

Abstract
The isolation and structure elucidation of three new secondary metabolites, chaetoglobosin-510 (1), -540 (2), and -542 (3), are described. These compounds were produced by cultures of the marine-derived fungus Phomopsis asparagi, challenged with the known F-actin inhibitor jasplakinolide. Chaetoglobosin-542 (3) displayed antimicrofilament activity and was cytotoxic toward murine colon and leukemia cancer cell lines.
The quest to exploit factors leading to the production of diverse molecular structures from cultured microorganisms represents a continuing challenge for natural products research.1 Noteworthy achievements in this area have been made through the application of insights derived from chemical genetics to manipulate synthases resulting in the isolation of previously unknown polyketide structures.2,3 The OSMAC (one strain many compounds) paradigm represents a less complex approach involving the systematic alteration of culture conditions in order to generate new metabolites.4 Encouraging results leading to the modification of secondary metabolites have been obtained by employing indigenous enzymes of fungi as biotransformation agents.5 Similarly, cytochrome P-450 enzymes have been used to selectively induce oxygenation in fungal-derived products.6 Other methodologies involve varying environmental factors such as nutrients4 or temperature7 to alter secondary metabolic processes during fungal culturing.
The increased attention being given to the culturing of marine-derived fungi has further expanded the diversity of structures that can be obtained. An interesting application of the OSMAC strategy is the production of new metabolites by varying seawater concentration during culturing of marine-derived fungal strains commonly encountered from terrestrial sources.8 In this regard, it is not surprising that the metabolite profiles of chemically prolific terrestrial fungal strains are modulated when regrown in culture medium using seawater as observed with Coriolus consors.9 Another milestone finding was the production of a new potent antibiotic, pestalone, from a vigorously growing seawater culture of marine-derived Pestalotia sp. inoculated with a marine-derived bacterium.10
Our previous attempts to induce a biosynthetic shift in the natural products observed from chemically prolific fungi by varying conditions of the seawater-based cultures have been only modestly successful. For example, the use of harsh conditions, such as the addition of Cu2+ salts to cultures of Myrothecium verrucaria, did not significantly alter its production of trichoverroids and related macrolides.11 As a next step, we considered challenging cultures with potent cytoskeletal inhibitors. A strain of Phomopsis asparagi obtained from a U.S. Virgin Islands collection of the sponge Rhaphidophlus juniperina was chosen for these studies. The Phomopsis genus includes both marine- and terrestrial-derived species, with several documented as plant pathogens.12 This genus is a rich source of secondary metabolites, diverse in their structures and biological activities.13 Recent examples include the phomoxanthones (with antimalarial, antitubercular, and cytotoxic properties) from Phomopsis sp.,13 phomopsidin (an antimicrotubule agent) from Phomospsis sp.,14 and chaetoglobosins (analogues of the cytochalasins known to target cytoskeletal processes)15 from Phomopsis leptostromiformis.16 Since none of these signature metabolites were observed in our seawater culture of P. asparagi, this strain seemed ideal for further study. In fact, the incubation of the fungus with the potent F-actin inhibitor jasplakinolide,17 first isolated18 from the sponge Jaspis splendens,19 resulted in the production of three new biologically active chaetoglobosin analogues, chaetoglobosin-510 (1), -540 (2), and -542 (3). The culture conditions used to isolate these compounds along with structure elucidation and an evaluation of their biological activities are described below.
Results and Discussion
Analyses of the LC-MS profiles of extracts obtained from small-scale (125 mL) cultures of P. asparagi (strain no. 031113a) were undertaken. These results, shown in Figure 1, clearly depict a dramatic shift in the profiles of the secondary metabolites stimulated by the addition of jasplakinolide. The untreated cultures appeared to be rich in polyketides of masses below 400 amu. While an exhaustive dereplication was not undertaken, examples of these compounds were provisionally identified as 4,6-dihydroxymellein (tR = 21.0 min),20 patulolide C (tR = 21.5 min),21 convolvulopyrone (tR = 24.0 min),22 culpin (tR = 24.5 min),23 phompsiol (tR = 28.0 min),24 and 4-hydroxymellein (tR = 34.0 min),25 as seen in the chromatogram A of Figure 1. Spiking the vigorously growing culture with jasplakinolide caused a dramatic shift in the biosynthetic profile, as shown by chromatogram B of Figure 1. A decrease in the intensity of the previously mentioned polyketides was observed, while new intense LC peaks appeared having [M + H]+ m/z values at 511, 541, and 543.
Figure 1.

Comparison of the ELSD trace of crude extract of P. asparagi culture conditions, untreated (A) vs inoculated with jasplakinolide (B).
To obtain milligram quantities of the new metabolites, scale-up cultures were grown (5 × 1 L) using methods and conditions outlined in the Experimental Section. The immediate goal was to characterize the new compounds and evaluate their biological properties. The MeOH-soluble portion of the combined broth and mycelia extract, shown by LC-MS to contain the compounds of interest, was purified by reversed-phase chromatography. This afforded the novel metabolites chaetoglobosin-510 (1), chaetoglobosin-540 (2), and chaetoglobosin-542 (3).
Chaetoglobosin-510 (1) has the molecular formula C34H42N2O2, requiring 15 degrees of unsaturation on the basis of the HRESIMS data, [M + H]+ 511.3374. Analysis of the 1H, 13C, and DEPT NMR data (Table 1) revealed the constitution of all 34 carbons: five methyls, two of which were vinylic; four methylenes; 17 methines, consisting of an ABCD aromatic system, two sets of trans-double bond coupled protons (H-13/14 J = 15.0 Hz and H-21/22 J = 15.5 Hz), three distinct vinyl resonances, and six aliphatics; and eight quaternaries with only one being sp3 hybridized (C-9, δC 69.0). These data were used to assemble the four substructures A–D, which revealed that 1 shared several structural similarities with the chaetoglobosin family of fungal metabolites16 and appeared to be the dimethyl derivative of prochaetoglobosin I (4).6 Consistent with this conclusion was the fact that 1 (as well as 2 and 3) had an intense ESIMS fragment ion m/z 144 (C10H10N) due to the extrusion of the indolyl-3-ethyl moiety, substructure A shown in Figure 2. The 2D NMR data provided the basis to verify the relationship of 1 to the known compound 4. Confirmation of substructure A was provided by complementary sets of data. Important gHMBC correlations were observed from H3-10′ to C-3, C-3′, and C-10 and from H-10 to C-2′, C-3, C-3′, C-3a′, and C-10′ (Table 1, Figure 2). The diagnostic 13C chemical shifts for the indole ring system were also identified (Table 1). Substructure B, comprised of atoms C5H3O2N, was visualized by assigning an NCH group (C-3, δC 59.3, δH 3.35) with gHMBC correlations to the amide carbonyl (C-1, δC 176.7) and to the quaternary sp3 carbon (C-9, δC 67.0). This proton (H-3) also displayed a vicinal coupling (J = 2.5) to H-4 (δH 2.95), which in turn exhibited gHMBC correlations to C-1, C-23 (δC 198.8, a conjugated ketone), and C-9. Substructure C was constructed using the allylic and homoallylic protons as a starting point. The methylene protons H2-11 exhibited gHMBC correlations to C-5 (δC 42.9), C-6 (δC 138.7), and C-11′ (δC 11.9) (Table 1, Figure 2). Other notable gHMBC correlations were observed from H-5 to C-6 and C-7, from H3-12 to C-5, C-6, and C-7, and from H-8 to C-7 (Table 1). Finally, the two sets of vicinally coupled spin systems interrupted by a vinyl methyl comprising substructure D were established through the TOCSY data (H-7 through H-17 and H2-19 through H-22) shown in Figure 3 and by the gHMBC and gCOSY correlations outlined in Figure 2.
Table 1. NMR Data for Chaetoglobosin-510 (1), -540 (2), and -542 (3) in MeOHa.
| position | δC | δH (J in Hz) | gHMBCb | δC | δH (J in Hz) | gHMBCb | δC | δH (J in Hz) | gHMBCb |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 176.6(C) | 173.7(C) | 175.7(C) | ||||||
| 3 | 59.3 (CH) | 3.35 (dd, 7.0, 2.5) | 1, 3′, 5, 9, 10 | 58.6 (CH) | 3.35 (t, 3.5) | 59.2 (CH) | 3.40 (dd, 6.5, 2.0,) | 1, 3′, 4, 10 | |
| 4 | 45.9 (CH) | 2.95 (dd, 5.5, 2.0) | 1, 5, 6, 9, 10, 23 | 44.3 (CH) | 3.25 (dd, 5.0, 3.5) | 46.6 (CH) | 2.86 (dd, 5.5, 2.0) | 1, 3, 5, 10, 23 | |
| 5 | 42.9 (CH) | 2.03 (m) | 4,6,7, 11 | 42.8 (CH) | 2.08 (m) | 3.1 (CH) | 2.08 (m) | ||
| 6 | 138.7(C) | 140.6(C) | 138.6(C) | ||||||
| 7 | 127.7 (CH) | 5.30 (m) | 125.9 (CH) | 5.28 (m) | 126.7 (CH) | 5.35 (m) | |||
| 8 | 46.2 (CH) | 2.80 (m) | 7, 13 | 47.0 (CH) | 2.55 (m) | 46.5 (CH) | 2.88 (m)c | 6, 9 | |
| 9 | 67.1(C) | 66.9(C) | 67.5(C) | ||||||
| 10 | 36.6 (CH) | 2.88 (p, 7.0) | 2′, 3, 3′, 3a′, 10′ | 35.8 (CH) | 3.10 (dq, 7.5, 3.5) | 10′ | 36.8 (CH) | 2.89 (p, 7.0) | 3′ |
| 10′ | 15.1 (CH3) | 1.22 (d, 7.0) | 3, 3′, 10 | 13.8 (CH3) | 1.20 (d, 7.5) | 3′, 3 | 15.6 (CH3) | 1.24 (d, 7.5) | 3′, 3, 10 |
| 11 | 20.4 (CH2) | 1.18 (m) | 5, 6, 11′ | 21.0 (CH2) | 1.60 (m) | 11′ | 20.5 (CH2) | 1.20 (m) | 11′ |
| 1.69 (m) | 5, 11′ | 1.90 (m) | 11′ | 1.78 (m) | 11′ | ||||
| 11′ | 11.9 (CH3) | 0.70 (t, 7.2) | 5, 11 | 12.2 (CH3) | 1.10 (t, 7.0) | 5, 11 | 12.0 (CH3) | 0.68 (t, 7.0) | 5, 11 |
| 12 | 18.2 (CH3) | 1.71 (d, 1.5) | 5,6,7 | 18.5 (CH3) | 1.80 (d, 1.5) | 5, 6, 7 | 18.2 (CH3) | 1.72 (br s) | 6, 7 |
| 13 | 128.5 (CH) | 6.09 (ddd, 15.0, 10.0, 2.0) | 130.7 (CH) | 5.94 (ddd, 15.0, 10.0, 1.5) | 128.4 (CH) | 6.00 (ddd, 15.5, 9.0, 1.5) | |||
| 14 | 133.2 (CH) | 5.12 (ddd, 15.0, 10.0, 3.0) | 15 | 130.8 (CH) | 5.08 (ddd, 15.0, 11.0, 4.0) | 133.4 (CH) | 5.12 (ddd, 15.5, 9.5, 3.0) | ||
| 15 | 40.0 (CH2) | 1.79 (m) | 17 | 41.7 (CH2) | 1.90 (m) | 16 | 39.9 (CH2) | 1.74 (m) | |
| 2.23 (m) | 17 | 2.21 (ddd, 13.5, 4.0, 3.5) | 2.18 (m) | ||||||
| 16 | 33.0 (CH) | 2.49 (m) | 31.8 (CH) | 2.43 (m) | 31.8 (CH) | 2.50 (m) | |||
| 16′ | 20.5 (CH3) | 0.9 (d, 6.6) | 15, 16, 17 | 19.8 (CH3) | 0.90 (d, 7.0) | 15, 16, 17 | 20.2 (CH3) | 0.90 (d, 6.5) | 15, 16 |
| 17 | 132.6 (CH) | 4.78 (dq, 10.0, 1.5) | 16, 19 | 139.9 (CH) | 5.51 (dq, 9.0, 1.5) | 19 | 134.1 (CH) | 4.80 (m)c | 19 |
| 18 | 130.8 (C) | 132.5(C) | 132.9(C) | ||||||
| 18′ | 15.1 (CH3) | 1.54 (d, 1.5) | 17 | 9.3 (CH3) | 1.30 (d, 1.5) | 17, 18, 19 | 12.0 (CH3) | 1.64 (d, 1.0) | 18, 19 |
| 19 | 37.0 (CH2) | 2.20 (m) | 18, 21 | 81.2 (CH) | 4.99 (s) | 17, 18, 18′, 20 | 79.2 (CH) | 4.05 (d, 3.5) | |
| 20 | 28.9 (CH2) | 2.32 (m) | 18, 21, 22 | 200.7(C) | 75.3 (CH) | 4.47 (ddd, 5.0, 3.5, 1.5) | |||
| 21 | 146.6 (CH) | 6.72 (dt, 15.5, 7.5) | 20, 22,23 | 131.0 (CH) | 6.20 (d, 17.0) | 19, 22, 23 | 146.1 (CH) | 6.65 (dd, 15.5, 5.0) | 20 |
| 22 | 126.8 (CH) | 6.58 (d, 15.5) | 20, 21 | 136.3 (CH) | 7.60 (d, 17.0) | 20,23 | 125.1 (CH) | 6.32 (dd, 15.5, 2.0) | 1 |
| 23 | 198.8(C) | 198.5 (C) | 199.5 (C) | ||||||
| 2′ | 121.8 (CH) | 6.97 (s) | 7a′, 2′,3a′ | 121.0 (CH) | 6.95 (s) | 3′ | 118.4 (CH) | 6.95 (s) | 3′, 7a′ |
| 3′ | 116.0(C) | 115.4(C) | 116.2(C) | ||||||
| 3a′ | 126.2(C) | 126.3(C) | 126.7(C) | ||||||
| 4′ | 118.3 (CH) | 7.42 (dt, 8.0, 1.0) | 7a′, 3a′, 5′, 6′ | 118.4 (CH) | 7.45 (dd, 8.0, 1.0) | 7a′, 5′ | 118.3 (CH) | 7.42 (brd, 8.0) | 6′, 7a′ |
| 5′ | 118.3 (CH) | 6.92 (ddd, 8.0, 7.0, 1.0) | 6′, 7′ | 122.3 (CH) | 6.94 (ddd, 8.0, 7.0, 1.0) | 3a′ | 122.0 (CH) | 6.96 (ddd, 8.0, 7.0, 1.0) | |
| 6′ | 120.9 (CH) | 7.05 (ddd, 8.0, 7.0, 1.0) | 7a′, 5′ | 118.4 (CH) | 6.96 (ddd, 8.0, 7.0, 1.0) | 7a′ | 120.9 (CH) | 7.15 (ddd, 8.0, 7.0, 1.0) | 7a′ |
| 7′ | 111.0 (CH) | 7.30 (dt, 8.0, 1.0) | 5′, 6′ | 111.2 (CH) | 7.20 (dd, 8.0, 1.0) | 6′ | 110.0 (CH) | 7.30 (brd, 8.0) | 2′ |
| 7a′ | 136.7 (C) | 136.9 (C) | 136.7 (C) |
Measured at 500 MHz (1H) and 125.7 MHz (13C).
Denotes proton correlating to carbon.
Determined by gCOSY and gHMBC correlation.
Figure 2.
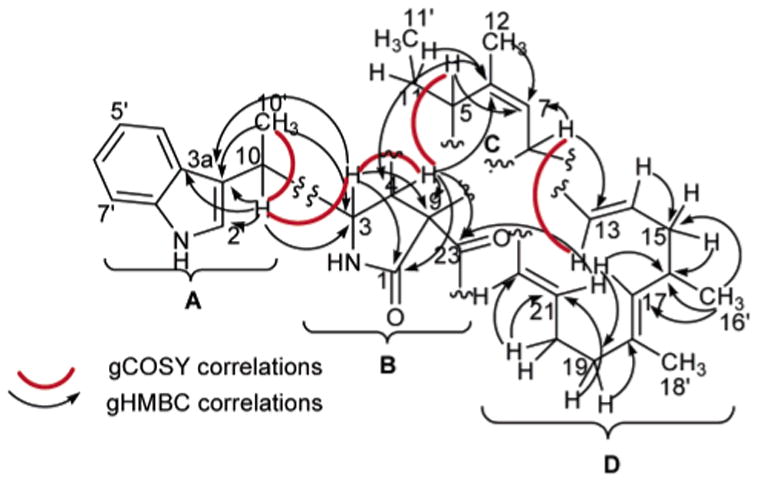
Substructures A–D with selected gHMBC and gCOSY correlations for 1.
Figure 3.

Portions of 1 confirmed by TOCSY.
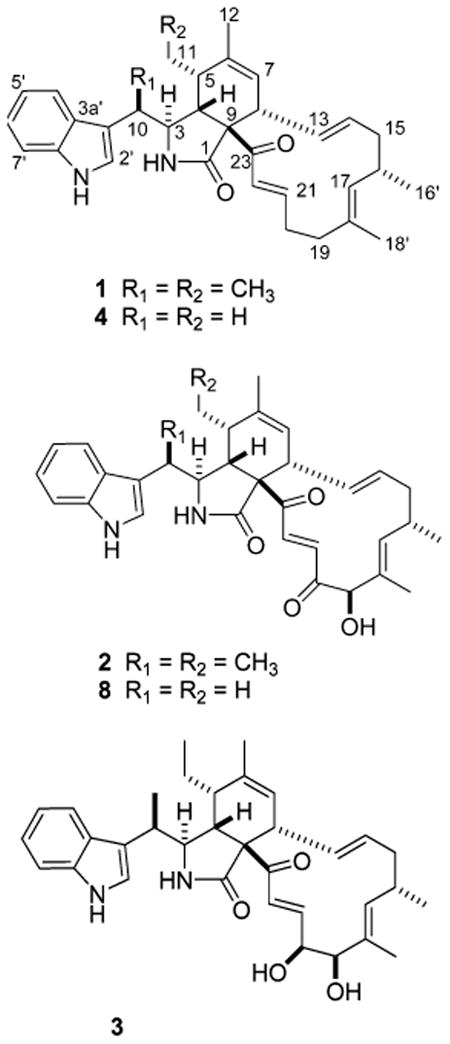
Two strategies were used to make connections between the four substructures A–D to complete the structure of 1. Dereplication based on search seeds consisting of substructure A and formulas C32-34H38-42N2O2-5 revealed 4 and 7 as relevant hits. The major differences in the NMR properties of 1 and 4 were in the regions of C-10 and C-11. Preliminary data to linking A, B, and C were based on gHMBC and gCOSY correlations. The most important included H-10 and H3-10′, both showing correlations to C-3, H-3 to C-5, and H-4 to C-10 and C-6 (Figure 2). The final fragment, D, could be added to complete the planar structure based on the gHMBC correlation from H-21 to C-23 (Table 1, Figure 2) and the gCOSY cross-peak between H-8 and H-13.
The configuration at C-3, C-4, C-5, C-8, C-9, and C-16 of prochaetoglobosin I (4)6 and chaetoglobosin A (5)26 was retained in 1. Data in support of this conclusion included parallel NMR chemical shifts for these three compounds and strong NOEs observed between H-5 and H-8 of 1. This result, along with the lack of observed NOEs between H-4 and H-8, could be explained by a boatlike conformation of the cyclohexene ring placing H-5 and H-8 in a proximal position, as also observed in 4.26,27 The irradiation of H-3 resulted in NOE correlations to H2-11a (δH 1.18) and H3-12 (δH 1.71). Additional NOEs were observed from H-16 to H-14, H-17, H-18′, and H2-19. The similar NMR chemical shifts in the vicinity of C-5, C-10, and C-11 for 1 (Table 1) as compared to that of chaetoglobosins K (6)27 and O (7)16 were used to assign the C-10 methyl and C-5 ethyl configurations shown in 1.
A similar approach, as described above, was used to assign the structure of chaetoglobosin-540 (2). Its molecular formula of C34H40N2O4, based on the HRESIMS [M + H]+ m/z 541.3085 and requiring 16 double bond equivalents, differed from that of 1 by 30 amu and can be rationalized by +O2 and −H2. Relative to the NMR data set of 1, the new features for 2 included an oxymethine (δC 81.2, δH 4.99 s) and a second unsaturated ketone (δC 200.7). Key gHMBC correlations were observed from H-19 to C-17, C-18, C-18′, and C-20 and from H-22 (δH 7.60 d, 17.0) to carbonyls C-20 and C-23. Another important observation was that 2 possessed two extra methyl groups relative to the structure of chaetoglobosin J (8).28 Once this comparison was made, it was evident that the stereocenters of 2 and 8 were identical, with the only differences in their NMR shifts occurring at C-10 and C-5 (Table 1). Comparison of the 13C NMR data to that of 1 formed the basis of the stereochemical assignments at these two chiral centers. The essential NOESY results were as expected and consistent with the configurations proposed at C-3, C-4, C-8, and C-16. The diagnostic NOE expected between H-5 and H-8 was also observed. Finally, the δ values for C-19 oxymethine (δC 81.2, δH 4.99) of 2 were nearly identical to those at the same position for 5 (δC 81.7, δH 5.01),26,29 9 (δC 81.4, δH 5.09),29 and 10 (δC 81.4, δH 5.09).29
The structural features of chaetoglobosin-542 (3), with a molecular formula of C34H42N2O4 based on HRESIMS [M + H]+ 543.3179, were similar to those of 2. The mass difference of 2 amu between these two compounds indicated an addition of two hydrogen atoms, with a vicinal diol now present at C-19–C-20. This was substantiated by the NMR data for C-19 (δC 79.2, δH 4.05) and C-20 (δC 75.3, δH 4.47) and by the fact that these protons were mutually coupled (J = 3.5 Hz). Also, the multiplet for H-20 (J = 5.0, 3.5, 1.5 Hz) included a 4J = 1.5 Hz coupling to H-22 (Table 1). Further diagnostic NMR features were gHMBC correlations from H3-18′ to C-19 and from H-21 (δH 6.65) to C-20. As noted above the chemical shifts at each chiral center, except C-19 and C-20, were used to show a similar mapping of chirality among 1–3. The 3J of 3.5 Hz between H-19 and H-20 reflects the β configuration proposed for both OH groups and is consistent with the expected syn diol relationship (calc H–H dihedral angle of 47.4°). This also extends the β C-19 OH theme observed in all other chaetoglobosins containing such substitution.
The members of the chaetoglobosin family possess structures that are chirally dense at six vicinal positions (C-3, C-4, C-5, C-8, C-9, C-16) and one remote site (C-10). Interestingly, the sign of the rotation is not sensitive to the minor structural changes. For example, the new chaetoglobosins-510 (1), -540 (2), and -542 (3) reported here all displayed negative specific rotations, [α]25D −100°, −118°, and −44°, respectively. Similarly, chaetoglobosins A (5),26 Q (9), R (10), and T (11),29 prochaetoglobosin I (4),6 and isoprochaetoglobosin I (12)6 all display a negative specific rotation, despite a large variance in structure at C-6, C-7, C-10, and C-20 and configuration at C-5 and C-8. Biogenetic insights previously published30,31 have suggested that the configuration at C-4, C-5, C-8, and C-9 is set through a stereospecific enzyme-mediated Diels–Alder reaction.31 The NMR parameters relevant for defining variations in the configuration in this region consist of 3J4-5 = 2.5 Hz, 3J7-8 = 2.0 Hz, and 3J8-13 = 10 Hz for the orientation present in 1-4. Reflecting the changes at these sites of isoprochaetoglobosin (12)6 are 3J4-5 = 6.3 Hz and 3J8-13 = 6.1 Hz.
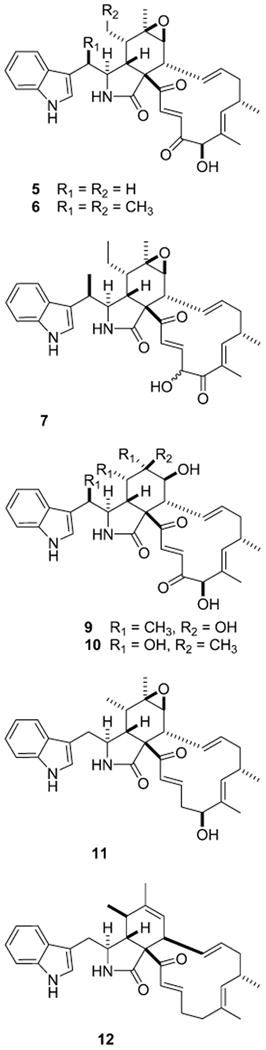
It has been shown that 2S,3R-3-methyltryptophan is the biosynthetic moiety incorporated in chaetoglobosin K (6) and as such is the origin of the 10R,3S configuration observed for compounds 6, 7, and 9.30,31 By analogy to this circumstance, we suggest an identical absolute configuration at C-3 and C-10 for compounds 1–3. Furthermore, the biogenetic similarity between the cytochalasins and the chaetoglobosins30 provides an additional handle to assign the absolute configuration of the multiple stereocenters. In particular, the X-ray results for isozygosporin A32 provide the basis for provisional assignment of centers 3S, 4R, 5S, 8R, 9R, 10R, and 16S for compounds 1–3.
The cytotoxicity profiles of 1, 2, and 3 observed against various murine cancer cell lines are given in Table 2.33 Compound 3 displayed the most cytotoxicity but nonselective activity against murine leukemia (L1210) and murine colon (C38) cancer cell lines; 3 was also toxic toward murine normal bone marrow (CFU-GM). Compound 2 was only moderately toxic toward the cell lines used, while compound 1 was virtually inactive. This activity pattern was also mirrored in the cytoskeletal assays, as 3 was the most potent and disrupted actin microfilaments at 1 μg/mL with eventual cell loss at 10 μg/mL (Table 2).34 On the basis of results in both the disk diffusion soft agar and the antimicrofilament assays, oxygenation at C-19 and/or C-20 appears to be required for maximal potency.
Table 2. Cytotoxicity and Antimicrofilament Data of 1, 2, and 3.
| cytotoxicitya,b | antimicrofilament activityc | ||||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||
| L1210 | C38 | CFU-GM | 1 μg/mL | 2.5 μg/mL | 5 μg/mL | 10 μg/mL | 15 μg/mL | 25 μg/mL | |
| 1 | 150 | +Δ | ++ΔΔ | ++++ΔΔ | |||||
| 2 | 250 | 350 | 250 | ND | ND | ND | ND | ND | ND |
| 3 | 500 | 400 | 500 | + | ++ | ++++ | |||
Cytotoxicity quoted in zone units.33 L1210 = murine leukemia, C38 = murine colon, CFU-GM = normal bone marrow.
Concentration of analyte 15 μg/disk.
Assays performed in A-10 cells (rat smooth muscle cells).34 + Loss of microfilament. ++Additional loss of microfilament. ++++Total loss of microfilament network. Δ Cell Loss. ND Not determined.
Conclusions
The chaetoglobosin family of fungal metabolites is a subset of the cytochalasins; they are well-known actin depolymerizers.15,35 The recent literature indicates that some members of the Phomopsis sp. and Chaetomium sp. are able to biosynthesize a variety of chaetoglobosins.16,29 The strategy of using an actin inhibitor to redirect a metabolite profile to produce chaetoglobosins is unprecedented. It is plausible that such measures may constitute a general approach to induce the production of novel metabolites from a variety of fungi. The biological properties shown here indicate the additional promise of C-19/C-20 oxygenated chaetoglobosins as molecular probes to study cytoskeletal proteins. In this regard, steps to broaden the array of active chaetoglobosins are ongoing in our laboratory. These involve the use of different actin inhibitors (e.g., latrunculin A)36 to shift secondary metabolite production in P. asparagi strains. The results of these studies will be disclosed in the future.
Experimental Section
General Experimental Procedures
All 1D and 2D NMR spectra were recorded in CD3OD except where noted on 500 MHz instruments. LC-MS was performed on a reversed-phase analytical column (4.6 × 250 mm, 5 μm) using a photodiode array (PDA) and evaporative light scattering detectors (ELSD) with an electrospray time-of-flight (ESI-TOF) mass spectrometer. High-resolution mass measurements were obtained on a benchtop ESI-TOF mass spectrometer. Optical rotation was determined on a Jasco DIP 370 polarimeter. UV spectra were recorded in MeOH on a Perkin-Elmer, Lambda 40 UV/vis spectrometer. Preparative and semipreparative HPLC were carried out on a C18 column (22 × 250 mm, 10 μm) or (10 × 250 mm, 4 μm). All HPLC solvents were of HPLC grade, while all other solvents were of ACS grade.
Biological Material
The fungus (strain no. 031113a) was isolated using previously described techniques11 from the sponge Rhaphidophlus juniperina (coll. no. 03111).37 This strain was phenotypically identified as Phomopsis sp. and confirmed as Phomopsis asparagi12 (<1% variance) on the basis of molecular analysis.11 The fungus is maintained as a cryopreserved glycerol stock at UCSC.
Preparative Scale Culture Conditions
The fungal strain was grown in a liquid medium. Cultures were prepared in (5 × 2 L) flasks, each containing 1 L of broth comprised of 3.5% Czapek-Dox and 0.5% yeast extract in Monterey Bay seawater adjusted to pH 7.40 (±0.2). The cultures were agitated at 150 rpm for 21 days at ambient temperature. Jasplakinolide (50 mg) was dissolved in DMF (2 mL) and used to inoculate the growing cultures after 2 days using a pulse feeding regiment (50% day 1, 50% day 2).
Extraction and Isolation
The broth and mycelia were extracted with EtOAc (3 × 1 L). The crude EtOAc extracts were combined and reduced in vacuo to yield a brown gum (1.05 g). After LC-MS analysis the crude extract was subjected to RP-HPLC (20–100% MeOH/0.1% formic acid–H2O), and 25 fractions were collected. Further purification of the three fractions eluted with 100% MeOH via reversed-phase chromatography yielded compounds 1 (15 mg), 2 (3 mg), and 3 (3 mg), as outlined in the isolation scheme (see Supporting Information S10).
Bioassay: Disk Diffusion Soft Agar Colony Formation Assay
An in vitro cell-based assay was employed to identify solid tumor selectivity in semipure fractions and pure compounds. The differential cytotoxicity33 is expressed by observing a zone differential between any solid tumor cell (Colon38, ColonH116, LungH125) and either leukemia cells (L1210 or CEM) or normal cells (CFU-GM). The sample is designated “solid tumor selective” if the zone unit of solid tumor–normal cell or leukemia cells is greater than 250 units.
Antimicrofilament
The microfilament-disrupting effects of the new chaetoglobosin analogues were evaluated in A-10 rat aortic smooth muscle cells. Cells were treated with the compounds and then fixed. The microfilaments were visualized using rhodamine-phalloidin.34
Chaetoglobolsin-510 (1)
white amorphous powder; [α]25D –100° (c 0.0245 MeOH); UV (MeOH) λmax nm (log ∊) 222 (4.37), 281 (3.57), 290 (3.50); HRESITOFMS m/z 511.3374 [M + H]+ (calcd for C34H43N2O2 511.3319); 1H NMR (500 MHz), see Table 1; 13C NMR (125 MHz), see Table 1.
Chaetogobosin-540 (2)
pale yellow amorphous powder; [α]25D –118° (c 0.009, MeOH); UV (MeOH) λmax nm (log ∊) 221 (4.43), 289 (3.36); HRESITOFMS m/z 541.3085 [M + H]+ (calcd for C34H41N2O4 541.3061); 1H NMR (500 MHz), see Table 1; 13C NMR (125 MHz), see Table 1.
Chaetoglobosin-542 (3)
white amorphous powder; [α]25D –44° (c 0.010, MeOH); UV (MeOH) λmax nm (log ∊) 222 (4.21), 281 (3.43), 290 (3.35); HRESITOFMS m/z 543.3179 [M + H]+ (calcd for C34H43N2O4, 543.3217); 1H NMR (500 MHz), see Table 1; 13C NMR (125 MHz), see Table 1.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health grant CA 47135 and the William Randolph Hearst Foundation. We thank B. Morinaka for providing samples of jasplakinolide used in this study.
Footnotes
Supporting Information Available: The 1H, 13C, and gCOSY NMR spectra for 1, 2, and 3 along with the isolation scheme and photographs of the disruption of the microfilament network in A-10 cells are provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Firn RD, Jones CG. Nat Prod Rep. 2003;20:382–391. doi: 10.1039/b208815k. [DOI] [PubMed] [Google Scholar]
- 2.(a) Cerda-Olmedo E. Crit Rev Micobiol. 1994;20:151–160. doi: 10.3109/10408419409113554. [DOI] [PubMed] [Google Scholar]; (b) Firn RD, Jones CG. Mol Microbiol. 2000;37:989–994. doi: 10.1046/j.1365-2958.2000.02098.x. [DOI] [PubMed] [Google Scholar]
- 3.Hutchinson CR. Proc Natl Acad Sci USA. 1999;96:3336–3338. doi: 10.1073/pnas.96.7.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.This approach has been named OSMAC: Bode HB, Bethe B, Höfs R, Zeeck A. ChemBioChem. 2002;3:691–627. doi: 10.1002/1439-7633(20020703)3:7<619::AID-CBIC619>3.0.CO;2-9.
- 5.(a) Preisg CL, Laakso JA, Mocek UM, Wang PT, Baez J, Byng G. J Nat Prod. 2003;66:350–356. doi: 10.1021/np020347a. [DOI] [PubMed] [Google Scholar]; (b) Okuno Y, Miyazawa M. J Nat Prod. 2004;67:1876–1878. doi: 10.1021/np034007g. [DOI] [PubMed] [Google Scholar]; (c) Zhang J, Cheng ZH, Yu BY, Cordell GA, Qiu SX. Tetrahedron Lett. 2005;46:2337–2340. [Google Scholar]; (d) Tian Y, Guo H, Han J, Guo D. J Nat Prod. 2005;68:678–680. doi: 10.1021/np049688+. [DOI] [PubMed] [Google Scholar]
- 6.Oikawa H, Murakami Y, Ichihara A. J Chem Soc, Perkin Trans. 1992;1:2949–2953. [Google Scholar]
- 7.De Croos JN, Bidochka MJ. Mycol Res. 2001;105:868–873. [Google Scholar]
- 8.For a recent review of the entire chemistry of marine-derived fungi (273 compounds discovered to 2002) see: Bugni TS, Ireland CM. Nat Prod Rep. 2004;21:143–163. doi: 10.1039/b301926h.
- 9.Wang GYS, Abrell LM, Avelar A, Borgeson BM, Crews P. Tetrahedron. 1998;54:7335–7342. [Google Scholar]
- 10.Cueto M, Jenson PR, Kauffman C, Fenical W, Lobkovsky E, Clardy J. J Nat Prod. 2001;64:1444–1446. doi: 10.1021/np0102713. [DOI] [PubMed] [Google Scholar]
- 11.(a) Amagata T, Rath C, Rigot JF, Tarlov N, Tenney K, Valeriote FA, Crews P. J Med Chem. 2003;46:4342–4350. doi: 10.1021/jm030090t. [DOI] [PubMed] [Google Scholar]; (b) Laurent D, Guella G, Roquebert MF, Farinole F, Mancini I, Pietra F. Planta Med. 2000;66:63–66. doi: 10.1055/s-2000-11110. [DOI] [PubMed] [Google Scholar]
- 12.(a) Mazars C, Rossignol M, Auriol P, Klaebe A. Phytochemistry. 1990;29:3441–3444. [Google Scholar]; (b) Farr DF, Castlebury LA, Rossman AY, Putnam ML. Mycol Res. 2002;106:745–752. [Google Scholar]
- 13.Isaka M, Jaturapat A, Rukseree K, Danwisetkanjana K. J Nat Prod. 2001;64:1015–1018. doi: 10.1021/np010006h. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi H, Meguro S, Yoshimoto T, Namikoshi M. Tetrahedron. 2003;59:455–459. [Google Scholar]
- 15.(a) Binder M, Tamm C. Angew Chem, Int Ed Engl. 1973;12:370–380. doi: 10.1002/anie.197303701. [DOI] [PubMed] [Google Scholar]; (b) Izawa Y, Hirose, Shimizu T, Koyama K, Natori S. Tetrahedron. 1989;45:2323–2335. [Google Scholar]; (c) Hirose K, Izawa Y, Koyama K, Natori S, Iida K, Yahara I, Shimaoka S, Maruyama K. Chem Pharm Bull. 1990;38:971–974. doi: 10.1248/cpb.38.971. [DOI] [PubMed] [Google Scholar]
- 16.Burlot L, Cherton JC, Convert O, Correia I, Dennetiere B. Spectroscopy. 2003;17:725–734.. The authors erroneously reported the isolation of chaetoblobosin O; the reported structure, although correct, was not the metabolite previously known as chaetoglobosin O: Ichihara A, Katayama K, Teshima H, Oikawa H, Sakamura S. Biosci Biotech Biochem. 1996;60:360–361. doi: 10.1271/bbb.60.360.
- 17.(a) Senderowicz AMJ, Kaur G, Sainz E, Laing C, Inman WD, Rodriquez J, Crews P, Malspeis L, Grever MR, Sausville EA, Duncan KLK. J Nat Cancer Inst. 1995;87:46–51. doi: 10.1093/jnci/87.1.46. [DOI] [PubMed] [Google Scholar]; (b) Bubb MR, Spector I, Beyer BB, Fosen KM. J Biol Chem. 2000;275:5163–5170. doi: 10.1074/jbc.275.7.5163. [DOI] [PubMed] [Google Scholar]
- 18.(a) Crews P, Manes LV, Boehler M. Tetrahedron Lett. 1986;27:2797–2800. [Google Scholar]; (b) Zabriskie TM, Klocke JA, Ireland CM, Marcus AH, Molinski TF, Faulkner DJ, Xu C, Clardy JC. J Am Chem Soc. 1986;108:3123–3124. [Google Scholar]
- 19.Sanders M, Diaz MC, Crews P. Mem Queens Mus. 1999;44:525–532. [Google Scholar]
- 20.Avantaggiato G, Solfrizzo M, Zazzerini A, Fanizzi FP, Visconti A. Nat Toxins. 1999;7:119–127. doi: 10.1002/(sici)1522-7189(199905/06)7:3<119::aid-nt49>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 21.Sekiguchi J, Kuroda H, Yamada Y, Okada H. Tetrahedron Lett. 1985;26:2341–2342. [Google Scholar]
- 22.Tsantrizo YS, Ogilvie KK, Watson AK. Can J Chem. 1992;70:2276–2284. [Google Scholar]
- 23.Johnson JH, Meyers E, O'Sullivan J, Phillipson DW, Robinson G, Trejo WT, Wells JS. J Antibiot. 1989;42:1515–1517. doi: 10.7164/antibiotics.42.1515. [DOI] [PubMed] [Google Scholar]
- 24.Tajima N, Nukina M, Kato N, Sassa T. Biosci Biotechnol Biochem. 2004;68:1125–1130. doi: 10.1271/bbb.68.1125. [DOI] [PubMed] [Google Scholar]
- 25.Cole RJ, Moore JH, Davis ND, Kirksey JW, Diener UL. J Agric Food Chem. 1971;19:909–911. [Google Scholar]
- 26.Silverton JV, Akiyama T, Kabuto C, Sekita S, Yoshihira K, Natori S. Tetrahedron Lett. 1976:1349–1350. [Google Scholar]
- 27.Springer JP, Cox RH, Cutler HG, Crumley FG. Tetrahedron Lett. 1980;21:1905–1908. [Google Scholar]
- 28.(a) Sekita S, Yoshihira K, Natori S, Kuwano H. Tetrahedron Lett. 1977;32:2771–2774. [Google Scholar]; (b) Sekita S, Yoshihira K, Natori S, Kuwano H. Chem Pharm Bull. 1982;30:1629–1638. doi: 10.1248/cpb.30.1609. [DOI] [PubMed] [Google Scholar]
- 29.Jiao W, Feng Y, Blunt JW, Cole ALJ, Munro MHG. J Nat Prod. 2004;67:1722–1725. doi: 10.1021/np030460g. [DOI] [PubMed] [Google Scholar]
- 30.Spöndlin C, Tamm C. Heterocycles. 1989;28:453–465. [Google Scholar]
- 31.Oikawa H, Murakami Y, Ichihara A. J Chem Soc, Perkin Trans. 1992;1:2955–2959. [Google Scholar]
- 32.Tsukuda Y, Koyama H. J Chem Soc, Perkin Trans. 1972;2:739–744. [Google Scholar]
- 33.Cytotoxicity bioassay procedures are described in: Valeriote F, Grieshaber CK, Media J, Pietraszkewicz H, Hoffmann J, Pan M, McLaughlin S. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x.
- 34.Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt T. Org Lett. 2001;3:3479–3481. doi: 10.1021/ol016515s. [DOI] [PubMed] [Google Scholar]
- 35.Mills JW, Pedersen FF, Walmod PS, Hoffman EK. Exp Cell Res. 2000;261:209–219. doi: 10.1006/excr.2000.5032. [DOI] [PubMed] [Google Scholar]
- 36.Groweiss A, Shmueli U, Kashman Y. J Org Chem. 1983;48:3512–3516. [Google Scholar]
- 37.Colin PI. Caribbean Reef Invertebrates and Plants. T. H. F Publications Inc Ltd; NJ: 1978. p. 68. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.