

Abstract
Recent studies have demonstrated the potential utility of antibodies for the treatment of Alzheimer’s disease (AD). In transgenic mouse models of AD, peripheral and intracerebral administration of Aβ-specific antibodies reduces amyloid burdens to varied extents. The mechanism may involve clearance of pre-existing amyloid plaques or prevention of new amyloid formation. Here we have used two transgenic models, the inducible CamKII-ttAxtetAPP/swe/ind (Line 107) and the APPswe/PS1dE9 (Line 85), to test the ability of intracerebral injection of Aβ antibodies to clear amyloid. Because the production of Aβ peptides in the Line 107 model is inducible, whereas production in Line 85 mice is constitutive, we could study the effects of antibody on pre-existing plaques versus continuous plaque formation. In Line 85, injection of antibody resulted in modest but statistically significant reductions in amyloid burden (average, 14–16%). However, injected antibodies had no effect on amyloid burden in Line 107 under conditions in which the production of Aβ was suppressed, indicating that pre-existing plaques are not rapidly cleared. These results indicate that, in these two models, intracerebral injection of Aβ antibodies produces modest reductions in amyloid deposition; and suggest that the mechanism may involve prevention of new amyloid deposits rather than clearance of pre-existing plaques.
Keywords: Alzheimer’s disease, AD, immunotherapy, Aβ, antibody, amyloid precursor protein, APP
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that leads to significant cognitive and behavioral impairments. AD is characterized by two histological hallmarks: amyloid plaques and neurofibrillary tangles in the hippocampus and cerebral cortex; along with loss of neurons and synapses. The predominant peptide found in amyloid plaques is β-amyloid peptide 1–42 (Aβ42), a highly fibrillogenic 4-kD peptide fragment produced by proteolytic processing of amyloid precursor protein (APP) (1, 2). The deposition of Aβ42 in amyloid plaques and diffuse deposits has been proposed as a causative factor in AD. Mutations found in familial AD lead to altered APP processing, with increased generation of Aβ42 and consequent deposition of this peptide into aggregates (3) (reviewed in (4)). Because of its clear connection to AD, the process of amyloid plaque formation has been considered a possible target for the treatment of AD.
A number of investigators have examined the potential of immunological approaches that target the Aβ42 peptide as therapeutics for AD. In 1999, Schenk et al. immunized PDAPP transgenic mice with the Aβ42 peptide and observed significant reduction of amyloid plaque levels in mice immunized either before or after amyloid plaque development. In this mouse model of AD, the authors observed colocalization of activated microglia and anti-Aβ antibodies, suggesting that microglia might be involved in removing the Aβ deposits (5). Subsequently, multiple studies have examined the effectiveness of active immunization, passive transfer and intracerebral (IC) injection of Aβ-specific antibodies in reducing amyloid plaque burden in PDAPP and Tg2576 transgenic mice (6–8). Immunotherapy has also been shown to improve working memory in transgenic mouse models (9–11). However, when active Aβ42 immunization was tested in a Phase II human trial (AN1792), ~6 % of the patients developed adverse inflammatory effects and the trial was stopped (12).
Intracerebroventricular injection of Aβ antibodies has been suggested as a safe and effective alternative to active immunization or peripheral transfer of antibodies in the treatment of AD (13–16). Several laboratories have reported rapid clearance, within 3–7 days, of brain amyloid after IC injection of Aβ antibodies (8, 17–20) or antibodies to oligomeric assemblies of Aβ (16). In some cases, the benefits of IC injection were only transient as amyloid plaques reductions approached reversal by 30 days (19). The extent of clearance achieved by this method varies substantially among these reports, ranging from what appears to be clearance throughout the central nervous system (CNS) (16) to very limited clearance of diffuse amyloid around the site of antibody injection (20). Therefore, the potential utility of intracerebral antibody administration in AD therapeutics is unclear.
In order to create a safe and effective immunotherapy for AD it is of great importance to determine the mechanism of amyloid reduction. It is presently unknown whether immunotherapy results in disintegration of amyloid plaques (by microglia or otherwise); whether the formation of amyloid plaques is prevented by Aβ antibodies, or both. It has been shown that Aβ antibodies are able to inhibit amyloid formation in vitro (21); hence, it is possible that the process of amyloid deposition is merely halted in the presence of Aβ-antibodies in vivo, provided enough antibody is available.
In the present study, we have examined the potential utility of IC injection of Aβ antibodies as a means to induce clearance of brain amyloid in two transgenic mouse models of AD that have unique features. One model, the CamKII-ttA × tetAPP/swe/ind (Line 107), expresses mutant APP via promoter elements regulated by doxycycline, allowing for studies of immunotherapy in a setting in which the effects of antibody on pre-existing amyloid plaques can be assessed while new amyloid formation is suppressed (22). The second transgenic mouse model co-expresses APPswe × PS1dE9 (Line 85) constitutively and has been bred to congenity to the C57BL6/J strain of mice (this model is distributed by the Jackson Laboratories). In studying the inducible Line 107 mice we wanted to address whether Aβ antibodies are able to clear previously generated amyloid plaques; and to determine the relative effectiveness of different antibody isotypes (IgG1 and IgG2a) in clearing Aβ. Our findings indicate that pre-existing amyloid plaques are not cleared by IC injection of Aβ antibodies in the Line 107 mice. In the congenic Line 85 mice, where Aβ production is constitutive, we detected modest reductions in amyloid burden after IC injection of Aβ antibody. We conclude that in these models of Alzheimer-type amyloidosis, it is not possible to stimulate rapid clearance of pre-existing amyloid by IC injection of Aβ antibodies. However, these antibodies may inhibit the deposition of new amyloid, accounting for the modest reductions in amyloid burden seen in the Line 85 mice.
MATERIALS AND METHODS
Antibodies
7B6 and IB3 are, respectively, IgG1 and IgG2a mouse monoclonal antibodies raised against amino acids 1–11 of human Aβ1–42 generated by our laboratory. 7B6 has slightly higher affinity than 6E10 for native human amyloid plaques, whereas IB3 is more comparable to 6E10. 7B6 and IB3 have higher affinity than 6E10 for monomeric synthetic Aβ (Table 1 and Supplementary data Fig. 1). 6E10 (Covance Research Products, Berkeley, CA) is an IgG1 mouse monoclonal raised against amino acids 1–16 of human Aβ1–42, with epitope specificity within amino acids 3–8 of Aβ1–42. The antibodies for intracerebral injection into all Line 107 mice and one cohort of Line 85 mice were prepared by diluting 5 μg of antibody stock in a final volume of 1.6 μL phosphate-buffered saline (PBS, pH=7.4) containing 0.4 μL India ink (1:5 final dilution) to mark the site of injection (BD Biosciences, San Jose, CA). The antibodies injected into the other cohort of Line 85 mice that received 2 μg 6E10 were prepared by suspending 2 μg of 6E10 in 2 μL PBS. Biotinylated 6E10 (bio-6E10) (Covance Research Products) was prepared by diluting 2 μg antibody in 2 μL PBS.
Table 1.
Antibodies used for intracerebral (IC) injection
| Antibody | Isotype | Binding to native Aβ plaques(+ = low, +++ = highest) | Affinity for monomeric Aβ(Dilution factor corresponding to 50% of A450max) |
|---|---|---|---|
| 7B6 | IgG1 | +++ | 5.7 × 10−7 |
| IB3 | IgG2a | ++ | 5.3 × 10−7 |
| 6E10 | IgG1 | ++ | 1.6 × 10−6 |
| Bio-6E10 | IgG1 | + | 4.9 × 10−6 |
Antibodies used for IC injection were compared in their ability to bind native human amyloid plaques and monomeric human Aβ. Antibody affinity for native human amyloid plaques was assessed by immunohistochemistry using serial dilutions of the antibodies on unfixed human tissue with amyloid plaques. The affinity of each antibody for native amyloid plaques is scored from very high (+++) to low (+) with 7B6 having the highest affinity for native amyloid plaques. Antibody affinity for monomeric human Aβ was compared by ELISA, and the affinity of each antibody is reported as the dilution factor that corresponds to ½ of the maximum absorbance value, with 7B6 having the highest affinity for monomeric Aβ.
Mice and Study Design
Heterozygous double-transgenic CamKII-ttA × tetAPP/swe/ind mice (Line 107) (22) were obtained by crossing CamKII-ttA single-transgenic mice with tetAPP/swe/ind mice (B6/C3 strain background). These mice begin to deposit amyloid plaques around the age of 3 months (22). Transgenic mice of both male and female gender were used. To examine whether Aβ antibodies cause disintegration of amyloid plaques, the mice were placed on doxycycline chow (Dox, 200 mg/kg, BioServ, Frenchtown, New Jersey) for 2 weeks prior to injection of antibody. Doxycycline chow suppresses expression of the APP transgene which minimizes novel amyloid plaque formation. Intracerebral injection was performed at the age of 6 months. The mice were divided into three cohorts (n=4 per cohort) that received 2 injections at one time of 5 μg 7B6, IB3 or 6E10 in a 2-μL volume as described in Table 2. The mice were maintained on doxycycline chow and sacrificed 7 days post-injection. Doxycycline chow was changed once weekly.
Table 2.
Experimental design of injected mouse cohorts.
| Mouse Line | N | Age | Dox | Antibody | Injection sites | Time between injection and sacrifice |
|---|---|---|---|---|---|---|
| Line 107 | 4 | 6 | + | 5 μg 7B6 | 2 | 7 days |
| Line 107 | 4 | 6 | + | 5 μg IB3 | 2 | 7 days |
| Line 107 | 4 | 6 | + | 5 μg 6E10 | 2 | 7 days |
| Line 85 | 6 | 15 | + | 5 μg 6E10 | 2 | 7 days |
| Line 85 | 5 | 12 | − | 2 μg 6E10 | 1 | 3 days |
| Line 85 | 3 | 12 | − | 2 μg bio-6E10 | 1 | 24 h |
| Line 85 | 3 | 12 | − | 2 μg bio-6E10 | 1 | 3 days |
Line 107 or Line 85 mice received intracerebral injections of Aβ antibodies in the presence or absence of doxycycline chow (Dox) which suppresses amyloid precursor protein expression in Line 107 mice, and were sacrificed 3 or 7 days after injection. Age is given in months. Bio-6E10 refers to the biotinylated form of the 6E10 antibody.
Heterozygous transgenic APPswe × PS1dE9 (Line 85) mice (23, 24) were generated by crossing the co-injected double-transgenic Line 85 mice with C57BL/6J females (Jackson Laboratory, Bar Harbor, ME). These mice had been backcrossed to the C57BL/6J strain for 8 generations. The onset of amyloid pathology in this mouse model occurs at approximately 6 months of age. Transgenic mice of both male and female gender were used. In order to determine if IC injection of Aβ antibody could induce amyloid plaque reduction in a model where Aβ production is constitutive, we injected 15-month-old transgenic APPswe × PS1dE9 (Line 85) mice (n= 6) in exactly the same manner as the Line 107 mice described above. These mice received two injections at one time of 5 μg 6E10 containing India ink to label the injection site. Although 6E10 has lower affinity than 7B6 for native human amyloid plaques, it was chosen for intracerebral injection because it has been previously demonstrated to be highly efficacious in clearing amyloid plaques (8). The mice were also placed on doxycycline chow 2 weeks prior to injection to control for any potential effect of doxycycline administration on amyloid reduction, and were maintained on this chow for 7 days post-injection, at which point they were sacrificed (Table 2).
To examine if the dose of antibody administered affected amyloid plaque reduction, five 12-month-old Line 85 mice were IC injected with 2 μg 6E10 in 2 μL according to previously published procedures (8, 18). These mice were sacrificed 72 h post-injection, as described in Table 2.
In order to monitor the spread of the injected antibody, six 12-month-old Line 85 mice were injected IC with 2 μg bio-6E10 in 2 μL and sacrificed at 24 h (n=3) or 72 h (n=3). The Line 85 mice who received biotinylated antibody were not pre-treated with doxycycline chow (Table 2). All animal procedures were performed in accordance with Johns Hopkins University Animal Care and Use Committee guidelines.
Intracerebral Injection of Antibody
The mice were anesthetized with 1–1.5% isoflurane (Penn Veterinary Supply, Lancaster, PA), placed in a stereotaxic apparatus, and the top of the skull was exposed by an incision along the midline. All injections were performed on the left hippocampus, with the right hippocampus serving as a contralateral control. In those animals with two injection sites, we used the following stereological coordinates measured from the bregma: −2 mm anteroposterior, 1.5 mm mediolateral, 1.5 mm dorsoventral (anterior injection); −3 mm anteroposterior, 3 mm mediolateral, 2.5 mm dorsoventral (posterior injection). In the Line 85 mice with one injection site, the stereological coordinates were: −2 mm anteroposterior, 1.5 mm mediolateral, 1.5 mm dorsoventral, measured from the bregma (25). A small hole was drilled in the skull at the site of injection using a dental drill, and the site of injection was cleaned with PBS. Antibody was injected through a hand-pulled glass capillary needle connected to a 5 μL Hamilton syringe using a syringe pump (New Era Pump System, Farmingdale, NY) at a rate of 0.5 μL/min (2 μL final volume). After injection of antibody, the incision was closed with sutures, and the wound was cleaned with 70% ethanol. Mice were observed regularly after injection, and appropriate measures were taken to minimize pain and discomfort. At the time of sacrifice, the mice were anesthetized with ether and transcardially perfused with cold PBS, pH=7.4 followed by perfusion with 4% paraformaldehyde. The brains were harvested and postfixed for 24 h in 4% paraformaldehyde, followed by 48 h in 30% sucrose at 4ºC for cryoprotection. The brains were then snap-frozen in 2-methylbutane at −20ºC. Serial coronal sections (40-μm thickness) were cut using a sliding microtome and stored in anti-freeze buffer (100 mM sodium acetate, 250 mM polyvinyl pyrrolidone, 40% ethylene glycol, pH=6.5) at −20ºC until used for immunohistochemical staining.
Affinity of Injected Antibodies for Monomeric Aβ (ELISA)
To determine the affinity of the injected antibodies for monomeric Aβ, Aβ1–42 (Bachem Bioscience Inc, King of Prussia, PA) was dissolved in hexafluoroisopropanol (HFIP) at 1 μg/μL, after which the HFIP was evaporated with nitrogen gas and the Aβ stored at −80°C until use. A 96-well ELISA plate was coated with 2.5 μg/mL human Aβ1–42 in 50 mM NaHCO3 (pH=9.6) overnight. The plate was washed in PBS with 0.1% Tween-20 (PBST) and blocked for two hours with 5% bovine albumin (Sigma, St. Louis, MO) in PBST. Aβ antibodies were diluted to 1 mg/mL in PBS and serially diluted in 5% BSA in PBST. Serial dilutions of the antibodies were applied in duplicate to the ELISA plate and incubated overnight at 4ºC. The plate was washed with PBST and secondary biotin-conjugated goat anti-mouse IgG was applied at 1:20,000 (Jackson immunoresearch, West Grove, PA) and incubated for 2 hours at room temperature. The plate was washed with PBST and streptavidin conjugated to horseradish peroxidase (Pierce, Rockford, IL) was added at 1:10,000 for 1 hour at RT. The plate was washed with PBST, and TMB substrate (KPL, Gaithersburg, MD) was added to each well, followed by an equal amount of phosphoric acid. The ELISA plate was read on a VersaMax ELISA plate reader at 450 nm. All antibodies displayed a one-site binding (hyperbola) curve with a maximum absorbance reading of 3.28–3.41. The curves were fitted to a Michaelis-Menten model and the affinity represented as the dilution that corresponds to ½ of maximum A450 nm.
Affinity of Injected Antibodies for Native Human Amyloid Plaques
To determine the affinity of 7B6, IB3, 6E10 and bio-6E10 for native human amyloid plaques, 20 μm unfixed cryostat sections from the inferior parietal lobe of a human case with diagnosed AD were used for immunohistochemical staining in the absence of formic acid pre-treatment. The antibodies were serially diluted in blocking buffer (4% normal goat serum in PBS with 0.2 % Triton X) starting at a 1 μg/μL concentration and incubated overnight at 4°C. The primary antibodies were detected with an anti-mouse Vector Lab Elite kit (Vector Laboratories, Burlingame, CA) and developed with 3,3′-diaminobenzidine (Vector Laboratories). The affinity of the antibodies for native plaques were assessed as high (+++) if considerable staining was detectable up to a dilution of 1:10−5, moderate (++) if staining was detectable up to a dilution of 1:10−4, and low (+) if staining was observed through a dilution of 1:10−3.
Immunohistochemical Staining
Five 40-μm coronal sections approximately 100 μm apart, spanning each of the two injection sites in Line 107 and one cohort of Line 85 mice, and seven sections approximately 100 μm apart, spanning the only injection site in the other Line 85 mice were selected for Aβ immunohistochemical staining and stereological assessment. The sections were washed in PBS and mounted on glass slides. Sections were treated with 88% formic acid, and immunohistochemical staining for Aβ was performed using rabbit anti-amyloid β-peptide antibody (Zymed Laboratories, San Francisco, CA) at a dilution of 1:500 in 4% normal goat serum in PBS with 0.2% Triton X. The immuohistochemical staining was completed as described above. Sections were counterstained with cresyl violet.
In order to measure the spread of the injected antibody in the brain of Line 85 mice that received biotinylated antibody (bio-6E10), the injected antibody was detected using a streptavidin-FITC conjugate (Pierce Biotechnology, Rockford, IL). A total of 14 sections spanning the injection site, approximately 200 μm apart, were selected for floating immunohistochemistry. The sections were incubated with Streptavidin-FITC at a dilution of 1:500. The spread of injected bio-6E10 was measured by finding the last section away from the site of injection that exhibited Streptavidin-FITC staining. The penultimate section (200 μm closer to the site of injection) was recorded as the end point of antibody spread. Two additional sections from each mouse that received bio-6E10 were incubated together with either the rabbit anti-amyloid β-peptide antibody (1:500 dilution) or rat anti-mouse I-A/I-E antibody (1:500 dilution, BD Biosciences, San Jose, CA) and a secondary antibody conjugated to Cy3. Immunofluorescent staining for MHC II was performed as described above on one section within the spread of injected antibody from each cohort. In order to detect the injected antibody in the brain of Line 107 and Line 85 mice, we performed free-floating immunohistochemical staining with anti-mouse-Cy3 antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) at 1:200 dilution on one section per mouse within the range of spread of the injected antibody. Sections were counterstained with Hoechst. A widefield fluorescent microscope (Zeiss, Thornwood, NY) was used to visualize immunofluorescent staining, and a laser confocal microscope (Zeiss) for double immunofluorescent staining. To control for non-specific immunoreactivity, primary antibody was omitted from one section in all staining procedures.
Three sections within the spread of injected antibody were selected from each mouse in the Line 85 cohort injected with 2 μg 6E10 or each Line 107 mouse injected with 5 μg 6E10 for staining with Gunther’s modification of thioflavin-S as a marker for β-pleated-sheet structures in cored amyloid plaques. Sections were incubated for 5 min in 0.5% KMnO4, 5 min in 1% K2S2O5 with 1% oxalic acid, and 8 min in 0.02% thioflavin-S, then washed twice in 80% ethanol and twice in distilled water. Non-overlapping images were taken at consistent locations at 20× magnification (200× final magnification) using a widefield fluorescent microscope (Zeiss) to cover approximately 75–80% of the hippocampus (1–4 images/section).
Stereology Image Analysis
We quantified the area fraction of Aβ immunoreactivity in coronal sections of the hippocampus using Stereo Investigator (MBF Bioscience). Each section was analyzed using a 40× objective. The hippocampus was outlined, and the Area Fraction Fractionator probe was used to systematically and randomly allocate sampling sites 300 μm apart. At each sampling site, a 100 × 80 μm counting frame was superimposed, containing markers equally spaced from one another at a distance of 12 μm. The markers that colocalized with Aβ immunoreactivity were labeled as positive, whereas remaining markers were labeled negative. The area fraction was calculated as the number of positive markers divided by the total number of markers. The stereological assessment was made in a blinded fashion with regard to treatment and mouse identification but not to which hemisphere was being counted. Fractional area of thioflavin-S staining was quantified by particle analysis using Image J (NIH, available at http://rsb.info.nih.gov/ij/). Thresholds were set to 100 (lower) and 240 (upper), pixel size limits were 1–999999, and analysis was performed with 20 bins.
Statistics
Paired t-tests of left versus right fractional area were performed using GraphPad Prism version 5.00 for Windows at a significance level of p<0.05 (GraphPad Software, San Diego, CA, www.graphpad.com).
RESULTS
In the present report, we set out to compare the degree of amyloid reduction after IC injection of Aβ antibodies in two transgenic mouse models of AD. The inducible CamKII-ttA × tetAPP/swe/ind (Line 107) mice express high levels of APP carrying the Indiana and Swedish familial AD-linked mutations, with onset of amyloid plaque deposition at 2 to 3 months of age (22). In the Line 107 model, expression of mutant APP is suppressed by doxycycline allowing for an assessment of antibody effects on pre-existing amyloid deposits in the absence of on-going amyloid formation. The APPswe × PS1dE9 (Line 85) mice exhibit lower levels of constitutive APP expression and form plaques beginning at approximately 5–6 months of age (23, 24, 26).
In order to compare the influence of Aβ antibody isotype on amyloid plaque clearance in the absence of Aβ peptide production, 5 μg of 6E10 (IgG1), IB3 (IgG2a) or 7B6 (IgG1) were injected IC into Line 107 mice at 6 months of age. Two weeks prior to injection and over the entire course of the experiment, the animals were given food containing doxycycline so that any reduction in amyloid that we observed would reflect the direct removal of previously generated Aβ deposits. At 7 days after injection, none of the injected antibodies reduced the fractional area of Aβ immunoreactivity in the injected hippocampus, when compared to the contralateral control (Fig. 1).
Fig. 1.

Intracerebral (IC) injection of Aβ antibodies does not clear amyloid plaques in Line 107 mice with suppressed production of Aβ peptides. A) Representative images of amyloid plaque burden by immunohistochemical staining for Aβ in the hippocampus of 6-month-old Line 107 mice in which amyloid precursor protein (APP) expression was suppressed by doxycycline administration. These mice received intracerebral injections of antibody 7B6 (IgG1), IB3 (IgG2a), or 6E10 (IgG1), as indicated. Arrows indicate India ink injected along with the antibody; the pattern of India ink marks the needle track but does not represent the spread of antibody. All images were taken at 2× magnification. B) IC injection of Aβ-specific antibody in Line 107 mice with suppressed APP expression does not reduce amyloid plaque burden. Images represent quantitation of fractional area of Aβ immunoreactivity by stereology of mice injected with 7B6 (top), IB3 (middle) or 6E10 (bottom). Each dot represents one mouse. Error bars indicate standard error of mean.
IC injection of Aβ antibody 6E10 in the Line 85 (APPswe/PS1dE9) transgenic mouse model, where production of Aβ peptide is constitutive, resulted in modest but statistically significant reductions in amyloid burden. The first cohort of Line 85 mice were injected according to previously published procedures (8, 18) At 72 h after injection of 2 μg 6E10, a significant reduction of 14% (range, 3% to 23%) in total Aβ was noted in the injected hippocampus of Line 85 mice (Fig. 2).
Fig. 2.

Intracerebral (IC) injection of Aβ antibodies results in limited amyloid plaque reductions in Line 85 mice. A) Representative images of amyloid plaque burden by immunohistochemical staining for Aβ in the hippocampus of 12-month-old Line 85 mice with constitutive Aβ production. The first cohort received intracerebral injections of 2 μg 6E10 (top). The second cohort was pretreated with doxycycline chow (+Dox) and received IC injections of 5 μg 6E10 with India ink (bottom), as indicated. A slight reduction in amyloid plaque burden is visible in the injected hippocampus (antibody) when compared to the contralateral control by Aβ immunoreactivity. Arrows indicate India ink injected along with the antibody; the pattern of India ink marks the needle track but does not represent the spread of antibody. All images were taken at 2× magnification. B) IC injection of Aβ-specific antibody in Line 85 mice results in statistically significant reductions in amyloid plaque burden. To determine if the dose of IC injected antibody affected plaque reduction, one cohort of Line 85 mice was given one IC injection of 2 μg 6E10, which resulted in a statistically significant reduction in amyloid plaque burden (average, 14%, top). To assess antibody-induced amyloid reduction in a model where Aβ production is constitutive and to control for any effect of doxycycline on amyloid clearance, Line 85 mice were injected in the same manner as Line 107 mice, with 5 μg 6E10 with doxycycline pretreatment (+Dox). In this cohort, IC injection of Aβ antibody resulted in a statistically significant reduction in amyloid burden (average, 16%, bottom). Images represent quantitation of fractional area of Aβ immunoreactivity by stereology. Each dot corresponds to one mouse, and error bars indicate standard error of mean.
To determine whether the absence of amyloid clearance we had seen in Line 107 mice could be attributed to some effect of doxycycline on antibody-mediated clearance mechanisms, we injected Line 85 mice given the same doxycycline treatment described above with 5 μg of 6E10. Since these mice are not inducible, they maintain constitutive production of Aβ peptide despite doxycycline treatment. Seven days after IC injection, there was a statistically significant reduction of 16% (range, 2% to 29%) in amyloid plaque burden on the injected side as compared to the contralateral control (p<0.04, Fig. 2). These results indicate that doxycycline does not interfere with the mechanisms involved in antibody-mediated reduction of amyloid plaques.
To determine the extent of antibody diffusion from the injection site, we injected Line 85 mice intracerebrally with 2 μg of biotinylated 6E10 (bio-6E10) followed by sacrifice at 24 h (n=3) and 72 h (n=3). The biotin tag allowed us to detect the injected bio-6E10 specifically with minimal non-specific immunofluorescence, revealing a localization of the injected antibody to amyloid plaques. Antibody could be detected approximately 700 μm anterior and 500 μm posterior to the injection site, on average, by conservative measurement at 24 h; this area encompasses the region of amyloid plaque quantification (+/− 300 μm from the only injection site in Line 85 mice) with a large margin of error. At 72 h, the staining for bio-6E10 was slightly reduced, spanning on average approximately 500 μm anterior and 400 μm posterior to the injection site (Fig. 3, supplementary data Fig. 2). Staining for injected bio-6E10 was not observed in the contralateral control hemisphere in any of the sections analyzed. To determine the distribution of non-biotinylated antibodies injected in animals shown in Figures 1 and 2, we stained sections from each animal with an anti-mouse IgG conjugated to Cy3. Antibody was readily detectable in the injected hippocampus but not in the contralateral control hippocampus in each animal (supplementary data Figure 3).
Fig. 3.

Intracerebral (IC) injection results in significant spread of the Aβ antibody, which is detectable at 24 h (top) as well as 72 h (bottom) after injection. A) Images represent immunofluorescent detection of injected biotinylated 6E10 (bio-6E10) by streptavidin conjugated to FITC (green) at 200 μm anterior to the injection site in Line 85 mice, and reveal a staining pattern that resembles amyloid plaques (arrows). There was no spread of bio-6E10 to the contralateral control hemisphere in any of the sections analyzed. Sections were counterstained with Hoechst stain (blue). All images are displayed at 5× magnification with 20× insets (upper corner, showing staining for bio-6E10 resembling an amyloid plaque). B) Conservative measurement of the spread of IC injected bio-6E10 reveals an anteroposterior spread of at least 1.2 mm at 24 h (top) and 0.9 mm at 72 h (bottom), indicating that the antibody spreads well beyond the region in which Aβ immunoreactivity is quantified. Each dot corresponds to one mouse.
Since the immunofluorescent detection of bio-6E10 displayed staining that resembled amyloid plaques, we performed double immunofluorescent staining to determine if the injected antibody indeed bound to amyloid plaques. We found that injected bio-6E10 frequently colocalized with Aβ immunostaining (Fig. 4). Previous studies have suggested that microglia contribute to the reduction of amyloid plaques after intracranial injection (8, 18). Staining for the microglial activation marker MHC II was seen throughout the injected hippocampus without visible clustering around areas that displayed Streptavidin-FITC staining for injected antibody (Fig. 4).
Fig. 4.

Intracerebrally injected bio-6E10 binds amyloid plaques in vivo. Streptavidin-FITC (green, labels injected antibody) colocalizes with Aβ immunofluorescent staining (Cy3, red, top). However, the microglial activation marker MHC II (Cy3, red, bottom) was not found clustering around areas that displayed Streptavidin-FITC staining (green, labels injected antibody). Images shown are from a Line 85 mouse sacrificed at 72 h, and are comparable to Line 85 mice that were sacrificed 24 h after injection. Images are shown at 20× magnification.
In order to examine whether microglia were differentially activated in the injected hippocampus as compared to the contralateral control, we stained sections from all mouse cohorts for MHC II. The relative number of amyloid plaques with MHC II staining was not different in the injected hippocampus compared to the contralateral control. There was no detectable increase in staining on the injected side, but microglia were clearly activated along the needle path, indicating that the microglia maintained the ability to become activated (Supplementary data Fig. 4).
Since we observed a modest reduction in Aβ immunoreactivity in Line 85 mice, we wanted to know if there was any reduction in the cored-plaques that contain highly aggregated material. To this end, sections from Line 85 mice that received 2 μg 6E10 and sections from Line 107 mice that received 5 μg 6E10 with doxycycline pretreatment were stained with thioflavin-S, which labels β-pleated sheet structures found in cored amyloid plaques (27). No reduction in thioflavin-S staining was observed in either cohort when the injected hippocampi and contralateral control hippocampi were compared (Fig. 5). Interestingly, the Line 107 mice displayed higher levels of thioflavin-S staining than did the Line 85 mice (0.8% and 0.4% in contralateral control, respectively, Fig. 5). Therefore, it appears that intracerebral injection of Aβ antibodies can modestly reduce the amount of Aβ immunoreactivity in Line 85 mice, but does not affect the levels of cored amyloid.
Fig. 5.
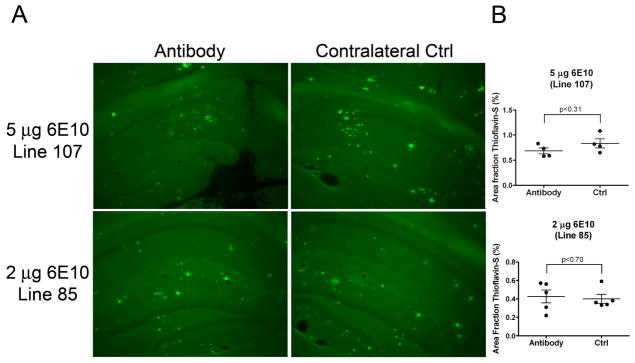
Intracerebral injection of Aβ-specific antibody does not reduce cored amyloid plaques in the injected hippocampus as compared with contralateral control. A) Cored amyloid plaques are not cleared in the injected hippocampus (antibody), as demonstrated by thioflavin-S which stains β-pleated sheet structures. Representative images of Line 107 mice that received 5 μg 6E10 with doxycycline pre-treatment (top) and Line 85 mice that received 2 μg 6E10 (bottom) at 10× magnification. Arrows indicate thioflavin-S positive amyloid plaques. B) Quantitation of thioflavin-S staining intensity reveals no reductions in cored amyloid plaques on the injected side (antibody) compared to contralateral control. Each dot corresponds to one mouse and error bars represent standard error of mean.
DISCUSSION
In the present study, we have utilized two AD mouse models that have not been extensively employed in immunotherapeutic studies to assess amyloid plaque clearance after IC injection of Aβ antibodies. One of the models used here (Line 107) expresses mutant APP via promoter elements regulated by doxycycline, allowing us to examine the efficacy of amyloid plaque clearance in a setting where new amyloid formation has been suppressed. In contrast to the Line 85 mice, where modest but statistically significant reductions in amyloid burden were noted, we found no evidence of amyloid clearance in the inducible (Line 107) model with any of the antibodies tested. The limited reductions in amyloid plaque burden could not be attributed to poor diffusion of the injected antibody, which was detectable 700 μm anterior and 500 μm posterior to the injection sites (amyloid burden was quantified in a region 200–300 μm on either side of the injection site). We conclude that in the inducible line of mice studied here, the injection of antibodies directly into the CNS is not effective in mediating clearance of pre-existing amyloid plaques. However, we did note reductions in amyloid burden in mice that constitutively express mutant APP (Line 85), though there were no statistically significant reductions in the number of mature, thioflavin-S positive, deposits. Together, these data suggest that there are circumstances in which mature, cored, deposits of amyloid are not effectively cleared by IC injection of Aβ antibodies.
Our outcome is similar to that of a recent study by Levites et al. (20) in the Tg2576 mouse model; which showed that some antibodies to Aβ can mediate reductions in amyloid burden, measured by immunohistochemistry, without effectively reducing the burden of mature, thioflavin-S positive, amyloid plaques. Yet, the reductions in amyloid burden that we observe are significantly smaller than those that have been previously reported in Tg2576 and 3xTg-AD transgenic mouse models (8, 17–19). This discrepancy can be attributed to a variety of factors. It is possible that the transgenic mouse models used here have certain characteristics that limit clearance of amyloid plaques. The strain background on which these transgenic animals are maintained may influence the phenotype and the immune response of the animal model. The transgenic Line 107 mice used in our study were on a C57B6/C3 mixed background, while the Line 85 mice were backcrossed to the C57BL/6J mouse strain. The 3xTg-AD transgenic mice, which exhibit at least a 60% reduction in Aβ immunoreactivity are on a mixed C57B6/129 background (19, 28). The Tg2576 mice used in the 2003 Wilcock et al. study where approximately 50% reduction in amyloid burden is reported are on a hybrid background of C57BL6, SJL, DBA and Swiss Webster strains (8, 29, 30). Since the strain backgrounds of the mouse models used in these studies are quite variable, it is possible that one model-specific factor that influences amyloid plaque clearance is the mouse strain.
Another variable that potentially affects the amount of amyloid reduction is the ratio of Aβ42 to Aβ40 formed in the brain. Tg2576 mice express a human APP transgene with the Swedish mutation, leading to concurrent increases in Aβ42 and Aβ40, such that the ratio of Aβ42:Aβ40 is approximately 1:3 at 7 months of age and at least 1:12 at 21 months of age in formic acid-extracted (insoluble) brain homogenate (31). In contrast, the concomitant expression of human APP with the Swedish mutation (K670N, M671L) with human presenilin-1 (PS1) with a deleted exon 9 (26) leads to a preferential increase in Aβ42 over Aβ40; with an approximate 2:1 Aβ42:Aβ40 ratio in formic acid-extracted brain homogenates of 12-month-old Line 85 mice (24). Moreover, the APP transgene construct used to produce the inducible Line 107 model encodes the “Swedish mutations” which augment total Aβ production, as well as mutations near the C-terminus of the Aβ domain, which specifically augment Aβ42 production (22). Thus, in both mouse models used here, the levels of Aβ42 would be expected to be very high. Since Aβ42 has a much higher propensity to aggregate than Aβ40 (32), it is possible that Aβ40 is more effectively cleared by antibody, leading to larger reductions of amyloid plaque burden in certain mouse models. In humans, the predominant Aβ peptide in amyloid plaques is Aβ42 (31, 33).
The abundance of amyloid present in the brain at the time of treatment with Aβ antibody may also influence Aβ clearance. Das and colleagues reported that active immunization of mice with moderate to high amyloid burden (2.8% to 5.6%, area fraction) produces only limited reductions in Aβ42 levels and no reduction in amyloid plaque burden (34). In our study, the amyloid burden in both the Line 107 and Line 85 mice was somewhat higher (approximately 9% and 6% area fraction measured by immunohistochemistry, respectively). Notably, statistically significant amyloid reductions were only observed in the Line 85 mice which exhibited the lower amyloid burden.
Furthermore, the physical state of the amyloid plaque could affect how easily it is cleared. Thioflavin-S is a good marker for the presence of β-pleated sheets, which predominate in the highly aggregated core of amyloid plaques (27). Similar to our study, Levites et al. observed significant reductions in Aβ immunoreactivity after IC injection of a variety of antibodies in the absence of concurrent reductions in the numbers of thioflavin-S positive deposits (20). Together, these studies indicate that under some circumstances, the intracerebral application of Aβ antibody is more effective at removing Aβ species that become part of diffuse deposits, rather than those within mature plaques.
The primary cells thought to respond to the injected antibody are the local immune cells of the central nervous system, microglia. Microglial clearance of Aβ has been suggested as a potential mechanism of Aβ reduction after immunotherapy (12). Wilcock et al. have shown that after IC injection, diffuse species of Aβ are removed through mechanisms independent of microglial activation, whereas microglial activation is necessary for reductions of compact, thioflavin-S positive plaques (8). We did not quantify microglial activation, but we did not observe a robust increase of activation around amyloid plaques in the injected hippocampus compared to the contralateral control by MHC II immunofluorescent staining, as would be expected if antibody induced microglial phagocytosis of existing plaques. Hence, regardless of staining intensity for MHC II, the role of the ipsilateral microglia in amyloid plaque clearance in these mice is questionable, as they did not appear to be homing to amyloid plaques in the presence of antibody. The microglial cells in these mice did maintain their ability to activate, since considerable MHC II staining was observed along the needle track. The Aβ antibodies used in this study are unlikely to be responsible for the lack of microglial activation, since 6E10 has been used in other IC injection studies in which microglial activation has been reported (8, 18). Therefore, it is likely that the density of the amyloid plaques, the type of the amyloid plaque, and the lack of significant microglial activation contributed to the lower amyloid reductions observed in this study compared to previous studies.
The data presented here raises questions about the mechanisms of amyloid clearance observed in transgenic mouse models of AD after immunotherapy. It is presently unknown whether amyloid reduction observed after Aβ immunotherapy is the result of plaque disintegration or prevention of plaque formation. To address this question, we suppressed APP expression in Line 107 mice, such that any reduction in amyloid pathology would be the result of direct clearance of pre-existing amyloid plaques. This experimental model simulates a patient receiving secretase-inhibitors to suppress the production of Aβ. In such a scenario, immunotherapy could potentially be used to remove the amyloid plaques that may already be present at the onset of treatment with the secretase-inhibitor. Since we did not observe any reduction in amyloid plaques in Line 107 mice in which APP expression had been suppressed, it is possible that antibodies are unable to remove pre-existing amyloid plaques. One explanation for the modest reduction observed in Line 85 mice is that antibody halts amyloid deposition for a number of days leading to a perceived reduction of amyloid plaques in the injected hemisphere. In Line 85 mice, this reduction could have reached a plateau by 3 days post-injection, leading to similar reductions at 3 and 7 days. However, since there was no trend toward a reduction of Aβ immunoreactivity (which encompasses both diffuse and cored amyloid) in Line 107 mice, the evidence is compelling that the amyloid plaques in Line 107 were too numerous and cored for antibody-induced clearance in the absence of production of Aβ peptide. Jankowsky et al. have shown that amyloid plaques do not spontaneously dissolve for six months after suppression of APP in Line 107 mice (22). The doxycycline chow did not interfere with the clearance of amyloid plaques, since a significant reduction was observed in Line 85 mice that received 5 μg 6E10 despite pretreatment with doxycycline.
It has been previously suggested that the molar ratio of injected antibody to Aβ is a limiting factor in Aβ clearance after immunotherapy (35). Based on our mathematical approximations, the amount of formic acid-extracted Aβ42, which represents the insoluble Aβ aggregates, is present in excess in Line 107 (24 Aβ42:1 IgG). This suggests that the antibody is limiting in clearing amyloid plaques in Line 107. In Line 85, the molar ratio of formic acid-extractable Aβ42 to oantibody is 1:1 for the mice injected with 2 μg antibody, whereas it is 1:3 for the mice that received 5 μg antibody. Interestingly, the reductions in area fraction of amyloid are comparable in Line 85 mice that received 2 or 5 μg of antibody (14 versus 16% amyloid reduction, respectively), even though the ratio of antibody to Aβ42 is 3-fold higher in the mice that received 5 μg antibody. Therefore, it is unlikely that the molar ratio of antibody to formic acid soluble Aβ42 influences the antibody-induced clearance, unless antibody-mediated amyloid clearance has reached a plateau at 2 μg antibody.
On the other hand, antibody is present in vast excess to the PBS soluble fraction of Aβ42 as well as total soluble Aβ (Aβ40 and Aβ42) in both Line 85 (1 Aβ42:>300 IgG) and Line 107 (1 Aβ42:160 IgG). This suggests that the injected antibody has a greater potential to clear the PBS-soluble forms of Aβ as opposed to deposited forms. Hence, the lack of production of Aβ species could explain why no amyloid reductions are observed in Line 107 where only pre-existing plaques exist, while reductions occur in the constitutively expressing Line 85 mice. Other factors may also influence the ability of antibody to access and clear Aβ from the brain, such as the surface area to volume ratio of the amyloid plaque. Nonetheless, we believe that it is likely that the injected antibody is acting on a on a pool of PBS-soluble Aβ species that contribute to the plaque formation (Supplementary data Table 1).
We have found that despite considerable spread of the injected antibody, IC injection of Aβ antibody results in modest reductions in amyloid plaque burden in Line 85 mice. As described above, a number of factors inherent to different transgenic mouse models could have contributed to these results. It has been suggested that intracerebroventricular injection of Aβ antibodies could be a safe therapeutic option in AD (13–16). However, our present study and at least one other (20) report circumstances in which IC injection of antibody results in relatively modest levels of amyloid clearance. Moreover, in some cases that report more significant clearance, amyloid plaques were found to reform in the injection region (19). It is possible that microglial activation is necessary to remove cored amyloid plaques after IC injection in mice (8); and that the strains of mice used here are less able to activate these cells to stimulate amyloid phagocytosis. Whether direct injection of Aβ antibody into the brains of human patients would induce reduction in pathology is uncertain. We note that we find good evidence for antibody binding to amyloid deposits; but whether IC injection of Aβ antibody in human patients could neutralize a toxic effect of amyloid plaques, or other Aβ oligomeric species, is similarly uncertain. Our studies raise a note of caution that under some circumstances, IC injection of Aβ antibody is not particularly effective in stimulating repair of pathologic lesions and suggest that other forms of immunotherapy, i.e. Aβ vaccination or passive administration of Aβ antibodies may be better suited for human therapy.
Supplementary Material
Acknowledgments
Support: This research was supported by NIH grants P5OAGO05146 and RO1NS47225, and a grant from the Alzheimer’s Association.
We would like to extend our sincere gratitude to Dr. Joanna Jankowsky for prodiving us with the CamKII-tTA× tetAPPswe/ind transgenic mouse model and Dr. Mohammed Farah for assistance with intracerebral injection techniques.
References
- 1.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 2.Kang J, Lemaire HG, Unterbeck A, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 3.Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 4.Tilley L, Morgan K, Kalsheker N. Genetic risk factors in Alzheimer’s disease. Mol Pathol. 1998;51:293–304. doi: 10.1136/mp.51.6.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 6.Lemere CA, Maron R, Selkoe DJ, et al. Nasal vaccination with beta-amyloid peptide for the treatment of Alzheimer’s disease. DNA Cell Biol. 2001;20:705–711. doi: 10.1089/10445490152717569. [DOI] [PubMed] [Google Scholar]
- 7.Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 8.Wilcock DM, DiCarlo G, Henderson D, et al. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 10.Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioral impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 11.Kotilinek LA, Bacskai B, Westerman M, et al. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16:599–606. doi: 10.1016/j.coi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization with anti-Abeta antibody in Tg2576. J Neurosci Res. 2003;74:142–147. doi: 10.1002/jnr.10721. [DOI] [PubMed] [Google Scholar]
- 14.Chauhan NB, Siegel GJ, Lichtor T. Effect of age on the duration and extent of amyloid plaque reduction and microglial activation after injection of anti-Abeta antibody into the third ventricle of TgCRND8 mice. J Neurosci Res. 2004;78:732–741. doi: 10.1002/jnr.20298. [DOI] [PubMed] [Google Scholar]
- 15.Chauhan NB, Siegel GJ. Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert Rev Vaccines. 2004;3:717–725. doi: 10.1586/14760584.3.6.717. [DOI] [PubMed] [Google Scholar]
- 16.Chauhan NB. Intracerebroventricular passive immunization with anti-oligoAbeta antibody in TgCRND8. J Neurosci Res. 2007;85:451–463. doi: 10.1002/jnr.21110. [DOI] [PubMed] [Google Scholar]
- 17.Wilcock DM, Alamed J, Gottschall PE, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilcock DM, Munireddy SK, Rosenthal A, et al. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis. 2004;15:11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Oddo S, Billings L, Kesslak JP, et al. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 20.Levites Y, Das P, Price RW, et al. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solomon B, Koppel R, Hanan E, et al. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci U S A. 1996;93:452–455. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jankowsky JL, Slunt HH, Gonzales V, et al. Persistent amyloidosis following suppression of Abeta production in a transgenic model of Alzheimer disease. PLoS Med. 2005;2:e355. doi: 10.1371/journal.pmed.0020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jankowsky JL, Slunt HH, Ratovitski T, et al. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, et al. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 25.Paxinos G, Franklin KBJ, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 2001. [Google Scholar]
- 26.Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 27.Urbanc B, Cruz L, Le R, et al. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci U S A. 2002;99:13990–13995. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 29.Duff K, Eckman C, Zehr C, et al. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 30.Frautschy SA, Yang F, Irrizarry M, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–317. [PMC free article] [PubMed] [Google Scholar]
- 31.Kawarabayashi T, Younkin LH, Saido TC, et al. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 33.Gravina SA, Ho L, Eckman CB, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43) J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 34.Das P, Murphy MP, Younkin LH, et al. Reduced effectiveness of Abeta1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging. 2001;22:721–727. doi: 10.1016/s0197-4580(01)00245-7. [DOI] [PubMed] [Google Scholar]
- 35.Levites Y, Smithson LA, Price RW, et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J. 2006;20:576–78. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.