

Abstract
Background
The Natural Killer Complex (NKC) is a genetic region of highly linked genes encoding several receptors involved in the control of NK cell function. The NKC is highly polymorphic and allelic variability of various NKC loci has been demonstrated in inbred mice, providing evidence for NKC haplotypes. Using BALB.B6-Cmv1r congenic mice, in which NKC genes from C57BL/6 mice were introduced into the BALB/c background, we have previously shown that the NKC is a genetic determinant of malarial pathogenesis. C57BL/6 alleles are associated with increased disease-susceptibility as BALB.B6-Cmv1r congenic mice had increased cerebral pathology and death rates during P. berghei ANKA infection than cerebral malaria-resistant BALB/c controls.
Methods
To investigate which regions of the NKC are involved in susceptibility to experimental cerebral malaria (ECM), intra-NKC congenic mice generated by backcrossing recombinant F2 progeny from a (BALB/c x BALB.B6-Cmv1r) F1 intercross to BALB/c mice were infected with P. berghei ANKA.
Results
Our results revealed that C57BL/6 alleles at two locations in the NKC contribute to the development of ECM. The increased severity to severe disease in intra-NKC congenic mice was not associated with higher parasite burdens but correlated with a significantly enhanced systemic IFN-γ response to infection and an increased recruitment of CD8+ T cells to the brain of infected animals.
Conclusions
Polymorphisms within the NKC modulate malarial pathogenesis and acquired immune responses to infection.
Introduction
Malaria is one the most serious infectious diseases of humans with ∼250 million clinical cases annually. Most cases of severe disease are caused by the blood stage of Plasmodium falciparum. [1]. Fatalities are associated with various disease syndromes including respiratory distress, metabolic acidosis, hypoglycaemia, renal failure, pulmonary oedema and cerebral malaria (CM) [2]. This disease syndrome, which accounts for nearly 1 million deaths every year [3] is characterised by seizures and coma. To avoid clearance in the spleen, mature forms of blood-stage malaria adhere to vascular endothelial cells. This process, known as parasite sequestration is thought to induce obstructions in blood flow resulting in hypoxia and haemorrhages [1] that are associated with the development of organ-specific syndromes such CM. Disease-association studies indicate that in addition to parasite sequestration, inflammatory responses also contribute to severe disease [4]. High TNF, IFN-γ and IL-1β levels have been associated with disease severity [4]. Inflammatory chemokines including MIP-1α, MIP-1β [5] and CXCL10 [6], [7] have been found to be associated with increased risk of CM mortality. Although parasite sequestration is the most common feature of patients succumbing to CM, post-mortem examination has also revealed intra and peri-vascular pathology including the presence of leukocytes within brain blood vessels [8]–[11]. These findings suggested that intravascular infiltration of host leukocytes might also contribute to the pathogenesis of some CM cases.
Much useful evidence on the inflammatory processes contributing to the induction of CM has been provided by the Plasmodium berghei ANKA model [12]. Like in humans, P. berghei ANKA parasitised red blood cells (pRBCs) have been found to bind to brain vascular endothelial cells [13]. Inflammatory responses mediated by the cytokines TNF [14] and IFN-γ [15] and the chemokine CXCL10 [16] have been shown to contribute to severe malaria in mice. Several leukocyte populations including macrophages, neutrophils, T cells, NK cells and platelets have been found in brain blood vessels of CM-affected mice during infection [17]–[21]. From those populations, CD8+ T cells are highly abundant and were shown to mediate CM in a perforin-dependent manner [17], [21].
Elements of the innate immune system have also been shown to contribute to the development of ECM [22]. CD1d-restricted NKT cells have been shown to play a protective role against P. berghei ANKA-mediated CM in BALB/c mice but induce early IFN-γ production and promote disease in C57BL/6 susceptible animals [23]. Like NKT cells, NK cells readily secrete IFN-γ in response to malaria in humans [24], [25] and mice [26]. Furthermore, NK cells were shown to facilitate the recruitment of CXCR3+ T cells to the brain of malaria-infected mice in an IFN-γ-mediated manner [19]. More specifically, NK cells stimulate the dendritic cell (DC)-mediated priming of CD8+ T cells in response to P. berghei ANKA [27].
NK and NKT cell function is controlled by surface receptors encoded within a genetic region called NKC [28]. Many of these genes encode type II integral membrane proteins that have inhibitory or activation function depending on the presence or absence of immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in their intracellular domains. Upon ligation, ITIMs become phosphorylated and recruit protein tyrosine phosphatases, which interferes with cell activation. In the mouse, inhibitory receptors include the Ly49 superfamily and NKG2 molecules, expressed as heterodimers with CD94. Ly49 receptors interact with MHC I molecules, and CD94/NKG2 complexes bind to Qa-1b, a non-classical MHC I receptor. Interaction of these receptors with their MHC I ligands induces inhibition of cytotoxic activity by NK cells. Under pathological conditions such as viral infections or tumors, the expression of MHC I molecules is down-regulated, resulting in loss of negative regulation by inhibitory receptors, NK cell activation and killing of target cells [29]. Some members of the Ly49 family lack ITIMs and have activation function. Stimulation of activation receptors such as Ly49D [30] and Ly49H [31] leads to IFN-γ secretion and cytotoxic activity. Other activation receptors encoded within the murine NKC are CD94/NKG2C, NKG2D and NK1.1. In humans, NK receptors are encoded by 2 regions: the NKC on chromosome 12 and the killer cell Ig-like receptor (KIR) region on chromosome 19. Control of NK cell function in humans is mediated by interactions between HLA I and KIR molecules. Thus mouse Ly49 and human KIR genes are functional homologues, which illustrates an intriguing example of convergent evolution [32].
The NKC on mouse chromosome 6 is a polymorphic region. Allelic variability of loci has been shown in inbred mice providing evidence for NKC haplotypes [33]. In malaria, C57BL/6 NKC alleles are associated with disease susceptibility, as congenic BALB.B6-Cmv1r, in which NKC genes from C57BL/6 mice were introduced in the BALB/c background showed increased cerebral pathology, pulmonary oedema, anaemia and death rates during P. berghei ANKA infection [34], compared to resistant BALB/c controls. To date, the specific NKC receptors involved in the induction of malarial pathogenesis have not been identified. To address this question in this study intra-NKC recombinant mouse strains bearing small intervals of the C57BL/6 NKC in the BABL/c background were infected with P. berghei-ANKA. Our results revealed that alleles at two locations in the NKC contribute to susceptibility to CM in this model.
Methods
Ethics Statement
This manuscript contains work carried out with experimental mice. In P. berghei ANKA-infected mice, clinical illness develops between days 6–10 p.i. During this period, P. berghei-infected mice were monitored 5 times at 8:00 AM, 11:00 AM, 3:00 PM and 6:00 PM and 10:00PM. Mice developing loss of self-righting reflex were humanely euthanized by CO2 inhalation or cervical dislocation. All experiments were performed in compliance with the Walter & Eliza Hall Institute Animal Ethics Committee requirements. The Walter & Eliza Hall Institute Animal Ethics Committee has approved this study.
Mice and infections
Eight to 12-week old BALB/c, C567BL/6, BALB.B6-Cmv1r, BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT1-2 and BALB.B6-CT-13 [35] were used throughout the study. All mice were bred in The Walter & Eliza Hall Institute animal facility. Groups of 10–20 mice were injected intraperitoneally (i.p.) with 1×106 freshly passaged P. berghei-ANKA pRBC. Parasitemia was assessed by counting 10 microscope fields from Giemsa-stained smears of tail blood prepared every 2–3 days. Mortality was checked daily. Mice were judged as developing CM if they displayed neurological signs such as ataxia, loss of reflex and hemiplegia.
In Vivo bioluminescence imaging
Mice were infected (1×105 pRBC, i.v.) with a transgenic P. berghei-ANKA line expressing luciferase and GFP under the control of the elongation factor 1-α promoter [36]. Luciferase-expressing pRBCs were visualized in the brain with an I-CCD photon-counting video camera and in vivo imaging system (IVIS 100; Xenogen, Alameda, CA). Bioluminescence generated by luciferase transgenic parasites in brain tissue was measured according to the manufacturer's instructions using the same regions of measurement for all samples being compared.
Flow cytometry
Spleen cells from BALB/c, C57BL/6, BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice were incubated with anti-CD16 antibody (Fc-block), washed and then stained with PE-conjugated anti-CD49b (DX-5) and PE-CY5-conjugated anti-TCR (H57-597) antibodies. Some samples were simultaneously stained with other antibodies such as FITC-conjugated anti-Ly49A (A1), Ly49D (4E5), Ly49G2 (Cwy-e), Ly49I (YLI-90), anti-NKG2A/C/E (20d5) or anti-NK1.1 (PK136) for 1 h on ice (all antibodies and conjugates are from BD Pharmingen, San Diego, CA, USA). The cells were then washed twice, resuspended in PBS and analysed in a FACScalibur cytofluorometer (BD Biosciences, NJ) using CellQuest software. Viable lymphocytes were gated by forward and side scatter.
ELISA for IFN-γ detection
Ninety-six-well plates were coated with capture antibody (R4-6A2) by overnight incubation at 4°C in Phosphate Buffer pH 9. Plates were then blocked with 1% BSA for 1 h at 37°C. Serum samples were diluted 1/5 and tested in triplicates by overnight incubation at 4°C. Plates were then incubated for 2 h at 20°C with the biotinylated antibody (XMG1-2) and then for 1 h with streptavidin-peroxidase conjugate (Pierce, Rockford, IL). Bound complexes were detected with tetramethyl-benzidine (KBL, Gaithersburg, MD) and H2O2. Absorbance was read at 450 nm. Cytokine concentration was calculated using recombinant IFN-γ for the preparation of standard curves.
Purification and analysis of brain-sequestered leukocytes
Brain-sequestered leukocytes were purified on day 7 p.i with P. berghei-ANKA as described before [19]. Briefly, euthanized mice were perfused to remove circulating leukocytes. Brains were then removed, crushed in RPMI medium and pushed through a cell mesh. The tissue extract was centrifuged at 200× g for 10 min and the pellet was dissolved in RPMI containing 0.05% Collagenase D (Worthington, Lakewood, NJ) and 2 U/ml DNAase I (Sigma). After 1 h incubation at 22°C, the mixture was filtered through a cell strainer, seeded on a 35% Percoll (Amersham Bioscience, Uppsala, Sweden) cushion and centrifuged at 400× g for 20 min at 22°C. The pellet was collected and erythrocytes were lysed with Tris-NH4Cl Buffer. After washing, recovered cells were incubated with anti-CD16 antibody, washed and stained with PE-anti-NK1.1 (PK136), FITC-anti-TCR (H57-597) and PerCP-Cy5.5-anti-CD8 (53-6.7). After washing, cells were resuspended in PBS and analysed by flow cytometry.
Statistical analysis
A one-way ANOVA was used for data evaluation. A Tukey's multiple comparison post-test was used to evaluate differences between individual mouse strains. Differences in mortality rates of P. berghei infected mice were assessed by Cox-Mantel logrank analysis.
Results
Differential expression of markers NKC in intra-NKC congenic mice
BALB.B6-Cmv1r mice are a congenic strain in which the NKC from C57BL6 mice has been introduced into the BALB/c background [37]. As the NKC encodes several different receptors, intra-NKC recombinant mouse strains were generated by backcrossing recombinant F2 progeny from a (BALB/c x BALB.B6-Cmv1r) F1 intercross to BALB/c mice [35]. These congenic strains are a valuable tool to facilitate mapping of phenotypically-defined loci [35]. The genotypes of congenic strains used in this study are described in Table 1. To determine the phenotypic properties of NK and NKT cells from wild-type and congenic mice, spleen cells from BALB/c, C57BL/6 BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice were stained with antibodies against the pan NK-NKT cell marker DX-5 and αβTCR. Figure 1A shows that spleens of all mouse strains tested have similar percentages of both NK and NKT cells. The expression of different NKC markers was then studied in all DX5 positive splenocytes from the 6 mouse strains. Antibodies directed to C57BL/6 NKC receptors such as NK1.1, Ly49D, Ly49G2 and Ly49I recognized these molecules on the surface of cells from wild-type C57BL/6 as well as BALB.B6-CT-12 mice (Figure 1B). None of these molecules could be detected in spleen cells from BALB/c wild-type or BALB.B6-CT-1 mice, which only express C57BL/6 alleles outside the NKC (Table 1). Recombinations in BALB.B6-CT-6 and BALB.B6-CT-13 mice divide the NKC interval expressed by the as BALB.B6-CT-12 strain in 2 regions (Table 1). DX5+ cells from BALB.B6-CT-6 mice expressed NK1.1 but were unable to be recognised by antibodies against members of the Ly49 superfamily. In contrast, cells from BALB.B6-CT-13 mice readily expressed Ly49D, Ly49G2 and Ly49I but did not react with an anti-NK1.1 antibody. Antibodies directed against NKG2A/C/E were able to detect these molecules on all mouse strains tested, suggesting a higher homology level (Figure 1B). Thus NK and NKT cells from intra-NKC congenic mice differ in the expression of NKC markers such as NK1.1 and members of the Ly49 gene superfamily.
Table 1. Genotypes of intra-NKC congenic strains.
| Cmv1r | CT-1 | CT-6 | CT-12 | CT-13 | |
| D6Mit108 | c | c | c | c | c |
| Tnfr1 | b | c | b | b | c |
| D6Mit52 | b | c | b | b | c |
| Nkrp1a | b | c | b | b | c |
| D6Mit135 | b | c | b | b | c |
| Cd69 | b | c | b | b | c |
| CD94 | b | c | c | b | b |
| Ly49a | b | c | c | b | b |
| Cmv1 (Ly49h) | b | c | c | b | b |
| D6Mit13 | b | b | c | c | c |
| D6Mit25 | b | b | c | c | c |
| D6Mit59 | c | c | c | c | c |
c: BALB/c allele, b: C57BL/6 allele.
Figure 1. Differential expression of NKC markers in NK/NKT cells from BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice.

(A) Spleen cells from C57BL6, BALB/c and BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice were stained with anti-CD49b (DX5) and anti-αβ TCR antibodies. The percentage of NK and NKT cells are indicated. (B) The expression of the NKC markers NK1.1, Ly49A, Ly49D, Ly49G2, Ly49I, and NKG2A/C/E was calculated on all DX5 positive cells from the 6 mouse strains. Representative histograms are shown.
C57BL/6 alleles at two locations in the NKC contribute to susceptibility to ECM
We have previously shown that BALB.B6-Cmv1r congenic mice display partial susceptibility to P. berghei ANKA-mediated severe malaria [34]. To investigate which regions of the NKC are involved in susceptibility to CM, intra-congenic mouse strains were infected with P. berghei-ANKA and survival monitored daily. Consistent with previous observations, BALB.B6-Cmv1r mice developed CM symptoms and 40% of the animals succumbed to diseased by day 9 post-infection (p.i.). The strain BALB.B6-CT-12, which bears most of the C57BL/6 NKC alleles but lacks a more distal region (defined by the microsatellite markers D6Mit13, D6Mit25 and D6Mit59) present in the original BALB.B6-Cmv1r congenic (Table 1), showed significant increased susceptibility to disease than both BALB/c (p = 0.0009) and BALB.B6-Cmv1r mice (p = 0.0264), reflected as 78% fatalities (Figure 2A). The increased penetrance of the susceptibility phenotype in BALB.B6-CT-12 compared to BALB.B6-Cmv1r mice suggests that C57BL/6 loci present in that distal region of BALB.B6-Cmv1r mice might contribute to resistance to severe malaria in this model. Similar to BALB/c wild-type controls, BALB.B6-CT-1 mice that express BALB/c alleles throughout most of the NKC region (Table 1) were highly resistant to P. berghei ANKA-mediated CM (Figure 2A). Recombinations in BALB.B6-CT-6 and BALB.B6-CT-13 mice split the NKC interval expressed by the BALB.B6-CT-12 strain in 2 regions (Table 1). Unlike BALB.B6-CT-12 mice, which were nearly as susceptible to disease as C57BL/6 controls, when BALB.B6-CT-6 and BALB.B6-CT-13 animals were challenged with P. berghei-ANKA (Figure 2A) they displayed partial susceptibility to ECM (p = 0.0356 between BALB.B6-CT-6 and BALB.B6-CT-12, p = 0.0037 between BALB.B6-CT-13 and BALB.B6-CT-12). Together these results suggest that C57BL/6 alleles at two locations in the NKC contribute to susceptibility to CM in this model.
Figure 2. Control of malarial pathogenesis by the NKC.
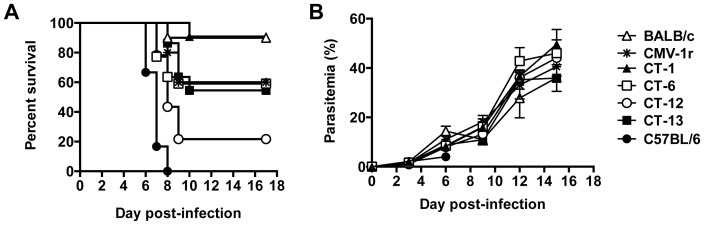
Groups of BALB/c, C567BL/6, BALB.B6-Cmv1r, BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT1-2 and BALB.B6-CT-13 mice (n = 10–20) were infected with P. berghei ANKA. (A) The percentage survival was monitored daily. (B) Parasitemia was assessed from Giemsa-stained blood smears. Each point represents the mean of 6–10 samples ± SD.
Parasite burdens were also evaluated at different time points p.i. Consistent with previous findings with BALB.B6-Cmv1r mice, parasitemia was not affected by the NKC genotype (Figure 2B). As P. berghei pRBC accumulate in various target organs, it has been suggested that peripheral parasitemia might not be an accurate estimation of parasite densities. To address this issue, intra-NKC congenic mice were infected with a transgenic P. berghei-ANKA line that constitutively expresses luciferase [36]. Following luciferin injection on days 3 and 6 p.i., mice were anesthetised and total parasite biomass was determined by calculating the bioluminescence emerging from living parasites. As expected parasite biomass increased as the infection developed. No significant differences were found in bioluminescence levels detected from parasites in NKC congenic mice (Figure 3 A, B). Thus these results indicate that control of malarial fatalities by the NKC cells does not operate through effects on parasite growth rates.
Figure 3. Control of malarial pathogenesis by the NKC does not affect total parasite biomass.
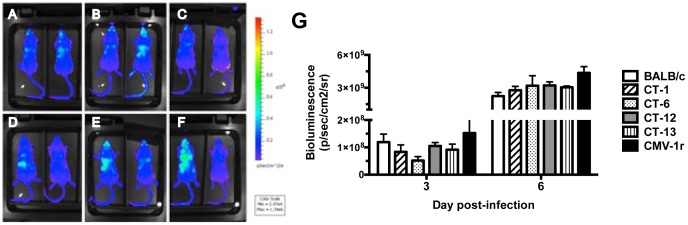
(A) BALB/c, (B) BALB.B6-Cmv1r, (C) BALB.B6-CT-1, (D) BALB.B6-CT-6, (E) BALB.B6-CT-12 and (F) BALB.B6-CT-13 were infected with a luciferase-expressing P. berghei ANKA transgenic strain. Whole body parasite biomass acquired on day 3 p.i is shown. Bioluminescence emerging from living parasites was calculated on days 3 and 6 p.i (G). Each point represents the mean of 3 samples ± SD. The experiment is representative of 2 separate infection experiments.
The NKC controls systemic IFN-γ in murine severe malaria
IFN-γ is a pro-inflammatory cytokine critical for CM pathogenesis in mice. We have previously found that the NKC differentially regulates systemic IFN-γ levels as well as the transcription of several IFN-γ-inducible genes during ECM [34]. To identify NKC loci responsible for these responses, intra-NKC congenic mouse strains were infected with P. berghei ANKA and IFN-γ levels were measured in sera collected on day 5 p.i. by capture ELISA. From all the congenic mouse stains evaluated, BALB.B6-CT-6 and BALB.B6-CT-12 mice secreted IFN-γ levels as high as those produced by C57BL/6 animals (Figure 4). This enhanced IFN-γ response by BALB.B6-CT-6 and BALB.B6-CT-12 mice was significantly higher than that obtained in BALB/c wild-type control mice as well as the CM-resistant BALB.B6-CT-1 congenic strain and BALB.B6-CT-13 animals (Figure 4). Thus together the data suggest that C57BL/6 alleles located within the NKC interval expressed by BALB.B6-CT-6 are required for optimal systemic IFN-γ responses to P. berghei ANKA.
Figure 4. The NKC controls systemic IFN-γ production during malaria infection.
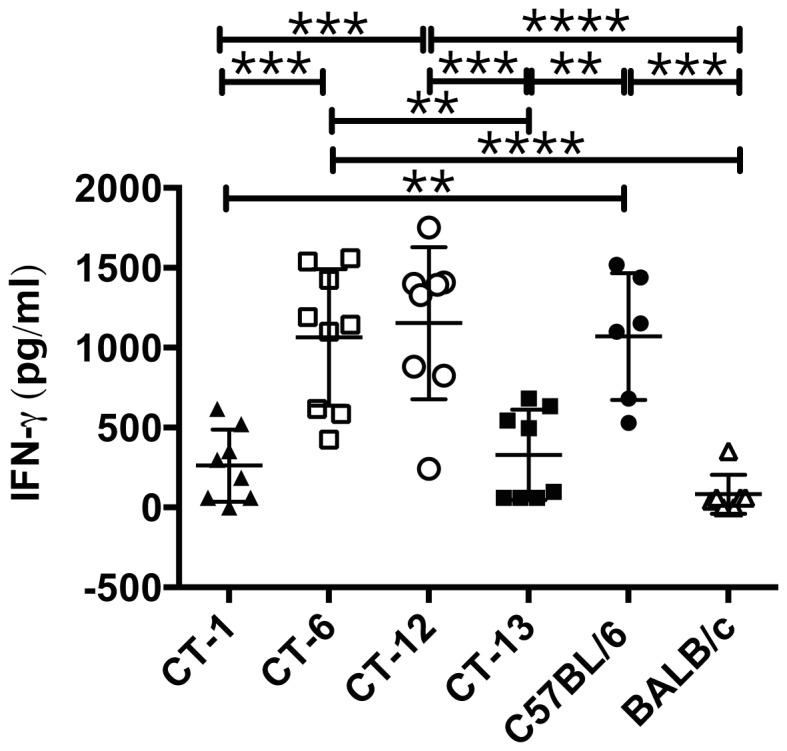
Groups of 4–5 BALB/c, C567BL/6, BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 were infected with P. berghei ANKA. IFN-γ levels in sera collected on day 5 p.i. were measured by capture ELISA. The graph shows pooled data from 2 separate infections, which gave similar results. **** P<0.0001, between BALB.B6-CT-12 and BALB/c and between BALB.B6-CT-6 and BALB/c; *** P<0.0005 between BALB.B6-CT-12 and BALB.B6-CT-1, between BALB.B6-CT-12 and BALB.B6-CT-13, between BALB.B6-CT-1 and BALB.B6-CT-6 and between C57BL/6 and BALB/c; ** P<0.005 between BALB.B6-CT-6 and BALB.B6-CT-13, between C57BL/6 and BALB.B6-CT-1 and between C57BL/6 and BALB.B6-CT-13.
C57BL/6 NKC alleles are required for optimal induction of brain-sequestered CD8+ T cells
The accumulation of CXCR3+ CD8+T cells in brains of P. berghei ANKA infected mice is a key contributing factor in ECM development. Previous work demonstrated that NK cells stimulate the DC-mediated priming of naïve CD8+ T cells in response to P. berghei ANKA [27] and facilitate the migration of these cells to the brain of infected mice [19]. To determine if different NKC receptors modulate this process, BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice were infected with P. berghei ANKA and the percentage and total number of brain-sequestered CD8+ T cells was determined on day 7 p.i. Consistent with their increased resistance to ECM, virtually no CD8+ T cells could be isolated from brains of BALB.B6-CT-1 mice (Figure 5 A,B). In contrast, 40–60% of total the brain-sequestered T cell pool consisted of CD8+ T cells in congenic mice expressing C57BL/6 NKC (Figure 5A). From these mice, the highly CM-susceptible BALB.B6-CT-12 strain had total numbers of brain-sequestered CD8+ T cells significantly higher than BALB.B6-CT-1, BALB.B6-CT-6 and BALB.B6-CT-13 mice (Figure 5B). This level of leukocyte sequestration in BALB.B6-CT-12 mice is similar to that previously reported in wild-type C57BL/6 animals [19]. BALB.B6-CT-6 and BALB.B6-CT-13 mice displayed intermediate levels of brain leukocyte sequestration, with absolute numbers of CD8+ T cells higher than CM-resistant BALB.B6-CT-1 animals, though only reaching significance for BALB.B6-CT-13 mice. Thus together the data suggest that C57BL/6 alleles in two locations are required for optimal recruitment of CD8+ T cells to the brain of P. berghei ANKA infected mice.
Figure 5. CD8+ T cells accumulate in brains of NKC congenic mice during P. berghei ANKA infection.

BALB.B6-CT-1, BALB.B6-CT-6, BALB.B6-CT-12 and BALB.B6-CT-13 mice were infected with P. berghei ANKA. Brains were collected on day 7 p.i after extensive perfusion of the euthanised animals. The BSL were isolated, stained anti-TCR and anti-CD8 antibodies and analysed by flow cytometry. The percentage (A) and absolute number (B) of CD8+ T cells were calculated. Representative dot plots are shown. Each bar represents the mean of 7–8 samples obtained over 2 separate infection experiments that gave similar results ± SEM, **** P<0.0001 between BALB.B6-CT-12 and BALB.B6-CT-1; ***P<0.0005 between BALB.B6-CT-12 and BALB.B6-CT-6; ** P<0.01 between BALB.B6-CT-12 and BALB.B6-CT-13; *P<0.05 between BALB.B6-CT-13 and BALB.B6-CT-1.
Discussion
Previous work demonstrated that the NKC is a significant genetic determinant of murine severe malaria, as congenic BALB.B6-Cmv1r mice showed increased cerebral pathology, pulmonary edema and death rates during P. berghei ANKA infection [23] compared to fully resistant BALB/c wild-type controls. In this study infection of intra-NKC congenic mice, generated by backcrossing recombinant F2 progeny from a (BALB/c x BALB.B6-Cmv1r) F1 intercross to BALB/c mice [35], revealed that C57BL/6 alleles at two locations in the NKC contribute to the development of ECM. The increased severity to malarial disease in intra-NKC congenic mice was not associated with higher parasite burdens but correlated with a significantly enhanced systemic IFN-γ response and an increased recruitment of CD8+ T cells to the brain of infected animals.
A large body of evidence indicates that IFN-γ plays a central role in CM pathogenesis [15], [38]–[43]. In vitro studies indicate that human NK cells produce IFN-γ in response to P. falciparum-pRBCs [24] and that the differential expression of human NK cell receptors modulates NK cell activation in response to blood stage malaria [44]. Moreover, the differential expression of the NKC-encoded and KIR-encoded receptors NKG2A, CD94, CD158α/KIR2DL1 was found to modulate P. falciparum-mediated IFN-γ responses by γδ-T cells [45]. Consistent with those finding, we found that the differential expression of NKC receptors in mice modulates IFN-γ responses to P. berghei ANKA and susceptibility to severe disease. Interestingly, although NKC loci at 2 separate locations were found to contribute to the development of ECM, C57BL/6 alleles located within the NKC interval expressed by BALB.B6-CT-6 appeared to be required for optimal systemic IFN-γ responses to infection. Several activation receptors including Ly49D [30], Ly49H [31], NK1.1 [46] and NKG2D [47] have been described to elicit IFN-γ secretion by NK and/or NKT cells upon stimulation. NK1.1 was found to be readily expressed by NK cells and NKT cells of BALB.B6-CT-6 congenic mice, raising the possibility that this receptor might modulate IFN-γ responses to infection. In support of that hypothesis, we have previously shown that cross-linking of NK1.1 preferentially induces IFN-γ and no IL-4 production by CD1d-restricted NKT cells [23], suggesting a TCR-independent pathway of pro-inflammatory responses to infection.
Although initially CD1d-restricted NKT cells appeared to be an important NKC receptor expressing cell lineage responsible for the increase susceptibility to disease in malaria-infected BALB.B6-Cmv1r mice [23], further evidence suggested that IFN-γ production by activated NK cells could also have an impact on the overall immunological status of BALB.B6-Cmv1r mice, influencing not only disease outcome but also innate and adaptive responses to infection [34]. Moreover in fully susceptible C57BL/6 mice, NK cells [19] were found to contribute to the development of ECM by stimulating the recruitment of CD8+ T cells to the brain of P. berghei ANKA infected mice. Similarly, the present study found using intra-NKC congenic mice (BALB.B6-CT-12) exerting 80% penetrance that NKC loci are required for that process. Thus the highly susceptibility level of congenic BALB.B6-CT-12 mice revealed novel aspects in the control of this response.
IFN-γ has been shown to participate in different responses involved in the development of ECM including, upregulation of receptors mediating parasite sequestration in the vascular endothelium, induction of CXCR3 chemokines responsible for the recruitment of T cells to the brain, etc [16], [48], [49]. Interestingly, although NKC alleles in the interval expressed by BALB.B6-CT-6 were found to be sufficient for the induction of systemic IFN-γ responses, the infection of this congenic strain resulted only in partial recruitment of CD8+ T cells to the brain. Similar results were also found after infection of BALB.B6-CT-13 mice that express incomplete C57BL/6 NKC intervals. Together these results are consistent with the notion that at least 2 NKC loci at different locations mediate different non-redundant responses required to fully recapitulate this phenotype. Prior to their CXCR3-mediated recruitment to the brain, the priming of naïve CD8+ T cells has been shown to be largely mediated by CD8α+ conventional DCs, which appears to be the main subset involved in cross-presentation of parasite-expressed antigens [20]. Interestingly, NK cells and DCs were shown to participate in a cross-talk which results in mutual activation whereby NK cells stimulate IL-12 output by CD8α+ conventional DCs required for optimal CD8+ T cell priming [19]. Further work is required to determine whether different NKC receptors participate in the induction of these 2 processes (T cell priming and chemokine secretion) required for efficient migration of inflammatory leukocytes to the brain of malaria-infected mice.
The NKC is conserved among species, with syntenic regions identified in rat and human chromosomes. Similar to our findings here, the differential expression of human NK cell receptors modulates NK cell activation in response to P. falciparum pRBC [44]. Using a rodent infection model, we established that polymorphisms within NKC loci regulate malarial pathogenesis and the induction of acquired immune responses to infection. Despite the important public health problem that human malaria infections poses worldwide, many immunological and genetic aspects of this disease are not fully understood. Thus further work is required to determine whether NKC receptors are associated with disease severity to malaria in human populations.
Funding Statement
This work was made possible through Victorian State Government Operational Infrastructure Support, Australian Government National Health and Medical Research Council IRIISS, and Project Grant 1031212. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Miller LH, Baruch DI, Marsh K, Doumbo OK (2002) The pathogenic basis of malaria. Nature 415: 673–679. [DOI] [PubMed] [Google Scholar]
- 2. White NJ, Ho M (1992) The pathophysiology of malaria. Adv Parasitol 31: 83–173. [DOI] [PubMed] [Google Scholar]
- 3. Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, et al. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379: 413–431. [DOI] [PubMed] [Google Scholar]
- 4. Schofield L, Grau GE (2005) Immunological processes in malaria pathogenesis. Nat Rev Immunol 5: 722–735. [DOI] [PubMed] [Google Scholar]
- 5. Ochiel DO, Awandare GA, Keller CC, Hittner JB, Kremsner PG, et al. (2005) Differential regulation of beta-chemokines in children with Plasmodium falciparum malaria. Infect Immun 73: 4190–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jain V, Armah HB, Tongren JE, Ned RM, Wilson NO, et al. (2008) Plasma IP-10, apoptotic and angiogenic factors associated with fatal cerebral malaria in India. Malar J 7: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Armah HB, Wilson NO, Sarfo BY, Powell MD, Bond VC, et al. (2007) Cerebrospinal fluid and serum biomarkers of cerebral malaria mortality in Ghanaian children. Malar J 6: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, et al. (2003) Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis 187: 461–466. [DOI] [PubMed] [Google Scholar]
- 9. Patnaik JK, Das BS, Mishra SK, Mohanty S, Satpathy SK, et al. (1994) Vascular clogging, mononuclear cell margination, and enhanced vascular-permeability in the pathogenesis of human cerebral malaria. American Journal of Tropical Medicine and Hygiene 51: 642–647. [PubMed] [Google Scholar]
- 10. Porta J, Carota A, Pizzolato GP, Wildi E, Widmer MC, et al. (1993) Immunopathological changes in human cerebral malaria. Clinical Neuropathology 12: 142–146. [PubMed] [Google Scholar]
- 11. Taylor TE, Fu WJ, Carr RA, Whitten RO, Mueller JS, et al. (2004) Differentiating the pathologies of cerebral malaria by postmortem parasite counts. Nat Med 10: 143–145. [DOI] [PubMed] [Google Scholar]
- 12. Hansen DS (2012) Inflammatory responses associated with the induction of cerebral malaria: lessons from experimental murine models. PLoS Pathog 8: e1003045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. El-Assaad F, Wheway J, Mitchell AJ, Lou J, Hunt NH, et al. (2013) Cytoadherence of Plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect Immun 81: 3984–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, et al. (1987) Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237: 1210–1212. [DOI] [PubMed] [Google Scholar]
- 15. Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, et al. (1989) Monoclonal antibody against interferon γ can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc Natl Acad Sci USA 86: 5572–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nie CQ, Bernard NJ, Norman MU, Amante FH, Lundie RJ, et al. (2009) IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog 5: e1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, et al. (2002) On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol 169: 6369–6375. [DOI] [PubMed] [Google Scholar]
- 18. Ma N, Hunt NH, Madigan MC, Chan-Ling T (1996) Correlation between enhanced vascular permeability, up-regulation of cellular adhesion molecules and monocyte adhesion to the endothelium in the retina during the development of fatal murine cerebral malaria. Am J Pathol 149: 1745–1762. [PMC free article] [PubMed] [Google Scholar]
- 19. Hansen DS, Bernard NJ, Nie CQ, Schofield L (2007) NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol 178: 5779–5788. [DOI] [PubMed] [Google Scholar]
- 20. Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, et al. (2008) Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A 105: 14509–14514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, et al. (2003) Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J Immunol 170: 2221–2228. [DOI] [PubMed] [Google Scholar]
- 22. Hansen DS, D'Ombrain MC, Schofield L (2007) The role of leukocytes bearing Natural Killer Complex receptors and Killer Immunoglobulin-like Receptors in the immunology of malaria. Curr Opin Immunol 19: 416–423. [DOI] [PubMed] [Google Scholar]
- 23. Hansen DS, Siomos MA, Buckingham L, Scalzo AA, Schofield L (2003) Regulation of murine cerebral malaria pathogenesis by CD1d-restricted NKT cells and the natural killer complex. Immunity 18: 391–402. [DOI] [PubMed] [Google Scholar]
- 24. Artavanis-Tsakonas K, Riley EM (2002) Innate immune response to malaria: rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. J Immunol 169: 2956–2963. [DOI] [PubMed] [Google Scholar]
- 25. Baratin M, Roetynck S, Lepolard C, Falk C, Sawadogo S, et al. (2005) Natural killer cell and macrophage cooperation in MyD88-dependent innate responses to Plasmodium falciparum . Proc Natl Acad Sci U S A 102: 14747–14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ing R, Stevenson MM (2009) Dendritic cell and NK cell reciprocal cross talk promotes gamma interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun 77: 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ryg-Cornejo V, Nie CQ, Bernard NJ, Lundie RJ, Evans KJ, et al. (2013) NK cells and conventional dendritic cells engage in reciprocal activation for the induction of inflammatory responses during Plasmodium berghei ANKA infection. Immunobiology 218: 263–271. [DOI] [PubMed] [Google Scholar]
- 28. Yokoyama WM, Plougastel BF (2003) Immune functions encoded by the natural killer gene complex. Nat Rev Immunol 3: 304–316. [DOI] [PubMed] [Google Scholar]
- 29. Karre K (2002) NK cells, MHC class I molecules and the missing self. Scand J Immunol 55: 221–228. [DOI] [PubMed] [Google Scholar]
- 30. Mason LH, Willette-Brown J, Mason AT, McVicar D, Ortaldo JR (2000) Interaction of Ly-49D+ NK cells with H-2Dd target cells leads to Dap-12 phosphorylation and IFN-gamma secretion. J Immunol 164: 603–611. [DOI] [PubMed] [Google Scholar]
- 31. Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, et al. (2001) Specific and nonspecific NK cell activation during virus infection. Nat Immunol 2: 951–956. [DOI] [PubMed] [Google Scholar]
- 32. Trowsdale J (2001) Genetic and functional relationships between MHC and NK receptor genes. Immunity 15: 363–374. [DOI] [PubMed] [Google Scholar]
- 33. Brown MG, Scalzo AA, Stone LR, Clark PY, Du Y, et al. (2001) Natural killer gene complex (Nkc) allelic variability in inbred mice: evidence for Nkc haplotypes. Immunogenetics 53: 584–591. [DOI] [PubMed] [Google Scholar]
- 34. Hansen DS, Evans KJ, D'Ombrain MC, Bernard NJ, Sexton AC, et al. (2005) The natural killer complex regulates severe malarial pathogenesis and influences acquired immune responses to Plasmodium berghei ANKA. Infect Immun 73: 2288–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Scalzo AA, Brown MG, Chu DT, Heusel JW, Yokoyama WM, et al. (1999) Development of intra-natural killer complex (NKC) recombinant and congenic mouse strains for mapping and functional analysis of NK cell regulatory loci. Immunogenetics 49: 238–241. [DOI] [PubMed] [Google Scholar]
- 36. Franke-Fayard B, Janse CJ, Cunha-Rodrigues M, Ramesar J, Buscher P, et al. (2005) Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc Natl Acad Sci U S A 102: 11468–11473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scalzo AA, Lyons PA, Fitzgerald NA, Forbes CA, Yokoyama WM, et al. (1995) Genetic mapping of Cmv1 in the region of mouse chromosome 6 encoding the NK gene complex-associated loci Ly49 and musNKR-P1. Genomics 27: 435–441. [DOI] [PubMed] [Google Scholar]
- 38. Hunt NH, Grau GE (2003) Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol 24: 491–499. [DOI] [PubMed] [Google Scholar]
- 39. Ho M, Sexton MM, Tongtawe P, Looareesuwan S, Suntharasamai P, et al. (1995) Interleukin-10 inhibits tumor necrosis factor production but not antigen-specific lymphoproliferation in acute Plasmodium falciparum malaria. J Infect Dis 172: 838–844. [DOI] [PubMed] [Google Scholar]
- 40. Ringwald P, Peyron F, Vuillez JP, Touze JE, Le BJ, et al. (1991) Levels of cytokines in plasma during Plasmodium falciparum malaria attacks. J Clin Microbiol 29: 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koch O, Awomoyi A, Usen S, Jallow M, Richardson A, et al. (2002) IFNGR1 gene promoter polymorphisms and susceptibility to cerebral malaria. J Infect Dis 185: 1684–1687. [DOI] [PubMed] [Google Scholar]
- 42. Yanez DM, Manning DD, Cooley AJ, Weidanz WP, van der Heyde HC (1996) Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J Immunol 157: 1620–1624. [PubMed] [Google Scholar]
- 43. Amani V, Margarida Vigario A, Belnoue E, Marussig M, Fonseca L, et al. (2000) Involvement of IFN gamma receptor-mediated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur J Immunol 30: 1646–1655. [DOI] [PubMed] [Google Scholar]
- 44. Artavanis-Tsakonas K, Eleme K, McQueen KL, Cheng NW, Parham P, et al. (2003) Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. J Immunol 171: 5396–5405. [DOI] [PubMed] [Google Scholar]
- 45. D'Ombrain MC, Hansen DS, Simpson KM, Schofield L (2007) gammadelta-T cells expressing NK receptors predominate over NK cells and conventional T cells in the innate IFN-gamma response to Plasmodium falciparum malaria. Eur J Immunol 37: 1864–1873. [DOI] [PubMed] [Google Scholar]
- 46. Arase H, Arase N, Saito T (1996) Interferon gamma production by natural killer (NK) cells and NK1.1+ T cells upon NKR-P1 cross-linking. J Exp Med 183: 2391–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ho EL, Carayannopoulos LN, Poursine-Laurent J, Kinder J, Plougastel B, et al. (2002) Costimulation of multiple NK cell activation receptors by NKG2D. J Immunol 169: 3667–3675. [DOI] [PubMed] [Google Scholar]
- 48. Campanella GS, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, et al. (2008) Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci U S A 105: 4814–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Miu J, Mitchell AJ, Muller M, Carter SL, Manders PM, et al. (2008) Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. J Immunol 180: 1217–1230. [DOI] [PubMed] [Google Scholar]