

Abstract
Objective
Anti-AMPAR encephalitis is a recently discovered disorder characterized by the presence of antibodies in serum or cerebrospinal fluid against the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. Here, we examine the antigenic specificity of anti-AMPAR antibodies, screen for new patients, and evaluate functional effects of antibody treatment of neurons.
Methods
We developed a fusion protein (FP)-based western blotting test for anti-AMPAR encephalitis antibodies. Antibody specificity was also evaluated using immunocytochemistry of HEK293 cells expressing deletion mutants of AMPAR subunits. Purified patient immunoglobulin G (IgG) or AMPAR antibody-depleted IgG was applied to live neuronal cultures; amplitude and frequency of miniature excitatory postsynaptic currents (mEPSCs) were measured to evaluate functional effects of antibodies.
Results
Using both immunocytochemistry and FP western blots, we defined an antigenic region of the receptor in the bottom lobe of the amino terminal domain. Additionally, we used FPs to screen 70 individuals with neurologic symptoms of unknown cause and 44 patients with no neurologic symptoms or symptoms of known neuroimmunological origin for anti-AMPAR antibodies. Fifteen of the 70 individuals had anti-AMPAR antibodies, with broader antigenic reactivity patterns. Using purified IgG from an individual of the original cohort of anti-AMPAR encephalitis patients and a newly discovered patient, we found that application of IgG from either patient cohort caused an AMPAR antibody-dependent decrease in the amplitude and frequency of mEPSCs in cultured neurons.
Interpretation
These results indicate that anti-AMPAR antibodies are widespread and functionally relevant; given the robust response of patients to immunomodulation, this represents a significant treatable patient population.
Introduction
Anti-AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor) encephalitis is one of several newly described autoimmune diseases that target synaptic surface proteins and cause psychiatric and neurologic symptoms.1,2 While most of these diseases were originally described in only a few individuals,3–8 the most prominent of them, anti-NMDAR (N-methyl-D-aspartate receptor) encephalitis, has since been diagnosed in hundreds of patients,9 and is one of the most common forms of immune encephalitis.10 Currently anti-AMPAR encephalitis has only been described in 16 patients over four case series,4,11–13 but even in those few cases certain trends, challenges, and ambiguities have emerged. Fifteen patients reported were women over the age of 30; 14 were diagnosed with limbic encephalitis; 13 had acute or subacute memory loss; eight had tumors; and six had seizures. These findings led to the broad characterization of anti-AMPAR encephalitis as a paraneoplastic disorder with seizures and memory deficits that primarily affects adult women. Antibodies from one anti-AMPAR encephalitis patient caused a decrease in surface and overall AMPAR levels4; given the well-established role of AMPARs in learning and memory14–18 and seizures,19–23 the symptoms of anti-AMPAR encephalitis could logically be mediated by decreases in AMPAR levels. Both anti-NMDAR and anti-AMPAR encephalitis resolve substantially with immunomodulation1; thus, these diseases represent treatable forms of neurologic and psychiatric impairment.
AMPARs are heterotetrameric receptors comprised of varying combinations of four subunits, GluA1-4. To date, anti-AMPAR encephalitis has been diagnosed using immunocytochemistry on HEK293 cells transfected with GluA1 and/or GluA2 subunits. The original patient cohort contained patients whose antibodies reacted with GluA1, GluA2, or both. Antibodies bind to an extracellular site on the receptor.4 AMPARs contain two extracellular domains: an amino-terminal domain (ATD) of roughly 400 amino acids (aa), and a ligand-binding domain made up of two sections of 120–150 aa, S1 and S2; three transmembrane domains (TM I, III, IV), and a transmembrane loop (TM II), and a C-terminal cytoplasmic tail that interacts with scaffolding proteins and signaling cascades.24
Here, we have used bacterial fusion proteins (FPs) composed of the ATD and ligand-binding domain of GluA1 and GluA2 to test for anti-AMPAR antibodies in patient serum or cerebrospinal fluid (CSF). This has also allowed us to investigate the antigenic location and differences in CSF and serum reactivity, explore anti-AMPAR reactivity in additional patients, and examine the physiological effects of anti-AMPAR antibodies from both cohorts of patients.
Materials and Methods
Patient material
CSF and serum were collected in accordance with the University of Pennsylvania Institutional Review Board guidelines and stored at −80°C. In immunocytochemistry experiments, patient CSF was used at a dilution of 1:10–1:100; control CSF was used at 1:10–1:20. For FP western blots, serum was used at 1:250–1:1000 and was precleared with uninduced bacteria. CSF was used at 1:100–1:1000; if 1:100 concentrations gave high levels of background, CSF was precleared with uninduced bacteria.
HEK293 transfection
HEK293 cells were cultured and transiently transfected as previously described.25 Briefly, cells were plated on poly-L-lysine-coated dishes in minimum essential medium containing 7.5% fetal bovine serum, 2.5% horse serum, 1% penicillin/streptomycin, and 1% l-glutamine, maintained at 37°C/5% CO2. Cells were transfected 1 day later using calcium phosphate. Transfection solution contained 2 μg of total DNA per mL of media.
Preparation of primary neuronal cultures
Neuronal cultures were prepared as previously described.25 Briefly, cortices of E17–19 rats were gently dissociated with trypsin and mechanical disruption, washed (1× Hank's Buffered Saline Solution), and seeded at variable densities on poly-D-lysine-coated plates. Cultures were maintained at 37°C/5% CO2 and fed with neurobasal medium supplemented with B27. All procedures were approved by the University of Pennsylvania and Children's Hospital of Philadelphia Institutional Animal Care and Use Committees.
IgG purification
A volume of 500 μL serum was incubated with 300 μL protein A/G agarose beads (30–60 min). Unbound material was collected and the beads were washed 3× phosphate buffered saline (PBS) + 0.1% Igepal and 3× PBS. Immunoglobulin G (IgG) was eluted (0.1 mol/L sodium citrate buffer, pH 2.7) directly into tubes containing 1.5 mol/L Tris pH 8.8 (10% of eluate). IgG was applied to neuronal cultures at 100 μg/mL for 24–36 h.
Mutant receptor and FP construction, solubilization, and preclearing/depletion
Receptors containing large deletions were made as previously described.26 FPs were constructed in a thioredoxin-His patch vector (Invitrogen, K102-01, Grand Island, NY) and expressed in BL21 or pLysS-BL21 Escherichia coli. External domain (ext dom) FPs and the GluA3-ATD were prone to protease degradation during production; therefore, the pLysS strain was used to minimize degradation. The GluA3-external domain and ATD continued to show high levels of breakdown and were excluded from further use. Bacteria were centrifuged (5000g, 4°C, 20 min) and stored in −20°C until solubilization.
FP bacterial pellets were solubilized (50 mmol/L potassium phosphate, 400 mmol/L NaCl, 100 mmol/L KCl, 10% glycerol, 0.5% TritonX-100, 10 mmol/L imidazole, 1:500 Calbiochem protease inhibitor cocktail III), sonicated, and centrifuged (15,000g, 4°C, 10 min). Supernatants were discarded; pellets, containing FP inclusion bodies, were resuspended in 1× sample buffer, boiled (10 min), and stored at −20°C until use.
Aliquots of uninduced bacteria for serum/CSF preclearing were solubilized by the same procedure, omitting TritonX-100. Inclusion body pellets were incubated in Tris-buffered saline with Tween (TBST) with patient serum or CSF (1 h, 4°C). Samples were centrifuged (13.2 K*g, 10 min, 4°C) to repellet inclusion bodies; TBST-serum/CSF supernatants were collected for use in western blotting. For depletion experiments, uninduced bacteria or bacteria expressing GluA2-ext dom or GluA2-S1 FPs were solubilized and incubated with patient or control IgG (4°C, 1–2 h or overnight; six rounds of incubation, three GluA2-ext dom and three GluA2-S1). Depletion was verified using treated IgG in western blotting of solubilized FPs, using an horseradish peroxidase (HRP)-linked protein standard as an internal exposure control (MagicMark; Invitrogen). Depleted IgG was applied to neuronal cultures at a volume equal to preabsorption 100 μg/mL.
Western blotting
Solubilized FPs were electrophoresed and transferred to nitrocellulose membranes. Blots were incubated with precleared serum or CSF, HRP-linked anti-human secondary antibody (1:3000–1:10,000) and developed. All FPs included an N-terminal thioredoxin tag; therefore, FP expression was assessed with anti-thioredoxin (Trx) antibody (1:5000; Invitrogen).
Immunocytochemistry
Immunocytochemistry was performed as described.27 Cells were incubated in commercial AMPAR or NMDAR antibody (GluA2: 07-598, 1:1000, Millipore, Billerica, MA; GluA1: AB1504, 1:500, Millipore; GluN1: 556308, 1:1000, BD Biosciences, San Jose, CA) and CSF (range 1:10–1:100) in 1% bovine serum albumin in PBS overnight, 4°C. Each experiment included CSF from at least one individual without anti-AMPAR encephalitis; none showed CSF staining of AMPARs. Staining intensity was quantified using ImageJ software (National Institutes of Health, Bethesda, MD) as previously described.27
mEPSC recordings
Primary rat cerebrocortical neurons (1–4 × 105 cells per 35 mm dish) were voltage-clamped at −60 mV as previously described28 using borosilicate glass pipettes (World Precision Instruments, Sarasota, FL), with resistances of 5–10 MΩ. Intrapipette solution contained 150 mmol/L potassium gluconate, 10 mmol/L HEPES, 10 mmol/L ethylene glycol tetraacetic acid, 2 mmol/L MgCl2, 1.4 mmol/L CaCl2, and 2 mmol/L Mg-ATP, pH 7.35, 310–315 mOsm. Miniature excitatory postsynaptic current (mEPSCs) were recorded at room temperature in extracellular solution (155 mmol/L NaCl, 3 mmol/L KCl, 3 mmol/L CaCl2, 10 mmol/L HEPES, 10 μmol/L bicuculline methiodide, 500 nmol/L tetrodotoxin, pH 7.35, 310–320 mOsm). Recording signals were amplified using an Axopatch-1D amplifier (Molecular Devices, Sunnyvale, CA), acquired at 10 kHz, filtered at 2 kHz, and saved using pClamp 10 for off-line analysis. A minimum of 1 min of traces were filtered at 1 kHz Bessel and quantified using Clampfit template match (Axon Instruments, Sunnyvale, CA).
Statistical analysis
Quantifications of immunocytochemistry images and electrophysiological measurements are expressed as mean ± standard error of the mean (SEM). Statistical comparisons were made using one-way analysis of variance (ANOVA) plus Tukey's or Dunnett's post hoc testing, as indicated in figure legends. All statistical analyses were performed with GraphPad Prism (GraphPad Software, La Jolla, CA), and values of P < 0.05 were considered significant.
Results
Anti-AMPAR patient antibodies bind the bottom lobe of the ATD
Using serum and/or CSF from patients previously diagnosed with anti-AMPAR encephalitis, we found that anti-AMPAR patient antibodies react robustly with FPs containing the external domains of GluA1 or GluA2 (Fig.1), indicating that antigenicity is preserved in denatured FPs. Similar results were found with all previously diagnosed anti-AMPAR encephalitis patients tested (16 total). Further, FP reactivity reflects both major and minor reactivity patterns: in a patient with primary reactivity to GluA2 and secondary reactivity to GluA1 in transfected HEK293 cells, the same pattern was observed in FP western blots (Fig.1A, lanes C and C′; 1C). These results suggest that this approach is sensitive and sufficiently linear to be used in the delineation of multiple antibody species within one sample.
Figure 1.
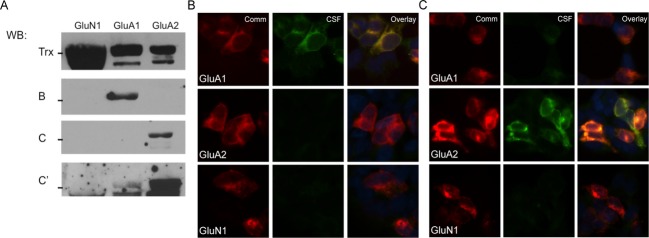
Patient CSF recognition of external domain fusion proteins is similar to staining of cells transfected with full subunits. (A) Patient CSF (blots B and C) recognizes a band of the correct size in transfected samples, indicating that native conformation may not be necessary for patient antibody recognition of AMPARs. GluA1, GluA2, and GluN1 all expressed well, with minimal proteolytic cleavage of the desired fusion protein when grown in pLyS BL21 Escherichia coli, as evidenced by blotting for the thioredoxin tag (Trx); even in this cell line, which minimizes protease activity, the GluA3 external domain fusion protein was almost entirely degraded during growth (data not shown). Two representative examples of patient reactivity are shown, as well as a darker exposure of the second patient to demonstrate minor GluA1 reactivity; all previously diagnosed anti-AMPAR encephalitis patients showed reactivity with fusion proteins. (B and C) CSF from two previously diagnosed anti-AMPAR encephalitis patients recognize GluA1 and/or GluA2-transfected HEK293 cells by immunocytochemistry in patterns that correspond to their observed GluA1/GluA2 external domain fusion protein reactivity (CSF from the same patient was used for (B) or (C)-labeled lanes in western blot in (A) and immunocytochemistry). (C) Example of one patient with primary GluA2 reactivity but minor GluA1 reactivity in transfected cells who showed the same differential reactivity with fusion proteins (A, bands C and C′; C, CSF panels). Comm, commercial antibody staining.
Therefore, we constructed additional FPs comprised of the individual external domains (ATD) or subdomains (top and bottom lobes of the ATD, S1, and S2) of GluA1 and GluA2, as well as the ATD of GluN1 and GluA3. The GluA3 ATD was almost entirely degraded by proteolytic cleavage and excluded from further analysis. We also assessed GluA1 and GluA2 subunits with immunocytochemistry of complementary deletions (mutants lacking the entire ATD, the top or bottom lobe of the ATD, S1, or S2) expressed in HEK293 cells using immunocytochemistry. Using CSF, both approaches suggest that the bottom lobe of the ATD is the main antigenic region of the receptor, independent of whether the primary reactivity is to GluA1 or GluA2. Deletion of the entire ATD or the bottom lobe in transfected HEK293 cells prevents antibody staining, while both the ATD and the bottom lobe FPs stain robustly on western blot (Fig.2A–C; Table1, original cohort). This reactivity matches the location of structural determinants for antigenicity in anti-NMDAR encephalitis,27 suggesting that the bottom lobe of the ATD in ionotropic glutamate receptors is particularly antigenic.
Figure 2.
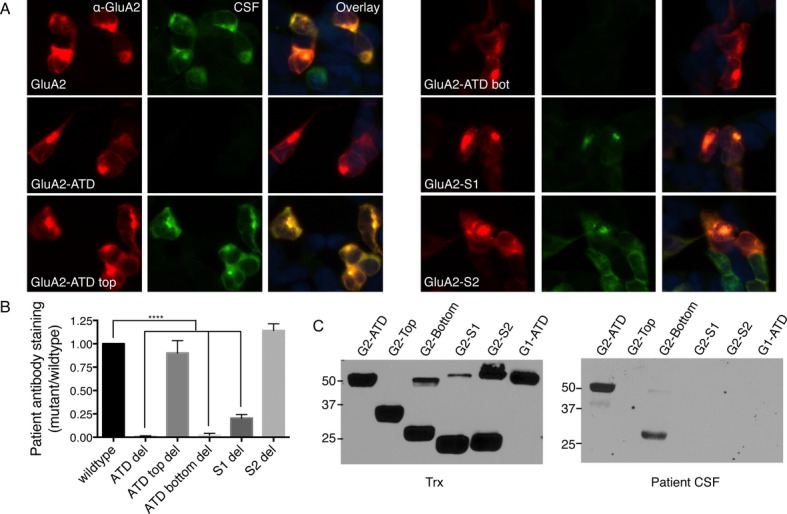
Antibodies from patients with anti-AMPAR encephalitis primarily recognize the bottom lobe of the ATD of GluA1 or GluA2. (A) Staining pattern of CSF from a typical anti-AMPAR encephalitis patient on HEK293 cells transfected with GluA2 deletion mutants shows a loss of reactivity with deletion of the bottom lobe of the ATD. (B) Quantification of deletion mutant CSF staining over four patients, three with primary GluA2 reactivity and one with primary GluA1 reactivity, using GluA2 or GluA1 mutants, respectively. (C) Reactivity with receptor domains on western blot. Left, fusion protein expression measured by anti-Thioredoxin antibody (Trx). Right, patient CSF reacts primarily with the ATD and the bottom lobe of the ATD. S1 deletion also decreases antibody binding, but does not appear to bind on western blot, possibly due to the proximity of the S1 domain to the bottom lobe of the ATD. *P < 0.05, ***P < 0.001, one-way ANOVA plus Dunnett's post hoc testing.
Table 1.
Antigenic reactivity of CSF and serum from the original cohort of anti-AMPAR encephalitis patients as well as newly identified patients with anti-AMPAR antibodies.
| Original cohort CSF | Original cohort serum | New cohort CSF | New cohort serum | |
|---|---|---|---|---|
| Main antigenic reactivity | ||||
| ATD | A1: 2/3 | A1: 0/3 | A1: 0/1 | A1: 1/7 |
| A2: 6/9 | A2: 7/10 | A2: 1/1 | A2: 5/7 | |
| Top lobe | A1: 0/3 | A1: 2/3 | A1: 0/1 | A1: 2/7 |
| A2: 1/9 | A2: 4/10 | A2: 1/1 | A2: 4/7 | |
| Bottom lobe | A1: 3/3 | A1: 2/3 | A1: 0/1 | A1: 0/7 |
| A2: 7/9 | A2: 5/10 | A2: 1/1 | A2: 1/7 | |
| S1 | A1: 1/3 | A1: 1/3 | A1: 0/1 | A1: 0/7 |
| A2: 1/9 | A2: 3/10 | A2: 1/1 | A2: 1/7 | |
| S2 | A1: 1/3 | A1: 3/3 | A1: 1/1 | A1: 6/7 |
| A2: 0/9 | A2: 3/10 | A2: 1/1 | A2: 1/7 | |
| GluN1-ATD | A1: 0/3 | A1: 0/3 | A1: 0/1 | A1: 0/7 |
| A2: 0/9 | A2: 1/10 | A2: 0/1 | A2: 0/7 | |
| Secondary antigenic reactivity | ||||
| ATD | A1: 0/3 | A1: 2/3 | A1: 0/1 | A1: 0/7 |
| A2: 1/9 | A2: 2/10 | A2: 0/1 | A2: 0/7 | |
| Top lobe | A1: 0/3 | A1: 1/3 | A1: 0/1 | A1: 0/7 |
| A2: 4/9 | A2: 3/10 | A2: 0/1 | A2: 0/7 | |
| Bottom lobe | A1: 0/3 | A1: 1/3 | A1: 0/1 | A1: 1/7 |
| A2: 2/9 | A2: 3/10 | A2: 0/1 | A2: 2/7 | |
| S1 | A1: 1/3 | A1: 1/3 | A1: 0/1 | A1: 1/7 |
| A2: 3/9 | A2: 5/10 | A2: 0/1 | A2: 2/7 | |
| S2 | A1: 2/3 | A1: 0/3 | A1: 0/1 | A1: 1/7 |
| A2: 6/9 | A2: 5/10 | A2: 0/1 | A2: 2/7 | |
| GluN1-ATD | A1: 1/3 | A1: 1/3 | A1: 0/1 | A1: 0/7 |
| A2: 1/9 | A2: 4/10 | A2: 1/1 | A2: 0/7 | |
ATD, amino terminal domain; CSF, cerebrospinal fluid; A1, GluA1; A2, GluA2.
While FPs and deletion mutants of the bottom lobe both suggest that the bottom lobe of the ATD is crucial for antibody binding, the role of the S1 domain is more ambiguous. Deletion of S1 reduces but does not abolish antibody staining in four patients (Fig.2B), while by western blot, the majority of patients have little to no reactivity to the S1 domain FP (Fig.2C; Table1). This may reflect the physical proximity of the S1 domain to the bottom lobe of the receptor: they are adjacent in primary structure, making it possible that deletion of the S1 domain alters the bottom lobe enough to reduce but not destroy antibody staining.
FP reactivity differs between serum and CSF and identifies additional patients with anti-AMPAR antibodies
Matched serum and CSF samples from 12 patients with anti-AMPAR encephalitis were tested on GluA1 or GluA2 subdomain FPs by western blot; two patients showed responses with serum but not CSF. While the primary response of CSF is to the bottom lobe of the ATD with lesser but considerable reactivity to the S1 domain and the ATD, serum reactivity is considerably more diverse, showing extensive reactivity to the top lobe as well as the S2 domains (Fig.3B and C, Table1). Interestingly, several patients showed some level of reactivity with GluN1 as well.
Figure 3.
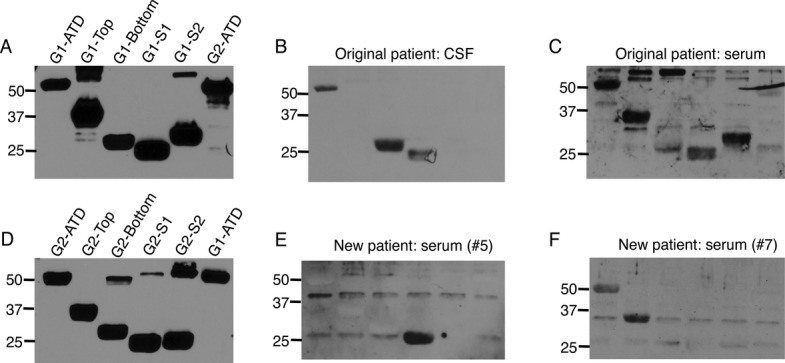
Fusion protein reactivity reveals differences in CSF and serum antibody responses and additional patients with anti-AMPAR antibodies in serum. (A and D) Fusion proteins of GluA1 (A) and GluA2 (D) subdomains (anti-Thioredoxin antibody). (B and C) While patient CSF binds the bottom lobe of the GluA1-ATD as well as the ATD and S1 domains, serum from the same patient shows broader reactivity. (E and F) Fusion protein reactivity of two individuals not previously diagnosed with anti-AMPAR encephalitis reveals reactivity with the GluA2-S1 domain (E) and the GluA2-ATD and top lobe (F).
Serum and/or CSF from 70 patients with neurologic symptoms including seizures and/or memory problems were tested on western blots with external domain FPs, as well as 15 control individuals with no known neurologic symptoms, 20 patients with anti-NMDAR encephalitis, and nine patients with Rasmussen's encephalitis. No control sample showed reactivity; three NMDAR encephalitis patients showed reactivity with the GluN1 external domain FP, and three Rasmussen's encephalitis patients showed GluA reactivity. Twenty-seven patients with neurologic symptoms showed potentially positive results. Sixteen of these patients were further analyzed with western blots of subdomain FPs, chosen for predominance of response as well as an even mix of GluA1 and GluA2 reactivity. One patient proved negative in this further analysis; eight showed primary reactivity with GluA1 and seven with GluA2; six patients had other evidence of immune system involvement (Table2). The majority of these patients were screened with serum only due to lack of CSF availability; one GluA1 patient was screened with both serum and CSF, and one GluA2 patient was screened with CSF only. Both CSF samples failed to recognize GluA1 or GluA2 in transfected HEK293 cells (data not shown). Overall, these 15 patients showed considerable diversity in their subdomain reactivity, with the largest population of responses showing a primary response to the ATD or S2 domains with additional reactivity toward ATD subdomains as well as the S1 domain (Fig.3E and F; Table1, new cohort). All newly identified positive patients showed symptoms compatible with decreased AMPAR activity, namely seizures and/or memory deficits (Table2). Men and women were virtually equally represented in this group, unlike the original, predominantly female cohort.
Table 2.
Clinical features of newly discovered patients with anti-AMPAR antibodies, screened with AMPAR subdomain fusion proteins on western blot.
| Case no. | Sex/age (year) | Symptom presentation | Evidence of immune involvement? | Main GluA subdomain reactivity |
|---|---|---|---|---|
| 1 | F/42 | Six month history of altered mental status (memory, behavior, verbal fluency) | Anti-thyroid antibodies | GluA2-all domains (CSF) |
| 2 | M/77 | Convulsions, memory loss, amnesia, limbic encephalitis | None | GluA1-S2 (CSF); GluA1- ATD, top lobe (serum) |
| 3 | M/62 | Convulsions, memory loss, amnesia, auditory hallucinations, limbic encephalitis | None | GluA1-S2 (serum) |
| 4 | M/30 | Convulsions, memory loss, amnesia, abnormal behavior | None | GluA1-S2 (serum) |
| 5 | M/43 | Mental status change | Improved with immunotherapy | GluA2-S1 (serum) |
| 6 | Limbic encephalitis | None | GluA1-S2 (serum) | |
| 7 | F/60 | Seizures, short-term memory loss, limbic encephalitis | None | GluA2-ATD, top lobe (serum) |
| 8 | F/23 | Schizophrenia, seizures | None | GluA1-S2, top lobe (serum) |
| 9 | F/55 | Schizophrenia, seizures | None | GluA1-S2 (serum) |
| 10 | F/7 | Memory loss, limbic encephalitis | CSF: 57 WBCs | GluA2-top lobe (serum) |
| 11 | F/78 | Encephalitis with increasing memory loss | None | GluA2-ATD, top lobe (serum) |
| 12 | F/37 | Altered mental status, encephalitis | Herpes encephalitis | GluA2-ATD, top, bottom, S2 (serum) |
| 13 | M/8 | Seizures, status epilepticus | None | GluA2-ATD (serum) |
| 14 | M/28 | Dizziness, ataxia, hyperthermia | CSF: 8 WBCs | GluA1-S2 (serum) |
| 15 | M/27 | Chorea, facial dyskinesia | CSF: 12 WBCs, onset with flu-like symptoms | GluA2-ATD (serum) |
IgG isolated from serum of patient 5 was used in electrophysiological experiments.
Similar electrophysiological effects of IgG isolated from original cohort anti-AMPAR patient and newly identified patient
While patient antibody reactivity differed between the original patient cohort and newly identified patients, both groups have neurologic symptoms consistent with decreased AMPAR activity4 (Table2). Therefore, we explored the effect of patient antibody application on mEPSCs in cultured neurons, using purified IgG from one patient previously diagnosed with anti-AMPAR encephalitis with predominant GluA2-bottom lobe antibodies (original cohort) and one patient with newly determined anti-AMPAR antibodies to the GluA2-S1 domain (new cohort; patient 5, Fig.3E, Table2). Twenty-four to 36 h application of antibody from either patient group increased the interevent interval and decreased the peak amplitude (Fig.4B and C) of mEPSCs compared to neurons treated with IgG from an individual without anti-AMPAR antibodies. The overall decrease in peak amplitude likely reflects a decrease in the number of large events, as is evidenced by the relatively greater proportion of smaller events in the cumulative frequency probability of patient antibody-treated cultures versus control antibody. To verify that these effects reflected the actions of anti-AMPAR antibodies, IgG from the new cohort patient or a control patient were incubated with solubilized uninduced bacteria (“uninduced”) or bacteria expressing GluA2-ext dom and GluA2-S1 FPs (“depleted”). FP lysates successfully depleted the anti-AMPAR antibodies from patient IgG as measured by western blot immunoreactivity. Uninduced patient IgG caused a similar decrease in mEPSC amplitude; depletion of anti-AMPAR antibodies abrogated this effect (Fig.4E–G).
Figure 4.

mEPSCs differ in cultured neurons treated with IgG from an individual with no anti-AMPAR reactivity (control IgG), an individual from the original cohort of anti-AMPAR encephalitis with GluA2 bottom lobe ATD reactivity (original patient IgG), and an individual with newly discovered anti-AMPAR antibodies in serum directed against the S1 domain (new patient IgG). (A) Example traces of mEPSCs from neurons treated with IgG from different patients. (B and C) Decreased average peak amplitude of mEPSCs in patient-treated neurons (B) is reflected in a decreased frequency of larger amplitude responses in the cumulative frequency histogram (C). (D) Increased interevent interval in neurons treated with patient material indicates a decrease in event frequency, which is more pronounced with original patient IgG treatment. (E) Incubation of patient IgG with GluA2 fusion proteins specifically depletes anti-AMPAR antibodies (Patient IgG, depleted), while incubation with uninduced bacteria does not (Patient IgG, uninduced; bands in GluA2-ext dom and GluA2-S1 lanes; compare to Fig.3E). Anti-Trx, Thioredoxin loading control. Standard, MagicMark internal exposure standard. (F and G) Patient IgG effect on mEPSC amplitude is dependent on the presence of anti-AMPAR antibodies: preincubation with uninduced bacteria (unind) maintains the patient-specific decrease in mEPSC amplitude (F) and decreased percentage of large events (G), while these effects are lost after AMPAR-specific IgG depletion (depl). n = 6–9 cells, P < 0.0001, one-way ANOVA plus Tukey's post hoc testing.
Discussion
Here, we present evidence that bacterial FPs of GluA1/2 can be used to test for anti-AMPAR antibodies in patient serum or CSF. We find that anti-AMPAR encephalitis antibodies recognize selected AMPAR domains and subdomains. This recognition is subunit-specific and largely matches the antibody staining in transfected cells. The main component of the CSF antibody response appears to be to the bottom lobe of the ATD in GluA1 or GluA2, although minor components may also be present. FPs also reveal differences in antibody populations between the serum and CSF of individual patients, and can be used to monitor changes in the antibody population over the course of disease progression. FPs were also used to test for the presence of AMPAR antibodies in serum or CSF of 70 additional individuals with neurologic symptoms, many with presumed immune involvement. Twenty-seven of these patients showed clear FP reactivity, 15 of which were verified with subdomain western blots. Application of IgG isolated from one of these patients as well as one patient from the original cohort showed similar effects on mEPSCs in cultured neurons, which was dependent on the presence of anti-AMPAR antibodies. Together, these results present a new tool to use in both the diagnosis and study of anti-AMPAR encephalitis, suggest that this disorder is more prevalent than currently appreciated, and provide the first evidence that patient antibodies have functional electrophysiological effects, reaffirming the crucial importance of carefully calibrated ionotropic glutamate receptor activity and protein level in proper nervous system function.
Even in the small anti-AMPAR encephalitis patient population diagnosed to date, there is considerable symptomologic heterogeneity, spanning memory loss, seizures, and acute psychosis.4,12 This presents significant practical difficulties: with such variability, it remains unclear what diagnostic guidelines merit testing for anti-AMPAR encephalitis, almost certainly resulting in underdiagnosis. For example, the initial descriptions of anti-AMPAR encephalitis as predominantly affecting women may be inaccurate; because autoimmune diseases tend to afflict more women than men,29 this may reflect ascertainment bias; however, given the small sample size, it may also be coincidental. In order to construct an accurate picture of anti-AMPAR encephalitis and develop guidelines for testing, it is important to screen a large database of patients with neurologic symptoms, particularly those with presumed immune involvement. This makes the development of a fast, robust test crucial. Because bacterial FPs express in large quantities and preclearing reduces background, FP western blots provide a simpler and more sensitive testing method for anti-AMPAR encephalitis than staining transfected cells. Additionally, antigenic diversity may explain some of the symptomologic differences observed. Several patients showed some degree of reactivity with the NMDAR ATD as well. The significance of this reactivity is not clear, although it is relatively common for autoimmune patients to develop a response to more than one target30; these additional antigenic populations may result in phenotypic differences.
In electrophysiological experiments, we found that patient IgG application decreased peak mEPSC amplitude and increased interevent interval. These results are compatible with immunocytochemistry results showing a decrease in the number, rather than intensity, of synaptic AMPAR clusters after patient antibody treatment.4 Both results suggest that patient antibody leads to a near-total loss of AMPARs from certain synapses, rather than a partial loss of AMPARs from all synapses. The increased interevent interval of mEPSCs may reflect a loss of AMPAR-containing synapses and a subsequent increase in so-called silent synapses, where quantal release of glutamate would not result in a measurable mEPSC. This would also result in fewer large-amplitude events, evidenced by the decrease in overall mEPSC amplitude and the increased percentage of small events in patient antibody-treated cultures. Why certain synapses are more susceptible to antibody-mediated AMPAR loss remains unclear, although EphrinB2 receptors can protect NMDARs from anti-NMDAR encephalitis antibodies31; a similar mechanism may exist in anti-AMPAR encephalitis.
While considerable work remains in the full mechanistic characterization of the effects of anti-AMPAR encephalitis antibodies, the first goal of anti-AMPAR encephalitis research is a more complete understanding of the clinical presentation of the disorder. The FP-based strategy presented here represents a fast, robust approach to achieving this goal, and provides valuable information as to the prevalence, antigenic variability, and electrophysiological mechanisms underlying this treatable neurologic disorder.
Contributions
A. J. G. designed, conducted, and analyzed experiments and wrote the manuscript. J. A. P. designed and conducted experiments and provided feedback on the manuscript. B. H. B. conducted experiments and provided feedback on the manuscript. J. D. supplied reagents, designed experiments, and provided feedback on the manuscript; D. L. designed experiments, interpreted data, and provided feedback on the manuscript.
Conflict of Interest
All authors have seen and agree with the contents of the manuscript, fulfill the ICMJE requirements for authorship, and believe it represents honest work. This submission is not currently under review at any other journals; none of the experiments described in this work have been published in other forms, outside of Dr. Gleichman's Ph.D. thesis.
References
- Lancaster E, Dalmau J. Neuronal autoantigens–pathogenesis, associated disorders and antibody testing. Nat Rev Neurol. 2012;8:380–390. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani L, Graus F, Giometto B, et al. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry. 2012;83:638–645. doi: 10.1136/jnnp-2011-301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61:25–36. doi: 10.1002/ana.21050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol. 2009;65:424–434. doi: 10.1002/ana.21589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M, Huijbers MG, Lancaster E, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9:776–785. doi: 10.1016/S1474-4422(10)70137-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster E, Huijbers MG, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol. 2011;69:303–311. doi: 10.1002/ana.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol. 2010;9:67–76. doi: 10.1016/S1474-4422(09)70324-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster E, Martinez-Hernandez E, Titulaer MJ, et al. Antibodies to metabotropic glutamate receptor 5 in the Ophelia syndrome. Neurology. 2011;77:1698–1701. doi: 10.1212/WNL.0b013e3182364a44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12:157–165. doi: 10.1016/S1474-4422(12)70310-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gable MS, Sheriff H, Dalmau J, et al. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis. 2012;54:899–904. doi: 10.1093/cid/cir1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataller L, Galiano R, Garcia-Escrig M, et al. Reversible paraneoplastic limbic encephalitis associated with antibodies to the AMPA receptor. Neurology. 2010;74:265–267. doi: 10.1212/WNL.0b013e3181cb3e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus F, Boronat A, Xifro X, et al. The expanding clinical profile of anti-AMPA receptor encephalitis. Neurology. 2010;74:857–859. doi: 10.1212/WNL.0b013e3181d3e404. [DOI] [PubMed] [Google Scholar]
- Wei YC, Liu CH, Lin JJ, et al. Rapid progression and brain atrophy in anti-AMPA receptor encephalitis. J Neuroimmunol. 2013;261:129–133. doi: 10.1016/j.jneuroim.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Kopec CD, Real E, Kessels HW, Malinow R. GluR1 links structural and functional plasticity at excitatory synapses. J Neurosci. 2007;27:13706–13718. doi: 10.1523/JNEUROSCI.3503-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makuch L, Volk L, Anggono V, et al. Regulation of AMPA receptor function by the human memory-associated gene KIBRA. Neuron. 2011;71:1022–1029. doi: 10.1016/j.neuron.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slipczuk L, Bekinschtein P, Katche C, et al. BDNF activates mTOR to regulate GluR1 expression required for memory formation. PLoS One. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Graebenitz S, Kedo O, Speckmann EJ, et al. Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain. 2011;134:2929–2947. doi: 10.1093/brain/awr202. [DOI] [PubMed] [Google Scholar]
- Lee WL, Hablitz JJ. Involvement of non-NMDA receptors in picrotoxin-induced epileptiform activity in the hippocampus. Neurosci Lett. 1989;107:129–134. doi: 10.1016/0304-3940(89)90804-5. [DOI] [PubMed] [Google Scholar]
- Menuz K, Nicoll RA. Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J Neurosci. 2008;28:10599–10603. doi: 10.1523/JNEUROSCI.2732-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorella A, Halonen T, Sahibzada N, Gale K. A crucial role of the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid subtype of glutamate receptors in piriform and perirhinal cortex for the initiation and propagation of limbic motor seizures. J Pharmacol Exp Ther. 1997;280:1401–1405. [PubMed] [Google Scholar]
- Weiczner R, Krisztin-Peva B, Mihaly A. Blockade of AMPA-receptors attenuates 4-aminopyridine seizures, decreases the activation of inhibitory neurons but is ineffective against seizure-related astrocytic swelling. Epilepsy Res. 2008;78:22–32. doi: 10.1016/j.eplepsyres.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hsu FC, Gleichman AJ, et al. Fyn-mediated phosphorylation of NR2B Tyr-1336 controls calpain-mediated NR2B cleavage in neurons and heterologous systems. J Biol Chem. 2007;282:20075–20087. doi: 10.1074/jbc.M700624200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova O, Kamberov E, Margolis B. Generation of deletion and point mutations with one primer in a single cloning step. Biotechniques. 2000;29:970–972. doi: 10.2144/00295bm08. [DOI] [PubMed] [Google Scholar]
- Gleichman AJ, Spruce LA, Dalmau J, et al. Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci. 2012;32:11082–11094. doi: 10.1523/JNEUROSCI.0064-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DR, Gleichman AJ, Cross SA, et al. NMDA receptor modulation by the neuropeptide apelin: implications for excitotoxic injury. J Neurochem. 2011;118:1113–1123. doi: 10.1111/j.1471-4159.2011.07383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitacre CC. Sex differences in autoimmune disease. Nat Immunol. 2001;2:777–780. doi: 10.1038/ni0901-777. [DOI] [PubMed] [Google Scholar]
- Pellkofer HL, Kuempfel T, Jacobson L, et al. Non-paraneoplastic limbic encephalitis associated with NMDAR and VGKC antibodies. J Neurol Neurosurg Psychiatry. 2010;81:1407–1408. doi: 10.1136/jnnp.2009.186494. [DOI] [PubMed] [Google Scholar]
- Mikasova L, De Rossi P, Bouchet D, et al. Disrupted surface cross-talk between NMDA and Ephrin-B2 receptors in anti-NMDA encephalitis. Brain. 2012;135(Pt 5):1606–1621. doi: 10.1093/brain/aws092. [DOI] [PubMed] [Google Scholar]