

Abstract
The delivery of nitric oxide (NO) has important applications in medicine, especially for procedures that involve the vasculature. We report photo-curable biodegradable poly(diol citrate) elastomers capable of slow release of NO. A methacrylated poly(diol citrate) macromonomer was prepared by polycondensation of citric acid with 1, 8-octanediol or 1, 12-dodecanediol followed by functionalization with 2-aminoethyl methacrylate. A miscible NO donor, diazeniumdiolated N, N-diethyldiethylenetriamine, was synthesized and incorporated into the polymer matrix. An elastomeric network was obtained via photo-polymerization of macromonomers upon UV irradiation within three minutes. Films and tubes of the NO-releasing crosslinked macromonomers exhibited strong tensile strength and radial compressive strength, respectively. They also exhibited cell compatibility and biodegradability in vitro. Sustained NO release under physiological conditions was achieved for at least one week. NO release enhanced the proliferation of human umbilical vein endothelial cells but inhibited the proliferation of human aortic smooth muscle cells. Photo-polymerizable NO-releasing materials provide a new approach for the localized and sustained delivery of NO to treat thrombosis and restenosis in the vasculature.
Keywords: photo-polymerization, elastomer, biodegradation, nitric oxide, cell proliferation
Introduction
Since the discovery of the role of nitric oxide (NO) in biological functions1–4, there has been significant interest in how to capture the benefits of this molecule in biomaterials and pharmaceuticals. NO is a gaseous diatomic free radical, which is synthesized from the amino acid L-arginine in a number of tissues by the NO synthases (NOS) with three distinct isoforms, neuronal NOS (nNOS), inducible NOS (iNOS) and endothelial NOS (eNOS)5. NO functions as a cellular messenger and mediator in many physiological and pathophysiological processes. In the cardiovasculature systems, NO plays an important role in regulation of blood pressure and vascular tone, inhibition of platelet aggregation and leukocyte adhesion, prevention of smooth muscle cell proliferation, and stimulation of endothelial cell proliferation6. These characteristics are relevant to the treatment of restenosis in stents and vascular grafts due to neointimal hyperplasia. In fact, the administration of NO at the site of vascular injury has been shown to inhibit both thrombosis and pathways that contribute to restenosis, thus promoting vascular health7, 8.
Over the years, researchers have developed different approaches to engineer biomaterials or devices that can release NO9, 10. Approaches include the use of diazeniumdiolates11, 12 or S-nitrosothiols13 as NO donor compounds that are physically mixed in polymeric matrix, or chemically bonded to the polymer backbone14, 15. For example, Smith and coworkers16 initially blended diazeniumdiolated diethylamine and spermine into poly(ethylene glycol) and polycaprolactone, grafted dipropylenetriamine onto a polysaccharide to enable diazeniumdiolation, and diazeniumdiolated cross-linked polyethyleneimine. The latter has been used to coat poly(tetrafluoroethylene) vascular grafts, which demonstrated less platelet deposition compared to traditional bare grafts. West, et al17, 18 imparted NO-release properties into hydrogels. They prepared the photo-polymerized polyethylene glycol hydrogels with covalently immobilized diazeniumdiolated lysine, diazeniumdiolated diethylenetriamine, and S-nitrosocysteine. The S-nitrosocysteine containing hydrogels inhibited neointima formation in a rat balloon-injury model by approximately 75 % at 14 days. However, hydrogels have limited mechanical properties, which limit their use and therefore the development of biomaterials with an increased range of mechanical properties that can be polymerized in situ and produce NO is warranted.
Our research in this field has been focused on biodegradable elastomers, which are promising materials for various tissue engineering and drug delivery applications19, 20. In particular, biodegradable elastomers may be useful for the replacement of soft tissues or organs, such as blood vessels or lung, which undergo cyclic deformation 21–24. Poly(diol citrate)s are a new class of biodegradable and biocompatible polyesters that have been proposed for use in several tissue engineering and regenerative medicine applications25, 26. We recently reported the development of nitric oxide-releasing poly(diol citrate) elastomers, which were fabricated through polycondensation of citric acid, aliphatic diols, and an ethylendiaminediol that allowed the formation of diazeniumdiolate functional groups27. Diazeniumdiolated poly(diol citrate)s were shown to release NO for 3 days in vitro and significantly reduce neointimal hyperplasia when implanted as perivascular wrap in a rat carotid artery injury model28. Although promising, a longer duration of NO release and the ability to rapidly cure the biomaterial in situ would open up new areas for the application of NO releasing technology. In this report, we describe a new approach to develop long-lasting NO-releasing elastomers based on photo-polymerizable poly(diol citrate)s blended with miscible diazeniumdiolated NO donors.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used as received unless otherwise indicated. All cells and media were purchased from Lonza Inc (Baltimore, MD). Fresh porcine aortas were obtained from local slaughter house, Park Packing Company (Chicago, IL). Nitric oxide reaction chamber was purchased from Parr Instrument Company (Moline, IL). Nitric oxide was obtained from Matheson Gas Products (Montgomeryville, PA).
1. Synthesis of methacrylated poly(diol citrate) macromonomers
The methacrylated poly(1, 8-octanediol citrate) and methacrylated poly(1, 12-dodecanediol citrate) macromonomers (MA-POC and MA-PDDC) were prepared by polycondensation of citric acid with 1, 8-octanediol or 1, 12-dodecanediol followed by a covalent coupling reaction with 2-aminoethyl methacrylate. Typically, under the nitrogen atmosphere, a mixture of citric acid (76.8 g) and 1, 8-octanediol (29.2 g) was melted at 165 °C and subsequently reacted at 140 °C for 40 min to obtain a viscous prepolymer poly(1, 8-octanediol citrate) (POC). The prepolymer was dissolved in ethanol and purified by precipitation of the ethanol solution into deionized water (Millipore water purification system) followed by freeze drying. 20 g of purified POC prepolymer was dissolved in 200 ml of N, N-Dimethylformamide (DMF), mixed with 22.7 g of N, N, N′, N′-Tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU, AnaSpec, Inc, Fremont, CA), 9.9 g of 2-aminoethyl methacrylate, and 15.5 g of N, N-diisopropylethylamine (DIEA) at 5 °C, and reacted for 5 hours at room temperature. The mixture was precipitated into water and subsequently lyophilized.
2. Synthesis of diazeniumdiolated N, N-diethyldiethylenetriamine (DEDETA/NO)
DEDETA/NO was prepared by treating N, N-diethyldiethylenetriamine (DEDETA) with nitric oxide in acetonitrile. A solution of DEDETA (0.5 g) in anhydrous acetonitrile (CH3CN, 15 ml) was placed in a standard Parr hydrogenation bottle, flushed with Argon and pressurized with NO gas at 70 psi for 5 days. After the reaction, the bottle was vented and flushed with Argon. The yellowish product was isolated by precipitating the compound into ether and dried under vacuum.
3. Photo-polymerization of poly(diol citrate) macromonomers
The photo-crosslinked poly(diol citrate) (cMA-POC or cMA-PDDC) was synthesized via UV polymerization of macromonomer (MA-POC or MA-PDDC) in ethanol (~ 60 wt%) with 1 wt% of 2-hydroxy-2-methylpropiophenone (Polyscience, Warrington, PA) as an initiator at 365 nm wavelength UV light (ELC-251 Ultraviolet Source, Electro-Lite Corporation). After three minutes exposure to UV light, cured materials were rinsed with ethanol and dried under vacuum. NO donor-containing polymer (cMA-POC/NO or cMA-PDDC/NO) were prepared via the polymerization of macromonomer MA-POC or MA-PDDC blended with the NO donor DEDETA/NO. Ethanol was used as a common solvent. Tubes were prepared by injecting the macromonomers into a mold fabricated with two silicone tubes with different diameters (Specialty Manufacturing Inc, Saginaw, MI) (thickness ~ 0.8 mm, outer diameter ~ 6 mm). The macromonomers were also coated and then polymerized onto the luminal surface of a porcine artery to assess the ability of the polymer to cure on tissue. The crosslink density of the materials were assessed using the equation derived from rubber elasticity theory: n = E0/3RT, where E0 is Young’s modulus, R is the universal gas constant 8.314 J/molK, T is the temperature in K19, 29.
4. Characterization of polymers and NO donors
The nuclear magnetic resonance (NMR) spectra were recorded with a Varian INOVA 500 MHz. Tensile tests and lateral compressive tests were conducted on an Instron 5544 mechanical tester equipped with 500N load cell. Bending tests were performed on a MTS Sintech 20/G Universal Testing Machine with three-point bend test apparatus and 8000 N load cell. Thermal properties of the elastomers were characterized with Perkin-Elmer DSC-7 differential scanning calorimeter. VARIAN Cary 50 Bio UV-visible spectrophotometer was used for wavelength scan and kinetics studies of NO donors.
5. In vitro degradation studies
In vitro degradation rate was tested by measuring the mass loss of the polymers after incubation at 37 °C in pH 7.4 phosphate buffered saline (PBS). Disk-shaped specimens (~ 6 mm in diameter, ~ 1 mm in thickness) of polymer films were placed in a test tube containing 10 ml of pH 7.4 PBS and incubated at 37 °C. Four samples were tested and every sample was measured in triplicate. Samples were collected at various time points, and then washed with distilled water and freeze-dried. Mass loss was calculated by comparing the initial mass of each sample with the mass measured at a given time point.
6. Assessment of NO release
Colorimetric analysis via the Griess reaction was used to indirectly monitor the NO release in pH 7.4 PBS at 37 °C. DEDETA/NO was dissolved in PBS and samples were collected at a given time point for measurements. Disk-shaped specimens (~ 6 mm in diameter, ~ 1 mm in thickness) of polymer films were placed in a test tube containing 1 ml of pH 7.4 PBS and incubated at 37 °C. The supernatant samples were collected at a given time point and fresh PBS was added after each collection. Four samples were tested and every sample was measured in triplicate. After the sequential incubation with Greiss reagents, sulfanilamide and N-1-napthylethylenediamine dihydrochloride, the absorbance of samples at 550 nm was measured on TECAN Safire microplate reader. Samples that did not contain NO donors were used as controls.
7. In vitro cell compatibility studies
The in vitro cell compatibility was assessed via cell culture with human umbilical vein endothelial cells (HUVECs) and human aortic smooth muscle cells (HASMCs). Polymer disc samples ~ 6 mm in diameter and ~ 0.5 mm in thickness were sterilized with 70 % ethanol solution and incubated in Dulbecco’s Modified Eagle’s Medium (DMEM) at 37 °C for 1 – 3 days prior to cell seeding. HUVEC and HASMC were seeded on the materials at a density of 10,000 – 30,000 cells/ml in the corresponding medium, endothelium growth medium (EGM-2) and smooth muscle cell growth medium (SmGM-2). Cultures were then maintained under 5 % CO2 at 37 °C. After 24 hours, cells were assessed for morphology and viability with a LIVE/DEAD Viability/Cytotoxicity Kit (Invitrogen, Molecular Probes, Eugene, OR) and imaged with an inverted light microscope (Nikon Eclipse, TE2000-U) equipped with a Photometrics CoolSNAP HQ camera (Silver Spring, MD). Cell proliferation was quantified by DNA concentration with Quant-iT PicoGreen ® dsDNA Reagent and Kits (Invitrogen, Molecular Probes, Eugene, OR). Tissue-culture polystyrene (TCP) was used as a control surface. Three samples were used for each study and every sample was measured in triplicate.
Results and Discussion
1. MA-POC and MA-PDDC can be cured within 3 minute via UV light
Polycondensation of citric acid with 1, 8-octanediol or 1, 12-dodecanediol can produce polyester prepolymers POC or PDDC (average molecular ~ 1000 mol/g)25. Acrylate and fumarate groups can be incorporated into the elastomer as a second type of crosslink to further modulate the mechanical properties of poly(diol citrate)30. However, efficiency of functionalization can be difficult to control when acrylate and fumarate groups are incorporated during the high temperature melt condensation reactions. In this study, we functionalize poly(diol citrate) via post modification of the prepolymers. Carboxyl groups at the ends and sides of prepolymer chains reacted with amino groups of methacrylated monomers 2-aminoethyl methacrylate resulting in the methacrylated poly(diol citrate) macromonomers (MA-POC and MA-PDDC) (Figure 1). The methacrylated prepolymers can be cross-linked by free radical polymerization to form a solid polymer network. MA-POC and MA-PDDC were cured via UV-polymerization with 1 wt% initiator of 2-hydroxy-2-methylpropiophenone within 3 minutes to form tough materials. The gel content of cMA-POC or cMA-PDDC is > 90 %, which is estimated as the gel fraction remaining after the cured film was soaked and rinsed with ethanol for several hours. Both MA-POC and MA-PDDC can be easily photo-polymerized into various shapes, such as films and tubes, and coated on surface of artery (Figure 1).
Figure 1.

Photo-crosslinked poly(diol citrate).
The structures of MA-POC and MA-PDDC macromonomers in DMSO were characterized by 1H NMR (Figure 2). The methylene groups in aliphatic diol were indicated at δ1.2 [-OCH2CH2(CH2)4CH2CH2O-], δ1.5 [-OCH2CH2(CH2)4CH2CH2O-], and δ4.0 ppm [-OCH2CH2(CH2)4CH2CH2O-]. The multiple peaks at δ2.7 ppm are from -CH2- in citric acid. The methylene groups in methacrylate were indicated at δ3.3 [-NHCH2CH2-] and δ4.0 ppm [-CH2CH2O-]. The a pair of peaks at δ5.6 and δ6.0 ppm are from –C(CH3)=CH2, and single peak at δ1.8 from –C(CH3)=CH2 in methacrylate. Based on the integration of peaks, ~ 70 % of carboxylic groups were methacrylated. MA-PDDC showed the similar spectra, in which the signal at δ1.2 increased due to the increasing methylene amount in MA-PDDC.
Figure 2.

1H NMR spectra of MA-POC (A) and MA-PDDC (B).
The cross-linking densities n (moles of active network chains per unit volume) of cMA-POC and cMA-PDDC were calculated to be 38.5 and 18.8 mol/m3 at 22°C, respectively. The DSC measurements demonstrated the glass transition temperature (Tg) to be 32.4 °C and 20.4 °C for UV-crosslinked polymers cMA-POC and cMA-PDDC, respectively. No crystallization and melting temperatures were observed in the detection range from −40 to 150 °C, suggesting that the cured polymers are amorphous.
Tensile tests of polymer films showed stress–strain curves with tough elastomeric characteristic (Figure 3a). At room temperature, dry cMA-POC in glassy state has a high tensile strength ~ 22 MPa and a short elongation ~ 18 %, displaying more plasticity. While cMA-PDDC above or close to its Tg exhibits more elasticity, i.e. lower tensile strength ~ 14 MPa and longer elongation ~ 45 %. After incubation in water for 28 hrs at 37°C, the polymers are hydrated and softer than the corresponding dry state due to the temperature being above Tg and the dissociation for hydrogen bonding in water. The tensile strength of wet cMA-POC and cMA-PDDC decreases to ~ 10.6 and 6.5 MPa and elongation increase to ~ 30.9 and 50.2 % respectively. The same tendency was observed for the lateral compression data of polymer tubes. cMA-POC is stiffer than cMA-PDDC in both dry and wet states. As displayed in Figure 3c, cMA-POC has higher compressive stress and shorter plateau region than that of cMA-PDDC. cMA-PDDC is more elastic, the lateral compressive deformation of tubular cMA-PDDC could extend to ~ 40 % at dry state and ~ 52 % at wet state when compared to those of cMA-POC ~ 30 % at dry state and ~ 45 % at wet state.
Figure 3.

Mechanical properties of 3-minute UV-cured cMA-POC, cMA-PDDC, cMA-POC/NO1.1 and cMA-PDDC/NO1.1.
A, B. Tensile tests, 22 °C, 500 mm/min.
C, D. Lateral compressive tests, 22 °C, 10 mm/min.
Both cMA-POC and cMA-PDDC degrade in aqueous solution. The in vitro degradation data for cMA-POC and cMA-PDDC disks in Figure 4 confirm ~ 19, 26, and 32 % of mass loss for cMA-POC and ~ 13, 23 and 26 % of mass loss for cMA-PDDC after 1, 3, and 6 months of incubation in pH 7.4 PBS at 37 °C, respectively.
Figure 4.

Mass loss of photo-crosslinked poly(diol citrate)s after incubation in pH 7.4 PBS, 37 °C. Data are mean ± standard deviation (n = 4).
2. Diazeniumdiolated N, N-diethyldiethylenetriamine (DEDETA/NO)
The reaction of DEDETA with gaseous NO results in a new diazeniumdiolate DEDETA/NO. This new diazeniumdiolate donor is based on structurally similar diazeniumdiolated diethylenetriamine (DETA/NO), which has a half-life of 20 hrs31. DETA/NO is white powder that cannot be dissolved in ethanol, the solvent used to dissolve the poly(diol citrate) prepolymer. The ethyl substitutes on DEDETA increase the solubility of the diazeniumdiolate in organic solvents. DEDETA/NO is easily dissolved in ethanol and miscible with poly(diol citrate) macromonomers to form a clear solution. The structure of DEDETA/NO was confirmed by 1H NMR in D2O 31, 32: δ1.3 ppm (6H, –CH3), δ2.9 (2H, t), δ2.95 ppm (2H, t), δ3.05 (2H, t), [(CH3CH2)2NCH2CH2NNONOCH2CH2NH2], and δ3.2 ppm [6H, N(CH2-)3] (Figure 5). Compared with the parent amine DEDETA, the proton chemical shifts of diazeniumdiolate DEDETA/NO moved downfield. Dissolving DEDETA/NO in both 0.01M sodium hydroxide aqueous solution and ethanol showed the characteristic UV absorption at 252 nm. Kinetic studies of the absorbance at 252 nm shows the half-life of DEDETA/NO in pH 7.4 PBS to be 25.4 hrs. The NO release profile obtained from the Griess assay showed that the NO release of DEDETA/NO was completed by 2 days, in accordance with the UV/Vis spectroscopy results (Figure 6a).
Figure 5.

1H NMR spectra of DEDETA (A) and DEDETA/NO (B).
Figure 6.

NO release in PBS (pH 7.4, 37 °C) as assessed by Griess reaction. Data are mean ± standard deviation (n = 4).
A. DEDETA/NO donor.
B. cMA-POC/NO1.1 and cMA-PDDC/NO1.1.
C. Effect of DEDETA/NO concentration in cMA-PDDC/NO.
3. Photo-crosslinked cMA-POC and cMA-PDDC containing DEDETA/NO slowly release NO
Lipophilic NO donors have been prepared to preferentially stay in the organic polymer phase via favorable partition coefficients33. After polymerization, DEDETA/NO was embedded in the polymer matrix to form DEDETA/NO-containing poly(diol citrate) materials cMA-POC/NO or cMA-PDDC/NO without visible phase-separation. Inclusion of the NO donor reduced the crosslink density. When DEDETA/NO was incorporated into the prepolymers at the feed ratio of 1.1 wt%, the cross-linking densities of cMA-POC/NO1.1 and cMA-PDDC/NO1.1 were 36.9 and 11.0 mol/m3 at 22°C, respectively.
The elastomeric properties of the polymers are maintained after incorporation of DEDETA/NO, although the tensile strength measured under dry and wet conditions are decreased and the elongation are increased (Figure 3b). The reduced compression stress and larger compressive deformation for the tubular materials are also shown in Figure 3d, indicating that cMA-POC/NO and cMA-PDDC/NO are more elastic than cMA-POC and cMA-PDDC, respectively, after curing for three minutes.
Upon distribution of DEDETA/NO into the polymeric matrix, the NO release under physiological conditions was significantly delayed. The sustained release of NO lasted at least one week (Figure 6b). The hydrophobic microenvironment of polymers protected the NO donor from water and delayed NO release. cMA-PDDC/NO showed more extended release than cMA-POC/NO. Increasing the content of the NO donor in the polymer matrix from 1.1 to 1.8 wt% increased the NO release rate and flux (Figure 6c). The maximum percent DEDETA/NO that could be incorporated in the pre-polymer was 2.4 wt%. Higher percentages require longer UV-irradiation times (> 3 minutes) to cure the polymer. NO release from polymer films is in the range to stimulate the desired cellular responses34.
NO release of DETA/NO indirectly calculated by Griess assay is lower than that detected by chemiluminescence. NO donor may be lost during the rinsing steps with ethanol to remove unreacted prepolymers after exposure to UV light and the drying process to remove the solvent. Also, diazeniumdiolates are photosensitive11, 35, 36. The NO donor may be dissociated during UV light initiation. A 3-minute UV light exposure on an ethanol solution of DEDETA/NO results in ~ 16 % decrease of the peak at 252 nm and appearance of a new peak at ~ 330 nm, indicating that UV photolysis and formation of nitrosamine11. However, this side reaction is significantly reduced (less than 5 %) when visible light initiators are used (data not shown).
4. NO-releasing cMA-POC and cMA-PDDC are compatible with cells
Biocompatibility of polymers was investigated by culturing HUVECs and HASMCs. After 24 hrs of incubation, green fluorescence (live cells) stains showed the characteristic cobblestone morphology for HUVECs (30,000 cells/well) and spindle-like morphology HASMCs (15,000 cells/well) on all materials, with or without NO donors. Few red fluorescence (dead cells) positive cells were found (Figure 7).
Figure 7.
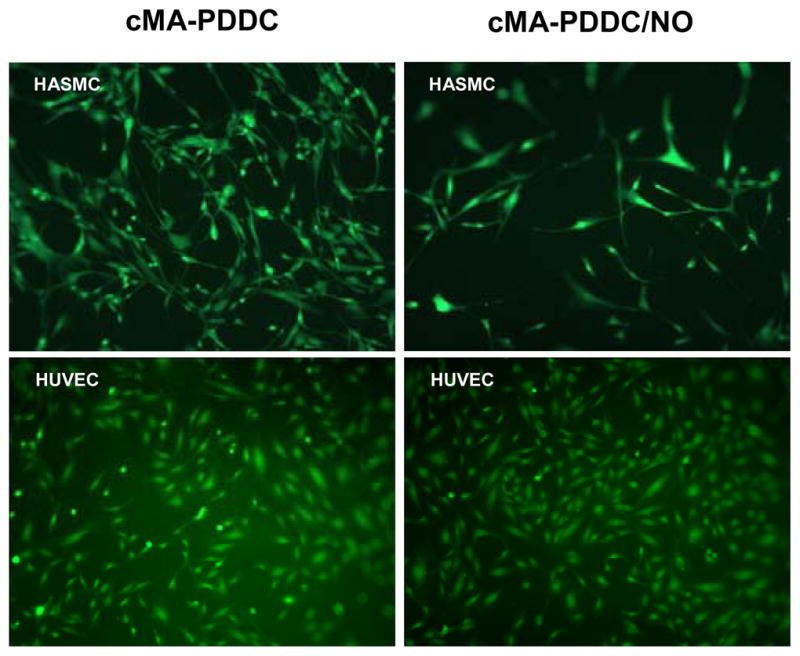
Viability and cytotoxicity of HASMCs (15,000 cells/well) and HUVECs (30,000 cells/well) on cMA-PDDC and cMA-PDDC/NO1.1 (24 hrs culture, 10x magnification).
The cellular effects of NO are dependent on the NO dose from the materials and cell type17, 28, 34, 37. Culturing the cells on cMA-PDDC/NO with the appropriate range of NO concentration, which was estimated from Figure 6, resulted in a cell response that is consistent with in vivo observations when cells are exposed to endogenous NO (e.g. decrease and increase in smooth muscle cell and endothelial cell proliferation, respectively). When total NO release in cell culture is ~ 3.6 nmol, HASMCs (15,000 cells/well) DNA content from cMA-PDDC/NO sample was significantly lower than that from the corresponding cMA-PDDC sample. For HUVECs (30,000 cells/well), DNA content from cMA-PDDC/NO sample was higher than that from cMA-PDDC sample with a total NO release in cell culture ~ 1.8 nmol (Figure 8).
Figure 8.

Effect of NO on cell proliferation on cMA-PDDC. Data are mean ± standard deviation (n = 3). * Student’s t-test, p < 0.05. DNA content from cells grown on cMA-PDDC/NO1.1 normalized to that from cells grown on cMA-PDDC.
A. 24 hrs culture with HASMCs (15,000 cells/well).
B. 24 hrs culture with HUVECs (30,000 cells/well).
Conclusion
We developed injectable and fast-curing materials based on poly(diol citrate)s (MA-POC and MA-PDDC) and a novel diazeniumdiolate (DEDETA/NO) that are biodegradable, cell compatible, mechanically strong upon curing and can provide long-term NO release. The NO-containing polymer network could be cured within three minutes upon exposure to UV light and released NO for at least one week. These materials may be useful in the prevention of restenosis and thrombosis after vascular interventions such as balloon angioplasty, stent deployment, and bypass grafting, as well as for the blood contacting surface of implantable devices.
Acknowledgments
This work was supported by National Institutes of Health Challenge Grant RC1HL100491 and Northwestern Memorial Foundation, Dixon Translational Priority Research Initiative Grant.
References
- 1.Furchgott RF, Zawadzki JV. Nature. 1980;288:373. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 2.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Proc Natl Acad Sci USA. 1987;84:9265. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ignarro LJ, Byrns RE, Buga GM, Wood KS. Circ Res. 1987;61:866. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 4.Palmer RM, Ferrige AG, Moncada S. Nature. 1987;327:524. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 5.Palmer RM, Rees DD, Ashton DS, Moncada S. Biochem Biophys Res Commun. 1988;153:1251. doi: 10.1016/s0006-291x(88)81362-7. [DOI] [PubMed] [Google Scholar]
- 6.Ahanchi SS, Tsihlis ND, Kibbe MR. J Vasc Surg. 2007;45(suppl A):A64. doi: 10.1016/j.jvs.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 7.Wu YD, Meyerhoff ME. TALANTA. 2008;75(3):642. doi: 10.1016/j.talanta.2007.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pearce CG, Najjar SF, Kapadia MR, Murar J, Eng J, Lyle B, Aalami OO, Jiang Q, Hrabie JA, Saavedra JE, Keefer LK, Kibbe MR. Free Radical Biol Med. 2008;44:73. doi: 10.1016/j.freeradbiomed.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keefer LK. Curr Top Med Chem. 2005;5:625. doi: 10.2174/1568026054679380. [DOI] [PubMed] [Google Scholar]
- 10.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1091. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 11.Hrabie’ JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 12.Maragos CM, Morley D, Wink DA, Dunams TM, Saavedra JE, Hoffman A, Bove AA, Isaac L, Hrabie’ JA, Keefer LK. J Med Chem. 1991;34:3242. doi: 10.1021/jm00115a013. [DOI] [PubMed] [Google Scholar]
- 13.Williams DLH. Acc Chem Res. 1999;32:869. [Google Scholar]
- 14.Varu VN, Tsihlis ND, Kibbe MR. Vasc Endovasc Surg. 2009;43:121. doi: 10.1177/1538574408322752. [DOI] [PubMed] [Google Scholar]
- 15.Frost MC, Reynolds MM, Meyerhoff ME. Biomaterials. 2005;26:1685. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Smith DJ, Chakravarthy D, Pulfer S, Simmons ML, Hrabie’ JA, Citro ML, Saavedra JE, Davies KM, Hutsell TC, Mooradian DL, Hanson SR, Keefer LK. J Med Chem. 1996;39:1148. doi: 10.1021/jm950652b. [DOI] [PubMed] [Google Scholar]
- 17.Bohl KS, West JL. Biomaterials. 2000;21:2273. doi: 10.1016/s0142-9612(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 18.Bohl Masters KS, Lipke EA, Rice EEH, Liel MS, Myler HA, Zygourakis C, Tulis DA, West JL. J Biomater Sci, Polym Ed. 2005;16:659. doi: 10.1163/1568562053783722. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Ameer GA, Sheppard BJ, Langer R. Nature Biotechnology. 2002;30:602. doi: 10.1038/nbt0602-602. [DOI] [PubMed] [Google Scholar]
- 20.Coneski PN, Rao KS, Schoenfisch MH. Biomacromolecules. 2010;11:3208. doi: 10.1021/bm1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bettinger CJ. Pure and Appl Chem. 2011;83(1):9. [Google Scholar]
- 22.Amsden B. Soft Matter. 2007;3:1335. doi: 10.1039/b707472g. [DOI] [PubMed] [Google Scholar]
- 23.Serrano MC, Chung EJ, Ameer GA. Adv Funct Mater. 2010;20:192. [Google Scholar]
- 24.Webb AR, Yang J, Ameer GA. Expert Opin Biol Ther. 2004;4:801. doi: 10.1517/14712598.4.6.801. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Webb AR, Ameer GA. Adv Mater. 2004;16:511. [Google Scholar]
- 26.Kibbe MR, Martinez J, Popowich DA, Kapadia MR, Ahanchi SS, Aalami OO, Jiang Q, Webb AR, Yang J, Carroll T, Ameer GA. J Biomed Mater Res. 2010;93A(1):314. doi: 10.1002/jbm.a.32537. [DOI] [PubMed] [Google Scholar]
- 27.Zhao H, Serrano MC, Popowich DA, Kibbe MR, Ameer GA. J Biomed Mater Res. 2010;93A(1):356. doi: 10.1002/jbm.a.32536. [DOI] [PubMed] [Google Scholar]
- 28.Serrano MC, Vavra AK, Jen M, Hogg ME, Murar J, Martinez J, Keefer LK, Ameer GA, Kibbe MR. Macromol Biosci. 2011;11(5):700. doi: 10.1002/mabi.201000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tran RT, Thevenot P, Gyawali D, Chiao JC, Tang L, Yang J. Soft Matter. 2010;6:2449. doi: 10.1039/C001605E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Ameer GA. J Appl Polym Sci. 2009;114(3):1464. [Google Scholar]
- 31.Hrabie’ JA, Klose JR, Wink DA, Keefer LK. J Org Chem. 1993;58:1472. [Google Scholar]
- 32.Zhou Z, Annich GM, Wu Y, Meyerhoff ME. Biomacromolecules. 2006;7(9):2565. doi: 10.1021/bm060361s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Batchelor MM, Reoma SL, Fleser PS, Nuthakki VK, Callahan RE, Shanley CJ, Politis JK, Elmore J, Merz SI, Meyerhoff ME. J Med Chem. 2003;46:5153. doi: 10.1021/jm030286t. [DOI] [PubMed] [Google Scholar]
- 34.Jun HW, Taite LJ, West JL. Biomacromolecules. 2005;6:838. doi: 10.1021/bm049419y. [DOI] [PubMed] [Google Scholar]
- 35.Srinivasan A, Kebede N, Saavedra JE, Nikolaitchik AV, Brady DA, Yourd E, Davies KM, Keefer LK, Toscano JP. J Am Chem Soc. 2001;123:5465. doi: 10.1021/ja002898y. [DOI] [PubMed] [Google Scholar]
- 36.Pavlos CM, Xu H, Toscano JP. Free Rad Biol & Med. 2004;37(6):745. doi: 10.1016/j.freeradbiomed.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 37.Kushwaha M, Anderson JM, Bosworth CA, Andukuri A, Minor WP, Lancaster JR, Jr, Anderson PG, Brott BC, Jun H-W. Biomaterials. 2010;31:1502. doi: 10.1016/j.biomaterials.2009.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]