

Abstract
Networks constructed from aggregated protein-protein interaction data are commonplace in biology. But the studies these data are derived from were conducted with their own hypotheses and foci. Focusing on data from budding yeast present in BioGRID, we determine that many of the downstream signals present in network data are significantly impacted by biases in the original data. We determine the degree to which selection bias in favor of biologically interesting bait proteins goes down with study size, while we also find that promiscuity in prey contributes more substantially in larger studies. We analyze interaction studies over time with respect to data in the Gene Ontology and find that reproducibly observed interactions are less likely to favor multifunctional proteins. We find strong alignment between co-expression and protein-protein interaction data occurs only for extreme co-expression values, and use this data to suggest candidates for targets likely to reveal novel biology in follow-up studies.
Keywords: protein-protein interaction, co-expression, bias, Gene Ontology, networks, multifunctionality
Introduction
The use of protein-protein interaction (PPI) data to study gene function is a topic attracting vigorous research interest, both from computational biologists who are developing techniques, and from biologists wanting to exploit the enormous quantities of data that have become available in the last decade. Multiple efforts have led to the collection of interactions from the literature and their aggregation into large databases [1]. These data are treated as a ready means of placing candidate genes into the context of existing information for the purpose of identifying biologically-interpretable patterns, for example from genetics studies. The networks are used to find commonalities among candidate genes (e.g., network based enrichment [2] ) or to hypothesize new functions for specific genes of interest using “guilt by association” [3]. There are many other types of data that are used for networks, but protein interactions form a basic component of most (if not all) network data integration approaches [4]. Their wide application makes understanding the properties of the data of increasing importance.
The convenience of interaction databases has enabled the comparatively uncritical use of protein interaction data. In most applications of which we are aware, a database of interactions is downloaded and used without much consideration given to the quality or reliability of the data. While interactions can be assessed for evidence quality [5], there is no gold standard way to do this. It is also common for bioinformatics researchers to proclaim their approaches to be “unbiased”, without adequately defining what is meant, much less explaining how such biases are measured [6, 7]. These problems form the motivation for the current work.
Experiments that generate protein interaction data typically involve one or more “bait” proteins used to identify “prey” interactors. Baits are typically selected by the investigator, while the pool of possible preys is limited by technical or biological factors. There are multiple types of bias that can arise from this situation. One is selection bias, which might be most obvious in the form of the choice of baits, but could also occur in the degree to which prey for those baits are accepted as valid. A second type of bias is what we call laboratory bias, referring to the specificity of a given result to a particular experiment, and are due to technical factors such as the choice of methodology and other less tangible causes. Laboratory biases are not as explicit as selection bias, but can be partly inferred from reproducibility across studies. The total number of unreplicated interactions provides a relative measure of laboratory bias, while the number of times a bait is studied is a measure of selection bias. We hypothesize that these two types of bias generally are traded off each other when examined in aggregated databases. Large studies that use many baits might publish results with less bias towards heavily-studied proteins, but then contribute a proportionately larger amount of data to public repositories, increasing laboratory biases (results specific to that study). In contrast small studies might select baits with a particular goal (and expected prey) in mind, but contribute smaller amounts of data to the aggregate, thus having less influence on overall laboratory bias. We predict that if one looks at the contribution to aggregated PPI databases from small and large studies, these biases and their tradeoff might be exposed.
We further hypothesize that this sense of bias tradeoff between large and small studies might be present as a tradeoff between biological and technological effects in the detected interactions (preys). Small studies may afford a greater possibility of examining the identified interactions individually by hand. This may increase the degree to which they are biased in favor of what is perceived by the investigator as interesting biology but should decrease the degree to which technical artifacts contribute to results. In contrast, in large studies, interactions that are “interesting” might not be specifically prioritized so we do not expect as much experimenter bias toward prior biology, but technical artifacts (e.g., due to protein stickiness) could have larger effects if less manual effort can be used to clean up the results. In other words, if one is attempting to be unbiased in the biological sense, it is more likely that technical artifacts will creep in since one is denied one way to identify them, namely using biological prior knowledge of what is reasonable.
These contrasting senses of bias hinge on the degree to which we are willing to use prior knowledge to select which observations are interesting; or to what degree we wish to use uninformative priors. An excellent example is provided by a set of networks created by Marcotte and colleagues [8–10] (these networks are not constructed only from protein interactions, but illustrate the point well). In their approach, interactions in the network are down-weighted if they conflict with prior biological knowledge as defined by the Gene Ontology (GO) [11], yielding a so-called “bias-reduced” network [8]. Thus connections between genes with similar GO annotations are favored. The benefit is that many technical artifacts are probably removed; the bias being reduced is the technical bias. At the same time, this approach increases the degree to which prior biological knowledge contributes to the network; the biological bias has been increased, leading to a potential “rich-get-richer” problem (a version of the Matthew effect) [12].
In this paper, we use two data sources as the basis of comparisons to protein interactions. The first is Gene Ontology annotations, which provide an invaluable way of characterizing what is interesting to biologists. It captures both the definitions of attributes biologists want to ascribe to genes (“functions”), as well as the knowledge linking genes to those functions. While GO is incomplete, it may be incomplete in interesting ways that reflect biases that show up in other contexts. Indeed, we have previously posited that the biases in GO capture real properties of the body of knowledge and how it is used [13]. In this context the most notable feature of GO is how the number of annotations varies from gene to gene, what we call multifunctionality bias. The fact that the yeast histone deacetylase Sin3 complex component RPD3 has over 200 directly annotated GO terms, while other complex members (SIN3 and UME1) have fewer than 30 GO terms each, is surely a reflection of “popularity” (with researchers and GO curators) as well as biological and technical factors (such as actual biological importance and ease of study). The impact of these disparities on computational analysis of networks is the topic of previous work [13, 14]. We also previously described how protein interaction databases are partly confounded with GO annotations, creating opportunities for invalid joint interpretations due to circular logic [15]. However, for the most part we have used protein interaction networks as monolithic entities with respect to their input experiments. The biases we hypothesize above have not been explicitly examined, although discordance among data sets has been previously explored [16, 17]. The second data type we use in this paper is RNA co-expression. Co-expression, or correlated expression, is often used as a means of identifying potential functional relationships based on the idea that expression of two genes at the same place and time reflects shared needs of the cell or co-regulation (guilt by association) [18]. While noisy, co-expression data is less biased by prior biological knowledge than protein interactions because it is typically measured simultaneously for all pairs of protein-coding genes [13].
Our goal in this paper is to examine some of the factors determining the properties of protein interaction network data, burrowing into some of the messy details to identify and quantify biases that have not previously been given much attention. We focus on protein interactions from Saccharomyces cerevisiae (budding yeast), due to the relatively comprehensive nature of the available data. Our strategy throughout is to dissect a large protein interaction database (BioGRID) [1] into subsets based on criteria about the underlying studies, and construct a protein interaction network using that subset. Then the incidence of a protein (number of interactions in which it appears) within that network (and its status as a bait or prey) is then quantified. We use data spanning multiple experimental types from a ten-year period, allowing us to examine trends over time and the effects of methodology. We believe many of the biases we observe involve trade-offs of various sorts, most of which are defensible, but should be made explicit. Our results can help guide the design of protein interaction studies, as well as the interpretation of the data and their use by other researchers.
Materials and Methods
Protein-protein interaction data was obtained from the Biological General Repository for Interaction Datasets (BioGRID) [1], version 3.2.100. The BioGRID -ALL-3.2.100.tab2.zip file was downloaded and extracted. The interactions between proteins from Saccharomyces cerevisiae (budding yeast) were mined from the file and only proteins from the same taxon were used (taxonomy reference 559292). The set of interactions were further filtered for only those labeled as “physical”. No further processing of the data was undertaken. This yielded a dataset of 125,009 interactions among 5,795 proteins (the data also include interactions of proteins with RNAs, which for the sake of simplicity we lump in with the rest). Using each interaction’s associated PubMed ID, the publication date was extracted using in-house R scripts and the “annotate” R library [19]. Two-hundred and thirty-eight interactions could not be resolved to a publication date at the month level and were removed. Removing self-connections and those not assessable in the microarray data described below yielded 114,736 connections across 5,457 genes.
Contaminant data from affinity-capture mass spectrometry (AC-MS) yeast experiments was obtained from the CRAPome database [20]. The file crap_db_v1_flat_file_yeast.xlsx was downloaded and the columns pertaining to the Entrez gene ID and spectral counts for the 17 documented experiments were extracted into a text file. This file contained 1,390 proteins, which mapped to 1,306 genes in the BioGRID data. Although the contaminant data contained spectral measures of each protein in each experiment, we used a binary measure of protein “crappiness”, i.e. the presence of the protein as a contaminant was enough to consider the protein a “crappy” result. A measure of study “crappiness” was then calculated as proportion by taking the number of interactions with either bait or prey that were in the contaminants list and dividing over the total number of interactions of that study (N). A measure of network crappiness was then taken as the mean crappiness of all studies of size N or smaller.
A gene co-expression network was created using the method of Gillis and Pavlidis (2011)[21]. Thirty microarray data sets generated from the Affymetrix Yeast Genome S98 Array (GPL90) were downloaded from GEO using the “GEOquery” R package [22]. For each expression data set, the data was quantile normalized with the “limma” R package [23]. The data was then log2 transformed, and filtered to retain only probes annotated to open reading frames. The platform contains 9,335 probes which mapped to 5,457 genes using the NCBI Saccharomyces cerevisiae.gene_info.gz data file. Probes with multiple genes were discarded. Expression level intensities from the same gene were aggregated, taking the median value. The Spearman correlation coefficient was calculated for each gene pair in the expression data set. The networks were then aggregated by summing [21] and the final matrix was re-ranked to obtain the final co-expression network. The names of the 30 data sets are available in the supplement.
GO annotations for yeast genes were obtained from the GO Consortium and multifunctionality calculated as described previously [13]. Briefly, the GO term annotations were propagated in the ontology graph (transitive closure), and multifunctionality is computed as
where Num_ini is the number of genes within GO group i, and Num_outi is the number of genes outside GO group i. Intuitively, a term contributes to the score for a gene in proportion to how specific the term is. This definition comes about from a proof (presented in [13]) that ranking genes by this scores provides a list that is optimal for a hypothetical task of predicting gene function: given any GO term, the multifunctionality ranking will give the highest mean performance in “predicting” genes annotated with that term (as measured by a receiver operating characteristic curve), averaged over all GO terms. Importantly, this ranking has a high explanatory value in the analyses of network algorithm behaviour presented in [13]. Also note that this ranking is very similar to the one yielded by simply counting the number of (propagated) annotations each gene has, so to a first approximation one can think of multifunctionality as proportional to the number of GO annotations. While more complex than the raw number of terms, the multifunctionality score corrects for the fact that some GO terms are associated with widely varying numbers of genes. In our results we open with using the number of GO annotations for simplicity of exposition, but switch to the more appropriate multifunctionality score once the basic concepts are clear.
Results
We hypothesized that larger studies (where size refers to the numbers of baits tested) are less biased with respect to biological knowledge. Our previous work showed that it is feasible to quantify such bias by using GO annotations [13]. Thus we take the degree of GO annotation of a gene (number of terms, or, in later sections, the multifunctionality score) to be a rough measure of how interesting the gene and thus how likely it is to be selected for study. The reasoning is that the more functions in which a gene is involved, the more likely any given investigator will deem it relevant to the functions they study. Thus if smaller protein interaction studies are more biased, then they should exhibit an elevation in the number of GO terms per bait. To measure this we characterize each study by the median number of GO terms attached to its baits, and compute the mean of these scores for studies of a given size (number of baits). The correlation of this score with the study size is −0.83 (Supplementary Figure 1, limiting ourselves to a bait size 1–10, the range in which there are sufficient data). As the large standard deviation for a given study size suggests (Supplementary Figure 1), the effect is fairly modest, although this is partially because most (80%) experiments use baits with more than the average numbers of GO annotations, regardless of study size. Importantly, the effect is not as dramatic (though still very significant) if we don’t average across studies, because the apparent bias is weak on a per-study basis (correlation −0.08, p<1E-7. Despite the weakness of this bias in any given study, the aggregate effects have important implications as we describe below.
Bias in the choice of which proteins to study is potentially important because we have shown that more highly annotated genes tend to have more interactions [13]. We confirmed this effect here: more interactions are found for baits with more GO terms in aggregate. The correlation is 0.09 (p~4E-8) across all studies with a weaker but still significant effect across small studies (20 or fewer baits, r=0.05, p,<0.01). This is calculated on a per protein level (i.e., allowing multiple studies to contribute interactions). Note that in simulations, we find that if a gene ranking has a correlation of just 0.1 with GO multifunctionality, enrichment analysis of that list yields hundreds of enriched GO terms (approximately 500 on average; manuscript in preparation). Similarly, even comparatively weak correlations with multifunctionality in network structure can be sufficient to drive the outcome of computational analyses of function [13]. In agreement with the observation above that the per-study effects are weak, if we switch to treating each bait use as distinct instead of aggregating across studies, there is almost no correlation between the number of preys reported and the number of GO terms (r=−0.01, not significant).
To summarize so far, smaller studies use more annotated baits on average and more annotated proteins yield more interactions in aggregate, but these effects are much less apparent (if at all) when looking at any individual study. This means that we would predict little correlation between the number of baits in a given experiment and the number of preys reported per bait. The correlation is −0.005 (p~0.7, again, restricting ourselves to studies of size 20 or less).
In the remainder of our analysis, we focus on biases in aggregated data (where aggregation is performed on different subsets of the data). This is appropriate because the downstream applications of protein interactions that motivated our study almost always use data aggregated across numerous studies (e.g. BioGRID), and as described above signals readily emerge in the aggregated data that are difficult to detect in single studies. In particular we focus on annotation bias, meaning the relationship between the numbers of interactions and degree of annotation. A protein interaction network where interaction number is very correlated with number of GO terms is said to be more biologically biased. This is a key parameter because drawing biological conclusions from the network almost always relies on its relationship to prior biological knowledge (e.g. GO), and the annotation bias is a major confound [13]. We also switch to using the more precise multifunctionality score instead of raw GO term counts.
Recasting the questions addressed above in the context of aggregated data, we hypothesize that an interaction network built only from small studies would show greater annotation bias than one where larger studies are included. Thus there should be a correlation between the number of interactions a bait has (“incidence”) and the GO multifunctionality of the proteins, but this correlation should be conditional on the size of the studies included. Taking a sliding threshold on the study size such that we analyze studies meeting a minimum number of baits (see schematic in Figure 1A), we find that the correlation with GO (the annotation bias) generally decreases as studies with fewer baits are excluded (Figure 1B, black line). That is, in small studies, researchers tend to find bait interactions when those baits are already more heavily annotated. We also wondered if we might see a similar effect in the prey (interactors); we hypothesized that large studies would tend to yield less-biased prey, essentially due to the same influences we predict for baits (more uniform treatment of results and less experimenter influence). In fact we observed the opposite: studies that use many baits end up with preys that are more biased towards highly-annotated proteins (Figure 1B, grey line). A possible explanation for this prey selection effect is provided by our analysis of the CRAPome, described later, in that contaminants tend to be more highly annotated.
Figure 1.

Relationships between study size and external knowledge. A: Schematic explaining the data and analysis shown in B and C (bait analysis illustrated). The number of studies each bait appears in was computed. Then the subset of studies at or below the threshold number of baits N was selected. Within this subset, the number of interaction each bait had was correlated with either multifunctionality (plotted in B) or co-expression node degree (plotted in C). This was repeated for each threshold of N up to 20 baits. B: Relation between study size (number of baits) and agreement with GO, for baits (black line) and preys (grey line). C: As in B but using co-expression instead of GO as the comparator.
It is still reasonable to ask to what extent this trend in the overall network is reflected in individual experiments. The most salient point in Figure 1B is the opposing pattern for preys and baits, and the fact it diminishes over modest increases in study size. This can be formally quantified on a per-study level by asking whether we can use the correlation with multifunctionality as a predictor of which proteins are baits in a given experiment. This yields significant predictive performance (AUROC~0.6, p<0.05 binomial test for each group of 100 experiments shown) and this performance significantly declines (Spearman r~−0.36, p<0.05, permutation test) over the range of study sizes shown (for which there is sufficient data; Supplementary Figure 2). Thus, the trends contributing to the network overall appear to be present in individual experiments.
We were interested in having an orthogonal reference data set to compare to the protein interactions. We choose RNA co-expression, which is noisy but not very biased by prior knowledge, especially when aggregated across studies [13]. Repeating the analysis of Figure 1B, but replacing GO multifunctionality with co-expression node degree, shows a weaker trend (Figure 1C; note the difference from 1B in y-axis scale). Small studies show a detectable bias towards baits with high co-expression node degree (this is unsurprising because co-expression node degree is also correlated with GO multifunctionality), but for prey the relationship flattens out in studies when considering studies that have more than one bait (Figure 1C, grey line).
We next asked if these biases are changing over time. Figure 2 shows that the alignment between protein interaction and GO is quite jumpy due to the effect of large studies. First, in Figure 2A we plot how many yeast protein interactions were added to BioGRID over time. The spikes are largely due to large studies added at that time point. A breakdown of these data by experiment type is given in Figure 2B (binned by quarters). On a per-interaction basis, affinity capture-mass spectrometry (AC-MS) data has had a very large effect, contributing 57,960 out of a total of 114,736 interaction reports; however, this is due to only 316 publications out of 6,368 in total (that is, ~5% of the publications contribute over half the interactions), indicating a substantial interplay between the magnitude of laboratory bias (as indicated by study-specific interactions) and experimental technique.
Figure 2.

Trends over time in protein-interaction data. A: Number of preys added over time. Large spikes mostly correspond to the contribution of single large studies to BioGRID. B: As in (A) with a coarser time resolution, showing the distributions of methodologies used. Key: AC: affinity capture; MS, mass spectroscopy; W, western blot; PCA: protein-fragment complementation assay; L: luminescence. C: Analysis as in Figure 1A, but resolved over time. D: As in (C), but using co-expression instead of GO. In C and D, the data in A is plotted in grey on an arbitrary scale to facilitate comparisons.
In Figure 2C and D, we plot correlations measured as outlined in Figure 1A, over time (without any stratification by study size). The data from Figure 2A is under-plotted in grey for ease of comparison. For GO multifunctionality, the correlation with the number of times a protein appeared generally rose until just prior to 2010, at which point it dropped and remained. Throughout the time course, jumps are both up and down. For co-expression, the correlation is both lower to begin with (note the difference in scales between 2C and 2D) and steadily declining till about 2009, with most large jumps being downward. We can see the contributions of large studies by tracing the number of interactions added to BioGRID over a very fine time interval. The large studies have clear and substantial contributions to our analysis of the network (that is, the large spikes in 2A often correspond to jumps in 2C).
The results thus far indicate that as we predicted, large studies generally decrease bias towards already highly-annotated proteins. This occurs despite their tendency to select for more highly-studied preys, because they more strongly reduce bait bias. The impact of larger studies can be emphasized by plotting how many interactions studies of different sizes contribute (Figure 3A). While the numerous small studies contribute much in aggregate (the peak between one and twenty interactions), a small number of large studies (providing 1,000 or more interactions each) dominate. There has also been a trend over time of an increase in the average number of interactions found per bait (Figure 3B). Again, this could reflect more sensitive methods (a type of laboratory bias), more informed choices of baits to favor those that will yield interactions (better use of prior knowledge) or more noisy results (more technical artifacts). We can see both biological and technical bias contribute to the data overall (Figure 3C) since small studies (expected to be more biology-biased) sometimes contribute very large numbers of interactions for a single bait (for example [24], which contributes hundreds of interactions for GIS2) while large studies (less biology-biased) are not as extreme (per bait) but still generally larger than the very small studies.
Figure 3.

Relationships between study size and relative contributions to the aggregate data. A: The number of interaction contributed per study is plotted relative to how many interactions are contributed to the database for all studies with that size of contribution. Thus studies contributing just one interaction contribute a total of about 1800 interactions; the number of studies per bin drops so there is only one study contributing to the right-most data point. B: The number of interactions per bait has increased over time. C: The subset of data used in A derived from bait-prey studies, but plotted on a per-study basis. Each point represents a single study. The data are slightly jittered to reduce overlap, and points further from the bulk of the data are larger to ensure visibility. D: At each time point, we calculate and plot the reproducibility of all studies conducted up to that time; reproducibility is dropping over time. Here reproducibility is defined as the number of times which an interaction is observed in the database (either before or after the study). The black dots show how often an interaction is reproduced in any study, while the grey dots are restricted to bait-prey studies.
The change in number of interactions per bait over time tracks the number of times those interactions are observed (either before or after when a study reports it) (Figure 3D). We term this the reproducibility and calculate its average for all studies up to a given date (the x-axis in Figure 3D). Reproducibility is tending to drop: that is, interactions which are currently being contributed to the aggregate database are increasingly likely to be novel. This effect is partially a reflection of the degree to which interactions observed long ago are more likely to have been observed again. However, together with the data in Figure 3B, it suggests that the additional interactions observed per bait have generally reflected changing experimenter standards, towards either novel interactions or weaker evidence. The data in Figure 3D isn’t particularly influenced by large studies since each study counts equally regardless of size. At the same time, we note that smaller studies tend to have higher reproducibility, with a mean of 4.6 for interactions from studies reporting only one interaction compared to 1.48 for interactions from any study.
We next considered in more detail how co-expression relates to protein interactions. High co-expression is very well aligned with the protein interaction at the extreme, but the trends are otherwise weak (Figure 4). More interestingly, the reproducibility of the interactions (as per Figure 3D) doesn’t seem to account for the trend: both “poorly-reproduced” and “highly-reproduced” interactions are represented among highly co-expressed genes. In Table 1 we offer a list of proposed interesting but poorly-studied baits based on those which have high co-expression and low GO multifunctionality.
Figure 4.

Relationship between co-expression and protein interactions. The distribution of co-expression score (rank) of each protein interaction is plotted. Different levels of reproducibility are shown in shades of grey. There are about 5,769 interactions that are strongly supported by co-expression data.
Table 1.
Suggested candidates for future protein-protein interaction studies. The genes were selected based on having many high-quality co-expression links (scoring in the top 1%), few GO annotations, and few previous reports of interactions. Their potential interactors based on coexpression are also shown.
| Gene Symbol | # Co-expression links | # Previous reports | Predicted Interactions (top 5) |
|---|---|---|---|
| YLR149C | 360 | 3 | YJL163C, YNL115C, USV1, TPS1, YLR312C |
| UGX2 | 355 | 2 | NDE2, DCS2, TMA17, ATG7, YHR138C |
| YGR127W | 304 | 1 | TFS1, RTC3, YGP1, SOD2, YKL091C |
| TSR4 | 400 | 2 | ELP3, NIP7, NOC2, TRM11, DBP9 |
| MSC1 | 329 | 1 | DCS2, GPX1, ECM4, PAI3, RTN2 |
| YNL195C | 237 | 0 | YNL194C, PHM7, USV1, TKL2, RTN2 |
| YER121W | 258 | 2 | UIP4, PBI2, ATG34, GPX1, STF2 |
The lack of a pattern in reproducibility (that is, reproducibility is not predicted by high co-expression) is highlighted when we look the effect of changing GO annotations (Figure 5). In this analysis we allow the GO annotations to vary over time, holding the protein interaction data constant (whereas in the analyses thus far we used a single recent version of the GO annotations while varying the protein interaction data available over time). This isolates the effects of changes in GO. Figure 5 shows correlations between the number of interactions present in the data for each protein and the protein multifunctionality, for a given GO version. We see that changes in GO result in dramatic shifts in overlap with interactions, suggesting that GO is not very safe to use as a gold standard. On the other hand, within each GO time point higher reproducibility is associated with a lower bias towards prior biological knowledge, confirming that the results of Figure 4 are not a quirk of a particular GO annotation set. Together these results indicate that an interaction is more likely to be seen again (reproduced) if it does not involve a highly multifunctional protein. The meaning of this is unclear, without knowing the number of times the proteins have been studied (counting them would require knowing about negative results, which are difficult to document in this context, and obviously not included in BioGRID). As it stands this effect could accurately reflect the robustness of results or could reflect experimenter biases toward novelty. The latter possibility would be consistent with the increasing trend of the grey line in Figure 1B. The importance of accounting for the number of times an interaction is tested in considering evidence is also borne out by previous reports that some interactions are found just a result of both proteins in a pair being studied more [25]. This makes placing too much emphasis on current estimates of reproducibility problematic, if one is attempting to filter out “technical artifacts”; part of what is more reproducible is the biological preferences of investigators or the technical artifacts in data.
Figure 5.

Correlations between current protein interaction incidence and GO multifunctionality assessed across versions of GO for different levels of reproducibility (reproduced 1,2,3,4 or 5 times). Changes in GO over time substantially alter the correlation to the network data, and reproducible interactions are less correlated with protein incidence in GO.
We identified some evidence that artifacts, and not just preference for interesting biology, play a role by assessing the network, using the recently published CRAPome [20]. The CRAPome documents proteins which are found in AC-MS studies as non-specific preys, and thus are likely to represent non-biologically-relevant artifacts. We find that networks constructed using studies of a given size or smaller become more likely to involve “crappy” proteins as the number of baits decreases (Figure 6A). This overlap between researcher preferences (hopefully reflecting biological interest) and crappiness can likewise be seen in the distribution GO terms for crappy genes; crappy genes very rarely have very few GO terms and have significantly more than non-crappy genes overall (Figure 6B; p<0.05, Mann-Whitney test). This result provides one possible explanation for the tendency of large studies to exhibit more prey bias toward highly-annotated proteins (Figure 1B): contaminants tend to be annotated, and large studies may be less selective in reporting preys (as we originally hypothesized). But where possible, researchers presumably respond to the influence of crappiness and prior biological knowledge and filter their data accordingly. While this tactic is often correct, our co-expression analysis (Figure 4) suggests it is also a way of missing out on results involving less-studied proteins.
Figure 6.
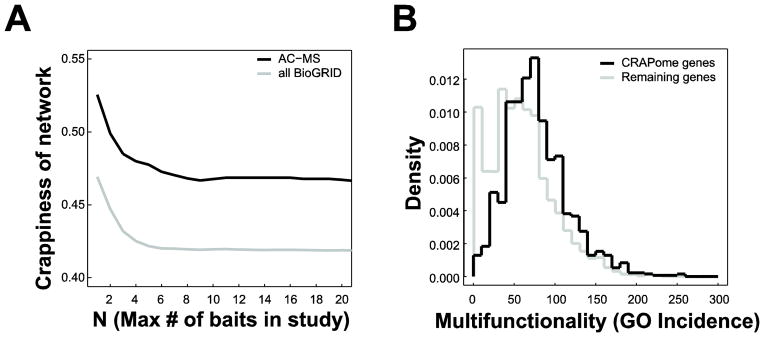
CRAPome proteins are not clearly removed by selection biases. A: Building networks from smaller studies (using data only below a given threshold) does not yield fewer interactions involving CRAPome members. B) The distribution of numbers of GO annotations per protein for CRAPome proteins (solid line) and all other proteins (dashed line).
Discussion
Our analysis of yeast protein interaction data provides support for the impact of two competing types of bias in the data. One bias is towards using prior biological knowledge in the selection of baits and preys; that is, the degree to which a bait or prey is likely to occur in a study (through direct selection or filtering). Small studies tend to have more bait selection bias but comparatively little prey selection bias (Figure 1B) while individual large studies cause drastic changes in network structure (Figure 2C). Biases present in protein interaction data have been previously discussed [26, 27]; however, the focus has been on methodological complementarities or overlaps among different data sources. Rather than simply considering these as problems to be cleaned from some idealized version of the data, we believe the competing demands which come into play in aggregating protein-protein interaction data makes it an ideal data set for the assessment of literature and researcher biases. These are pressing concerns, particularly in genomics research [28], where protein-protein interaction data finds extensive use.
Regardless of the precise cause of the trends we observe, they add to a body of evidence making it increasingly clear that the attempts of researchers to turn to protein interaction networks to interpret their own data are deeply problematic. It is well-documented that protein interaction data are perceived as noisy and incomplete. The generally proposed solution to the noise problem is reproducibility (weeding out false positives), while data integration is an answer to the incomplete nature of any single data source (reducing false negatives). But these approaches, applied uncritically, have tradeoffs. Our experiments in particular highlight the tradeoff between laboratory and selection biases. Using prior knowledge to remove technical artifacts is also a way to accidentally remove previously unknown biology. We argue that GO and co-expression data are two useful ways to measure these effects. There are good agreements between co-expression and protein interactions. Because co-expression is relatively unbiased with respect to prior biological knowledge, we propose it is a way to help prioritize baits for study.
The comparison of GO to the protein interactions suggests that prior knowledge has had somewhat complex and perhaps perverse effects on reproducibility. The data indicate that researchers use prior knowledge to prioritize or de-prioritize baits and preys, but it is not completely clear how or why this happens. The data at hand (especially on the inner workings of the research groups that submit data to BioGRID) are too limited to allow a clear interpretation.
In this paper we have used BioGRID data “as is”, without any filtering or processing. It is reasonable to ask whether biologist’s applications of the PPI data are as naïve. Our experience suggests that uncritical use of PPI data is prevalent. One prominent example (published after completion of our analysis) is illustrative of what we observe to be general trends, and also shows how the biases we have identified can play a critical role. Gulsuner and colleagues (2013) attempted to characterize the properties of de novo DNA sequence variants in schizophrenic individuals [29]. Genes harboring such variants might play a role in schizophrenia, but the false positive rate in detecting them is high, as such variants occur at a similar rate in unaffected individuals (outside of rare nonsense variants). To identify properties of these genes that would more clearly distinguish them from those mutated in controls, Gulsuner et al. downloaded all the physical interactions present in a particular tool’s database (GeneMANIA, [30]), which obtains data from other databases (e.g., Pathway Commons[31]) which in turn obtains their data from other databases, including BioGRID. In this manner, Gulsuner et al. found 753,875 unique interactions across 12,010 proteins; no filtering or other processing was applied. Gulsuner et al. report higher connectivity in this network for their schizophrenia candidate genes compared to genes found mutated in controls, and interpret this as pointing to the clinical relevance of the mutations. What would we expect if the biases we have identified are important? A simple way to examine the effect of selection bias is to look at the number of their candidate genes which are in the network at all; this turns out to be only 18 out of 54. That is, selection bias within the protein interaction data leads to the majority of candidate genes being uncharacterized. This extreme bias and its overlap with selection biases in other domains (e.g. the Gene Ontology) have profound implications for the interpretation of the result reported by Gulsuner et al. In addition, we noted that the vast majority of the 750K interactions were subsequently removed from GeneMANIA (but well before Gulsuner et al.’s work was published), reducing the number to 93K unique interactions. For example, data derived from a study of the human autophagy system [32] went from erroneously adding interactions for every protein pair tested — nearly 200K interactions — to under 700. Other large sources of interactions, such as more than 150K entries from [33], were completely removed for reasons that are less clear. While anecdotal, the example of Gulsuner et al. is not unique. Many biologists use PPI data “as is”, and are apparently unaware of the potential volatility of the data and the biases present. Our results should help highlight the potential for these problems to impact research.
Assuming for the sake of argument that at least some of the factors leading to bias are under direct control of investigators, our work leads to some recommendations. Researchers who generate PPI data should be cognizant of the tradeoffs between selection and laboratory bias when designing their studies and post-processing the results. For example, filtering results for what is deemed likely to be most replicable is risky if replicability is defined in any way using prior biological knowledge. Ideally researchers would document the procedures they use to do final data filtering, and if possible standardize them. A good first step would be to provide both unfiltered and filtered data, which would allow more direct measurement of the effects of prior knowledge. Currently to our knowledge resources such as BioGRID do not provide the ability to include confidence or quality measures with submissions. This would be a valuable addition, especially if the measures were standardized. We have also uncovered signs of data sets that have both high selection and laboratory bias: single yeast baits that have hundreds of interactors are outliers. Such studies greatly alter the network properties of the individual protein in question, which can cause serious problems for later interpretation. For users of the PPI data, removing such studies would probably be beneficial.
There are some caveats and limitations to our study that suggest avenues for future work. We only examined the yeast interactions in BioGRID. We hypothesize the same trends would be detectable in other species and/or databases. We emphasize that these trends are present in the face of much heterogeneity in the data, especially among small studies; there are plenty of exceptions to the trends. This noisiness, as well as limits to the annotations of the data, makes it difficult to definitively map the trends we see onto conscious or unconscious actions of researchers. We hope our analysis stimulates further research in this area.
Supplementary Material
Supplementary Figure 1. Number of GO terms per bait declines as more baits are used. For each study of bait size X, we calculated the median number of GO terms associated with its baits, and then averaged across all studies of size X (bars show standard deviations). While the changes are not dramatic relative to the variation present across all studies, the average effect is pronounced (r=−0.83, p<0.01). We stop at 10 baits per study because past this point the number of studies of a given bait size becomes sparse (and there is often only single studies for a given bait size).
Supplementary Figure 2. The influence of bait and prey correlations with multifunctionality in individual protein interaction experiments. For each of 4,584 unique PubMed IDs generating prey-bait data, we calculated the correlation between the prey genes and the gene multifunctionality in GO, and the bait genes and the gene multifunctionality in GO. We then used that to predict which genes were bait and which were prey (separately, for each experiment) with ROC (probability of successful ranking) shown. Dots represent averages of 100 experiments. The initial learnability is significant (p<0.05, binomial test) and the decline with increasing bait number is also significant (Spearman r~−0.36, p<0.05, permutation test). Higher values on the x-axis (not shown) exhibit extreme trends towards learnability for a few experiments (likely due to the drastic difference in the number of baits available to make predictions).
Highlights.
There are substantial tradeoffs between selection and laboratory biases
Small studies tend to have more selection biases
Larger studies tend to produce more laboratory bias
Reproducibility of protein interactions is confounded by use of prior knowledge
Significance.
Protein-protein interaction data finds particularly heavy use in the interpretation of disease-causal variants. In principle, network data allows researchers to find novel commonalities among candidate genes. In this study, we detail several of the most salient biases contributing to aggregated protein-protein interaction databases. We find strong evidence for the role of selection and laboratory biases. Many of these effects contribute to the commonalities researchers find for disease genes. In order for characterization of disease genes and their interactions to not simply be an artifact of researcher preference, it is imperative to identify data biases explicitly. Based on this, we also suggest ways to move forward in producing candidates less influenced by prior knowledge.
Acknowledgments
We thank Adriana Sedeño for assistance with the Gene Ontology data. We thank an anonymous reviewer for valuable comments.
Funding
PP was supported by NIH Grant GM076990 and the Canadian Institutes for Health Research. JG and SB were supported by a grant from T. and V. Stanley. No funding source played any role in the design, in the collection, analysis, and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Footnotes
Author contributions
JG performed the experiments with contributions from SB. The manuscript was written by JG and PP. All authors approved the final draft.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Jesse Gillis, Email: JGillis@cshl.edu.
Sara Ballouz, Email: sballouz@cshl.edu.
Paul Pavlidis, Email: paul@chibi.ubc.ca.
References
- 1.Chatr-Aryamontri A, Breitkreutz BJ, Heinicke S, Boucher L, Winter A, Stark C, et al. The BioGRID interaction database: 2013 update. Nucleic acids research. 2013;41:D816–23. doi: 10.1093/nar/gks1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poirel CL, Owens CC, 3rd, Murali TM. Network-based functional enrichment. BMC Bioinformatics. 2011;12 (Suppl 13):S14. doi: 10.1186/1471-2105-12-S13-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wren JD. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25:1694–701. doi: 10.1093/bioinformatics/btp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res. 2002;12:37–46. doi: 10.1101/gr.205602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaefer MH, Fontaine JF, Vinayagam A, Porras P, Wanker EE, Andrade-Navarro MA. HIPPIE: Integrating protein interaction networks with experiment based quality scores. PLoS One. 2012;7:e31826. doi: 10.1371/journal.pone.0031826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ren G, Liu Z. NetCAD: a network analysis tool for coronary artery disease-associated PPI network. Bioinformatics. 2013;29:279–80. doi: 10.1093/bioinformatics/bts666. [DOI] [PubMed] [Google Scholar]
- 7.von Eichborn J, Dunkel M, Gohlke BO, Preissner SC, Hoffmann MF, Bauer JM, et al. SynSysNet: integration of experimental data on synaptic protein-protein interactions with drug-target relations. Nucleic acids research. 2013;41:D834–40. doi: 10.1093/nar/gks1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee I, Li Z, Marcotte EM. An improved, bias-reduced probabilistic functional gene network of baker’s yeast, Saccharomyces cerevisiae. PLoS One. 2007;2:e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee I, Blom UM, Wang PI, Shim JE, Marcotte EM. Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res. 2011;21:1109–21. doi: 10.1101/gr.118992.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim WK, Krumpelman C, Marcotte EM. Inferring mouse gene functions from genomic-scale data using a combined functional network/classification strategy. Genome biology. 2008;9 (Suppl 1):S5. doi: 10.1186/gb-2008-9-s1-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merton RK. The Matthew Effect in Science: The reward and communication systems of science are considered. Science. 1968;159:56–63. [PubMed] [Google Scholar]
- 13.Gillis J, Pavlidis P. The impact of multifunctional genes on “guilt by association” analysis. PLoS One. 2011;6:e17258. doi: 10.1371/journal.pone.0017258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gillis J, Pavlidis P. “Guilt by association” is the exception rather than the rule in gene networks. PLoS Comput Biol. 2012;8:e1002444. doi: 10.1371/journal.pcbi.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillis J, Pavlidis P. Assessing identity, redundancy and confounds in Gene Ontology annotations over time. Bioinformatics. 2013;29:476–82. doi: 10.1093/bioinformatics/bts727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta S, Wallqvist A, Bondugula R, Ivanic J, Reifman J. Unraveling the conundrum of seemingly discordant protein-protein interaction datasets. Conf Proc IEEE Eng Med Biol Soc. 2010;2010:783–6. doi: 10.1109/IEMBS.2010.5626490. [DOI] [PubMed] [Google Scholar]
- 17.Yook SH, Oltvai ZN, Barabasi AL. Functional and topological characterization of protein interaction networks. Proteomics. 2004;4:928–42. doi: 10.1002/pmic.200300636. [DOI] [PubMed] [Google Scholar]
- 18.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome biology. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nature methods. 2013;10:730–6. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gillis J, Pavlidis P. The role of indirect connections in gene networks in predicting function. Bioinformatics. 2011;27:1860–6. doi: 10.1093/bioinformatics/btr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23:1846–7. doi: 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- 23.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3:Article3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 24.Sammons MA, Samir P, Link AJ. Saccharomyces cerevisiae Gis2 interacts with the translation machinery and is orthogonal to myotonic dystrophy type 2 protein ZNF9. Biochemical and biophysical research communications. 2011;406:13–9. doi: 10.1016/j.bbrc.2011.01.086. [DOI] [PubMed] [Google Scholar]
- 25.Ivanic J, Wallqvist A, Reifman J. Evidence of probabilistic behaviour in protein interaction networks. BMC Syst Biol. 2008;2:11. doi: 10.1186/1752-0509-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu HF, Huang FL, Lin CJ. Dual coordinate descent methods for logistic regression and maximum entropy models. Mach Learn. 2011;85:41–75. [Google Scholar]
- 27.Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nature biotechnology. 2002;20:991–7. doi: 10.1038/nbt1002-991. [DOI] [PubMed] [Google Scholar]
- 28.Ioannidis JP. This I believe in genetics: discovery can be a nuisance, replication is science, implementation matters. Frontiers in genetics. 2013;4:33. doi: 10.3389/fgene.2013.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulsuner S, Walsh T, Watts Amanda C, Lee Ming K, Thornton Anne M, Casadei S, et al. Spatial and Temporal Mapping of De Novo Mutations in Schizophrenia to a Fetal Prefrontal Cortical Network. Cell. 2013;154:518–29. doi: 10.1016/j.cell.2013.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic acids research. 2010;38:W214–20. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerami EG, Gross BE, Demir E, Rodchenkov I, Babur O, Anwar N, et al. Pathway Commons, a web resource for biological pathway data. Nucleic acids research. 2011;39:D685–90. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lleres D, Denegri M, Biggiogera M, Ajuh P, Lamond AI. Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO reports. 2010;11:445–51. doi: 10.1038/embor.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Number of GO terms per bait declines as more baits are used. For each study of bait size X, we calculated the median number of GO terms associated with its baits, and then averaged across all studies of size X (bars show standard deviations). While the changes are not dramatic relative to the variation present across all studies, the average effect is pronounced (r=−0.83, p<0.01). We stop at 10 baits per study because past this point the number of studies of a given bait size becomes sparse (and there is often only single studies for a given bait size).
Supplementary Figure 2. The influence of bait and prey correlations with multifunctionality in individual protein interaction experiments. For each of 4,584 unique PubMed IDs generating prey-bait data, we calculated the correlation between the prey genes and the gene multifunctionality in GO, and the bait genes and the gene multifunctionality in GO. We then used that to predict which genes were bait and which were prey (separately, for each experiment) with ROC (probability of successful ranking) shown. Dots represent averages of 100 experiments. The initial learnability is significant (p<0.05, binomial test) and the decline with increasing bait number is also significant (Spearman r~−0.36, p<0.05, permutation test). Higher values on the x-axis (not shown) exhibit extreme trends towards learnability for a few experiments (likely due to the drastic difference in the number of baits available to make predictions).