamo N. A tin oxide transparent electrode provide the means for rapid time-resolved pH measurements: Application to photoinduced proton transfer of bacteriorhodopsin and proteorhodoposin. Photochem Photobiol. 2009;85:578–589. doi: 10.1111/j.1751-1097.2008.00520.x. [DOI] [PubMed] [Google Scholar]
- 32.Brown LS, Váró G, Hatanaka M, Sasaki J, Kandori H, Maeda A, Friedman N, Sheves M, Needleman R, Lanyi JK. The complex extracellular domain regulates the deprotonation and reprotonation of the retinal Schiff base during the bacteriorhodopsin photocycle. Biochemistry. 1995;34:12903–12914. doi: 10.1021/bi00039a053. [DOI] [PubMed] [Google Scholar]
- 33.Needleman R, Chang M, Ni B, Váró G, Fornes J, White SH, Lanyi JK. Properties of Asp212→Asn bacteriorhodopsin suggest that Asp212 and Asp85 both participate in a counterion and proton acceptor complex near the Schiff base. J Biol Chem. 1991;266:11478–11484. [PubMed] [Google Scholar]
- 34.Cao Y, Váró G, Klinger AL, Czaikowsky DM, Braiman MS, Needleman R, Lanyi JK. Proton transfer from Asp-96 to the bacteriorhodopsin Schiff base is caused by a decrease of the pKa of Asp96 which follows a protein backbone conformational change. Biochemistry. 1993;32:1981–1990. doi: 10.1021/bi00059a015. [DOI] [PubMed] [Google Scholar]
- 35.Moltke S, Krebs MP, Mollaaghababa R, Khorana HG, Heyn MP. Intramolecular charge transfer in the bacteriorhodopsin mutants Asp85→Asn and Asp212→Asn: Effects of pH and anions. Biophys J. 1995;69:2074–2083. doi: 10.1016/S0006-3495(95)80078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oesterhelt D, Stoeckenius W. Isolation of cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 37.Morgan JE, Vakkasoglu AS, Gennis RB, Maeda A. Water structural changes in the L and M of bacteriorhodopsin as revealed by time-resolved step-scan Fourier transform infrared (FTIR) spectroscopy. Biochemistry. 2007;46:2787–2796. doi: 10.1021/bi0616596. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braiman MS, Ahl PL, Rothschild KJ. Millisecond Fourier-transform infrared difference spectra of bacteriorhodopsin's M412 photoproduct. Proc Natl Acad Sci USA. 1987;84:5221–5225. doi: 10.1073/pnas.84.15.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Provencher SW, Vogel RH. Regularization Techniques for Inverse Problems in Molecular Biology. In: Deufhard P, Hairer E, editors. Numerical Treatment of Inverse Problems in Differential and Integral Equations. Birkhäuser; Boston, MA: 1983. pp. 304–319. [Google Scholar]
- 40.Chizhov I, Chernavskii DS, Engelhard M, Mueller KH, Zubov BV, Hess B. Spectrally silent transitions in the bacteriorhodopsin photocylce. Biophys J. 1996;71:2329–2345. doi: 10.1016/S0006-3495(96)79475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rödig C, Chizhov I, Weidlich O, Siebert F. Time-resolved step-scan Fourier transform infrared spectroscopy reveals differences between early and late M intermediates of bacteriorhodopsin. Biophys J. 1999;76:2687–2701. doi: 10.1016/S0006-3495(99)77421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown LS, Dioumaev AK, Needleman R, Lanyi JK. Connectivity of the protonated Schiff base to Asp85 and Asp96 during the bacteriorhodopsin photocycle: the local access model. Biophys J. 1998;75:1455–1465. doi: 10.1016/S0006-3495(98)74064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zscherp C, Schlesinger R, Tittor J, Oesterhelt D, Heberle J. In situ determination of transient pKa changes of internal amino acids of bacteriorhodopsin by using time-resolved attenuated total reflection Fourier transform infrared spectroscopy. Proc Natl Acad Sci USA. 1999;96:5498–5503. doi: 10.1073/pnas.96.10.5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf S, Freier E, Gerwert K. How does a membrane protein achieve a vectorial proton transfer via water molecules? ChemPhysChem. 2008;9:2772–2778. doi: 10.1002/cphc.200800703. [DOI] [PubMed] [Google Scholar]
- 45.Rammelsberg R, Huhn G, Lübben M, Gerwert K. Bacteriorhodopsin's intramolecular proton-release pathway consists of a hydrogen-bonded network. Biochemistry. 1998;37:5001–5009. doi: 10.1021/bi971701k. [DOI] [PubMed] [Google Scholar]
- 46.Garczarek F, Wang J, E-Sayed MA, Gerwert K. The assignment of the different infrared continuum absorbance changes observed in the 3000—1800- cm-1 region during the bacteriorhodopsin photocycle. Biophys J. 2004;87:2676–2682. doi: 10.1529/biophysj.104.046433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sasaki J, Shichida Y, Lanyi JK, Maeda A. Protein changes associated with reprotonation of the Schiff base in the photocycle of Asp96→Asn bacteriorhodopsin. J Biol Chem. 1992;267:20782–20786. [PubMed] [Google Scholar]
- 48.Hessling B, Souvignir G, Gerwert K. A model-independent approach to assigning bacteriorhodopsin's intramolecular reactions to photocycle intermediates. Biophys J. 1993;65:1929–1941. doi: 10.1016/S0006-3495(93)81264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith SO, Braiman MS, Myers AB, Pardoen JA, Courtin JML, Winkel C, Lugtenburg J, Mathies RA. Vibrational analysis of the all-trans retinal chromophore in light-adapted bacteriorhodopsin. J Am Chem Soc. 1987;109:3108–3125. [Google Scholar]
- 50.Maeda A, Sasaki J, Pfefferle JM, Shichida Y, Yoshizawa T. Fourier transform infrared spectral studies on the Schiff base mode of all-trans bacteriorhodopsin and its photointermediates, K and L. Photochem Photobiol. 1991;54:911–921. [Google Scholar]
- 51.Hessling B, Herbst J, Rammelsberg R, Gerwert K. Fourier transform infrared double-flash experiments resolve bacteriorhodopsin's M1 and M2 transition. Biophys J. 1997;73:2071–2080. doi: 10.1016/S0006-3495(97)78237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Headrick JM, Diken EG, Walters RS, Hammer NI, Christie RA, Cui J, Myshakin EM, Duncan MA, Johnson MA, Jordan KD. Spectral signatures of hydrated proton vibrations in water clusters. Science. 2005;308:1765–1769.55. doi: 10.1126/science.1113094. [DOI] [PubMed] [Google Scholar]
- 53.McCunn LR, Roscioli JR, Johnson MA, McCoy AB. An H/D isotopic substitution study of the HO•Ar vibrational predissociation spectra: Exploring the putative role of Fermi resonances in the bridging proton fundamentals. J Phys Chem B. 2008;112:321–327. doi: 10.1021/jp075289m. [DOI] [PubMed] [Google Scholar]
- 54.Petkova AT, Hu JG, Bizounok M, Simpson M, Griffin RG, Herzfeld J. Arginine activity in the proton-motive photocycle of bacteriorhodopsin: Solid-state NMR studies of the wild-type and D85N proteins. Biochemistry. 1999;38:1562–1572. doi: 10.1021/bi981968z. [DOI] [PubMed] [Google Scholar]
- 55.Xiao Y, Braiman M. Modeling amino acid side chain in proteins: 15N NMR of guanidine groups in nonpolar environments. J Phys Chem B. 2005;109:16953–16958. doi: 10.1021/jp051279e. [DOI] [PubMed] [Google Scholar]
- 56.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 Å resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 57.DeLano W. PYMOL. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 58.Hatanaka M, Sasaki J, Kandori H, Ebrey TG, Needleman R, Lanyi JK, Maeda A. Effects of arginine-82 on the interactions of internal water molecules in bacteriorhodopsin. Biochemistry. 1995;34:6308–6312. doi: 10.1021/bi952973v. [DOI] [PubMed] [Google Scholar]
- 59.Liu XM, Sonar S, Lee CP, Coleman M, RajBhandary UL, Rothschild KJ. Site-directed isotope labeling and FTIR spectroscopy: assignment of tyrosine bands in the bR → M difference spectrum of bacteriorhodopsin. Biophys Chem. 1995;56:63–70. doi: 10.1016/0301-4622(95)00016-q. [DOI] [PubMed] [Google Scholar]
- 60.Nevskaya NA, Chirgadze UN. Infrared spectra and resonance interactions of amide-I and II vibrations of α-helix. Biopolymers. 1976;15:637–648. doi: 10.1002/bip.1976.360150404. [DOI] [PubMed] [Google Scholar]
- 61.Barth A, Zscherp C. What vibrations tells us about proteins. Quart Rev Biophys. 2002;35:369–430. doi: 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- 62.Tanimoto T, Shibata M, Belenky M, Herzfeld J, Kandori H. Altered hydrogen bonding of Arg82 during the proton pump cycle of bacteriorhodopsin: a low-temperature polarized FTIR spectroscopic study. Biochemistry. 2004;43:9439–9447. doi: 10.1021/bi049368p. [DOI] [PubMed] [Google Scholar]
- 63.Uhmann W, Becker A, Taran C, Siebert F. Time-resolved FT-IR absorption spectroscopy using a step-scan interferomer. Appl Spectrosc. 1991;45:390–397. [Google Scholar]
- 64.Maeda A, Tomson FL, Gennis RB, Kandori H, Ebrey TG, Balashov SP. Relocation of internal water in bacteriorhodopsin during the photoreaction of M at low temperatures: An FTIR study. Biochemistry. 2000;39:10154–10162. doi: 10.1021/bi000190q. [DOI] [PubMed] [Google Scholar]
- 65.Hatanaka M, Kashima R, Kandori H, Friedman N, Sheves M, Needleman R, Lanyi JK, Maeda A. Trp86 → Phe replacement in bacteriorhodopsin affects a water molecule near Asp85 and light adaptation. Biochemistry. 1997;36:5493–5498. doi: 10.1021/bi970081k. [DOI] [PubMed] [Google Scholar]
- 66.Metz G, Siebert F, Engelhard M. High-resolution solid state 13C NMR of bacteriorhodopsin: characterization of [4-13C] resonances. Biochemistry. 1992;31:455–461. doi: 10.1021/bi00117a022. [DOI] [PubMed] [Google Scholar]
- 67.Száraz S, Oesterhelt D, Ormos P. pH-induced structural changes in bacteriorhodopsin studied by Fourier transform infrared spectroscopy. Biophys J. 1994;67:1706–1712. doi: 10.1016/S0006-3495(94)80644-7. [DOI] [PMC free article] [PubMed] [Google Scholar]