

Abstract
Background
Janus kinases (JAK) are regulators of signaling through cytokine receptors. The importance of JAK1/3 signaling on Th2 differentiation and development of lung allergic responses has not been investigated.
Objective
To examine a selective JAK1/3 inhibitor (R256) on differentiation of Th subsets in vitro and on development of ovalbumin (OVA)-induced airway hyperresponsiveness (AHR) and inflammation in an experimental model of asthma.
Methods
A selective JAK1/3 inhibitor was used to assay the importance of this pathway on induction of Th1, Th2, and Th17 differentiation in vitro. In vivo, the effects of inhibiting JAK1/3 signaling were examined by administering the inhibitor during the sensitization or during allergen challenge phases in the primary challenge model or just prior to provocative challenge in the secondary challenge model. Airway inflammation and AHR were examined after the last airway challenge.
Results
In vitro, R256 inhibited differentiation of Th2 but not Th1 or Th17 cells, associated with downregulation of STAT6 and STAT5 phosphorylation. However, once polarized, Th2 cells were unaffected by the inhibitor. In vivo, R256 administered during the OVA sensitization phase prevented development of AHR, airway eosinophilia, mucus hypersecretion, and Th2 cytokine production without changes in Th1 and Th17 cytokine levels, indicating that selective blockade of Th2 differentiation was critical. Inhibitor administration after OVA sensitization but during the challenge phases in the primary or secondary challenge models similarly suppressed AHR, airway eosinophilia, and mucus hypersecretion without any reduction in Th2 cytokine production, suggesting the inhibitory effects were downstream of Th2 cytokine receptor signaling pathways.
Conclusions
Targeting the Th2-dependent JAK-STAT activation pathway represents a novel therapeutic approach for the treatment of asthma.
Clinical Implications
Targeting JAK1/3 signaling pathways provides a novel intervention for preventing allergen-induced alterations in lung function.
Capsule Summary
JAK1/3 signaling pathways are essential for initiation of Th2 differentiation and the development of lung allergic responses.
Keywords: JAK1/3, asthma, Th2
Introduction
Cell signaling through the Janus kinases (JAK)-signal transduction and activator of transcription (STAT) pathway plays a critical role in the function of cytokines, growth factors, and hormones (1). The JAK tyrosine kinase family members are activated following receptor ligation, which leads to phosphorylation of the STATs, events central to DNA transcription and cell activation (2, 3). Activation of receptor-associated JAKs by phosphorylation of specific residues leads to the recruitment of specific STATs which are then tyrosine phosphorylated. The activated STATs are released from the receptor, dimerize and translocate to the nucleus where they bind to members of cytokine response elements (4, 5). As most proinflammatory cytokines and growth factors utilize this pathway, the JAK-STAT pathway is linked to various immunodeficiency and autoimmune diseases (1, 6-8).
There are four members of the JAK family JAK1, JAK2, JAK3, and Tyk2. JAK1 associates with a variety of cytokine and growth factor signaling pathways (1). JAK3 expression is restricted to hematopoietic cells and co-localizes with JAK1, transmitting signals through the common γ chain of the cytokine receptors for IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21 (2). As a result, JAK1 and JAK3 play critical roles in the initiation of inflammation and have been investigated as potent therapeutic targets in a variety of inflammatory diseases. A number of JAK inhibitors have been developed for clinical use (1, 9). Ruxolitinib, a selective JAK1 and JAK2 inhibitor, has been used in the treatment of myelofibrosis and shown to impair dendritic cell functions (10). Tofacitinib, a pan-JAK inhibitor, which targets JAK1 and JAK3 and to a lesser extent JAK2, has been effective in rheumatoid arthritis (11, 12) as well in preliminary studies of other autoimmune diseases, inflammatory bowel disease, transplant rejection, or hematopoietic disorders such as myelofibrosis and polycythemia vera (8, 9, 13, 14).
Asthma is chronic inflammatory respiratory disease associated with Th2-dominant immune responses (15). Despite advances in therapy, asthma prevalence remains high in industrial countries and the socio-economic burden caused by disease exacerbations or steroid refractory asthma is significant (16). Therefore numerous strategies have been attempted, but to date, they have failed to sustain in the clinic (17). Since the JAK/STAT pathways play a critical role in the differentiation of T helper (Th) cells including Th2 cells, JAK inhibitors were studied in animal models of asthma. Tofacitinib, a pan-JAK inhibitor, was shown to reduce allergen-induced airway eosinophilia and IL-13 levels in mice (18). Administration of the pan-JAK inhibitor, pyridone 6, reduced airway hyperresponsiveness (AHR), airway eosinophila, and airway mucus hyperproduction when mice were treated only during the challenge phase (19). Although non-selective JAK inhibitors showed benefit in experimental asthma models, the targeting of JAK2 function with these reagents may be of concern including suppression of T regulatory cell activity or interference with other important pathways involving JAK2.
R256 is a selective ATP-competitive JAK1/3 inhibitor identified through cell-based assays. In this study, we determined the effects of R256 on the differentiation of Th subsets in vitro and the development of allergen-induced AHR, airway inflammation, and cytokine production in the airways. The results demonstrated that selective inhibition of JAK1/3 prevented Th2 differentiation without altering Th1 and Th17 differentiation. When added to differentiated cells, no effects were observed. In an animal model, in mice which received R256 during the sensitization phase, the development of AHR, airway eosinophilia, and mucus hypersecretion were prevented. On the other hand, when mice received R256 after allergen sensitization, but during either primary allergen challenge or a single provocative secondary allergen challenge, after allergen-induced airway inflammation and AHR were established, AHR, airway eosinophilia, and mucus hypersecretion were reduced but without any modification of Th2 cytokine production. These results suggest that R256 has important activities both during the allergen sensitization phase as well as the allergen challenge phase, attenuating development of Th2-dependent asthma.
Methods
Animals
Wild-type (WT) female BALB/c, OT-2 TCR transgenic, and C57BL/6 mice aged 6-8 weeks old were obtained from Jackson Laboratories (Bar Harbor, ME). All mice were maintained under specific pathogen-free conditions. All experiments were conducted under a protocol approved by the Institutional Animal Care and Use Committee of the National Jewish Health.
Cell-based selectivity assays of R256 activities
The activity of R256 (Rigel Inc.) was assessed in a panel of cell-based assays. R256 is a selective inhibitor of JAK1/3-dependent signaling. Eotaxin production induced by IL-13 (25 ng/ml, Peprotech, Rocky Hill, NJ) or IL-4 (5 ng/ml, Peprotech) in normal human lung fibroblasts (NHLF, Lonza, Allendale, NJ) was measured by ELISA (R&D Systems) (20-23). STAT6 phosphorylation induced by IL-13 (50 ng/ml) or IL-4 (10 ng/ml) in NHLF was measured by intracellular FACS (anti-pY641-STAT6 AlexaFluor488; BD Biosciences, San Jose, CA). IL-2-dependent human primary T cell proliferation was assessed using Promega CellTiter-GloTM Luminescent Cell Viability Assay (Promega, Madison, WI) in the presence of 40 units/ml IL-2 (R&D Systems, Minneapolis, MN) (24). STAT5 phosphorylation induced by IL-2 in human primary T cells was measured by intracellular FACS analysis (anti-pY694-STAT5 AlexaFluor488; BD Biosciences). The erythropoietin (EPO, 1 unit/ml, R&D Systems) -dependent survival of cultured human erythroid progenitor cells (CHEPs) was determined using Promega's CellTiter- GloTM Luminescent Cell Viability Assay (25, 26). Surface ICAM-1 (anti-ICAM-1-APC, BD Biosciences) expression induced by IFNγ (10 ng/ml, Peprotech) on U937 cells was measured by FACS (27). CHEPs were differentiated from CD34+ cord blood cells in the presence of IL-3 (10 ng/ml), IL-10 (10 ng/ml) and SCF (25 ng/ml) (Peprotech) for 9 days and with addition of EPO for the last day (28). The enzymatic activity of tryptase released by human cultured mast cells upon stimulation with IgE was quantified by cleavage of the synthetic fluorescent peptide substrate Z Ala Lys-Arg-AMC.2TFA (MP Biomedicals, Solon OH) in tryptase buffer (29). B-cell receptor-dependent Erk1/2 phosphorylation was measured in Ramos cells by intracellular FACS (human anti-IgM 5 μg/ml, Jackson Imunoresearch Labs, West Grove, PA; anti-pT202/pY204-ERK1/2-AlexaFluor488; BD Biosciences). Human primary T cell activation was assessed by measuring IL-2 production by ELISA (R&D Systems) following plate-bound anti-CD3 (1 μg/ml) and anti-CD28 (5 μg/ml) stimulation (anti-human CD3, BD Biosciences; anti-human CD28, Immunotech, Pasadena, CA). Human umbilical vein endothelial cells (HUVEC, LONZA) were stimulated with VEGF and VEGFR2 phosphorylation was assessed by ELISA (100 ng/ml VEGF165; R&D Systems; Rabbit anti-phospho-VEGFR2 mAb, Cell Signaling Technology) (30). EGFR phosphorylation was measured in HeLa cells following EGF stimulation by staining permeabilized cells with a phospho-specific EGFR antibody and quantified by chemiluminescence (0.2 μM EGF, Peprotech; Phospho-EGFR Tyr1173, Cell Signaling Technology, Danvers, MA).
Generation of Th1, Th2, and Th17 cells and R256 treatment
CD4+CD45RB+ naive Th cells were isolated from OT-2 TCR transgenic mouse spleen cells by flow cytometry (Mo-FLO XDP; Beckman Coulter, Inc.). Isolated naive Th cells were cultured with rmIL-2 (20 ng/ml; R&D Systems, Inc.), rmIL-12 (5 ng/ml; Peprotech), rmIFN-γ (1 ng/ml; Pepro Tech EC Ltd.), and anti-IL-4 mAb (10 μg/ml; eBioscience) for Th1 differentiation (31), rmIL-2 (20 ng/ml), rmIL-4 (1 ng/ml; Peprotech), anti-IFN-γ mAb (10 μg/ml; eBioscience), and anti-IL-12p40 mAb (10 μg/ml; eBioscience) for Th2 differentiation (32), or rhIL-6 (50 ng/ml; Pepro Tech EC Ltd.), rhTGF-β1 (2 ng/ml; Pepro Tech EC Ltd.), rmIL-23 (10 ng/ml, R&D Systems, Inc.), anti-IL-4 mAb (10 μg/ml), and anti-IFN-γ mAb (10 μg/ml) for Th17 differentiation (33), in the presence of mitomycin C-treated splenocytes of WT C57BL/6 and 5 μg/ml OVA323-339 peptide (AnaSpec, Fremont, CA). Each Th subset was re-stimulated with OVA323-339 peptide 48 hrs later under the same conditions. Cells were collected and washed after incubation for 6-8 days and restimulated with plate-coated anti-CD3 and anti-CD28 (eBioscience) for 6 hrs with or without brefeldin A (BFA) (Sigma). The supernatants from cultures without BFA were assayed for cytokine production by ELISA, and the cells from cultures with BFA were assayed by intracellular cytokine staining. For intracellular staining, 6 hrs after the stimulation the cells were fixed in 4% paraformaldehyde and permeabilized with 0.1% saponin buffer prior to intracellular cytokine staining and flow cytometry using a FACSCalibur with FlowJo software (Tree Star, Ashland, OR). The following antibodies were used: APC-conjugated anti-IL-4 mAb, PE-conjugated anti-IL-13 or IL-17 mAbs, FITC-conjugated IFN-γ mAb (eBioscience).
During differentiation from days 0-6, or after polarization was complete from days 6-8 of Th1, Th2, and Th17 cell cultures, R256 (25, 50, and 100 nM) was added daily. In control cultures, DMSO alone was added.
Flow cytometric analyses of STAT phosphorylation
On day 6 of the Th1, Th2, and Th17 cell cultures, cells were washed and fixed by the addition of 4% paraformaldehyde for 15 minutes and permeabilized with cold 90% methanol for 30 minutes on ice. Cells were stained with Alexa Fluor 647-conjugated p-STAT1, p-STAT3, p-STAT4, p-STAT5, or p-STAT6 mAb (BD Biosciences). Flow cytometry was carried out using FACSCalibur with FlowJo software.
Development of allergen-induced airway inflammation and treatment with R256
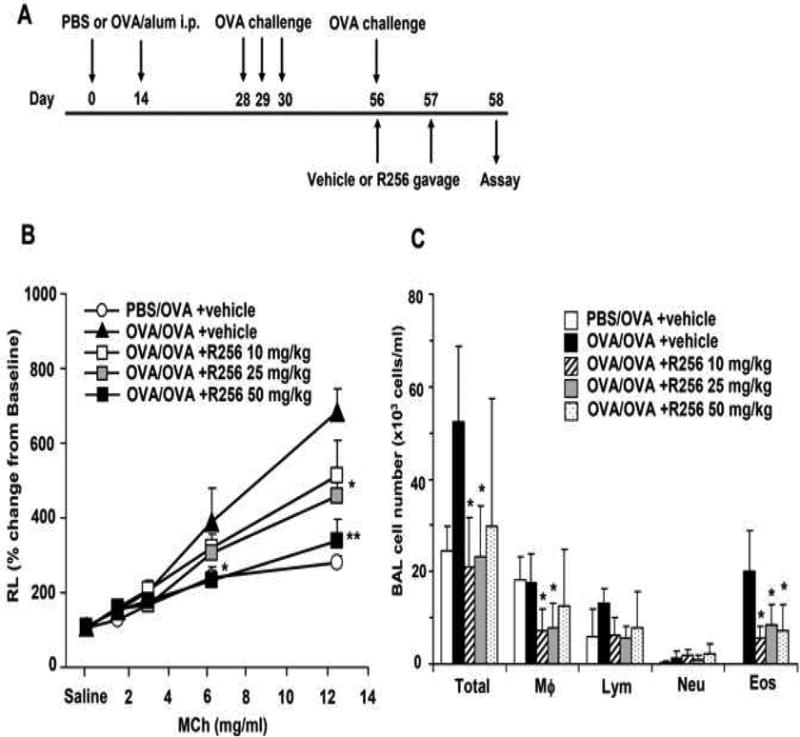
Mice were sensitized intraperitoneally with 20 μg of OVA (Fisher Scientific) emulsified in 2 mg of alum (Imject Alum, Pierce) in a total volume of 100 μl on days 0 and 14. Mice were subsequently challenged via the airways by inhalation exposure to aerosols of OVA (1% in PBS) for 20 minutes on days 28-30 (primary allergen challenge model). To further investigate the effects of the inhibitor on established allergen-induced airway disease, sensitized and previously challenged mice received a single, provocative allergen challenge on day 56 (secondary allergen challenge) (34). OVA aerosols were produced by an ultrasonic nebulizer (model NE-U07; Omron Healthcare, Kyoto, Japan). Airway function was measured after the last allergen challenge (day 32 or 56) as described below, followed by collection of samples for further analyses.
The mice were administered R256 orally twice daily at each time point either during the OVA-sensitization phase or during the primary or secondary OVA-challenge phases. In the sensitization phase (Fig. 2A), R256 (50 mg/kg/time) was administered from days 0-4 and from days 14-18, twice a day. In the primary challenge phase (Fig. 3A) and the secondary challenge phase (Fig. 4A), mice were treated with R256 (10, 25, and 50 mg/kg/time) on days 28-31 or days 56-57, respectively, twice a day prior to and subsequent to each OVA challenge. In control groups, only vehicle (0.25% HPMC) was administered.
Figure 2.
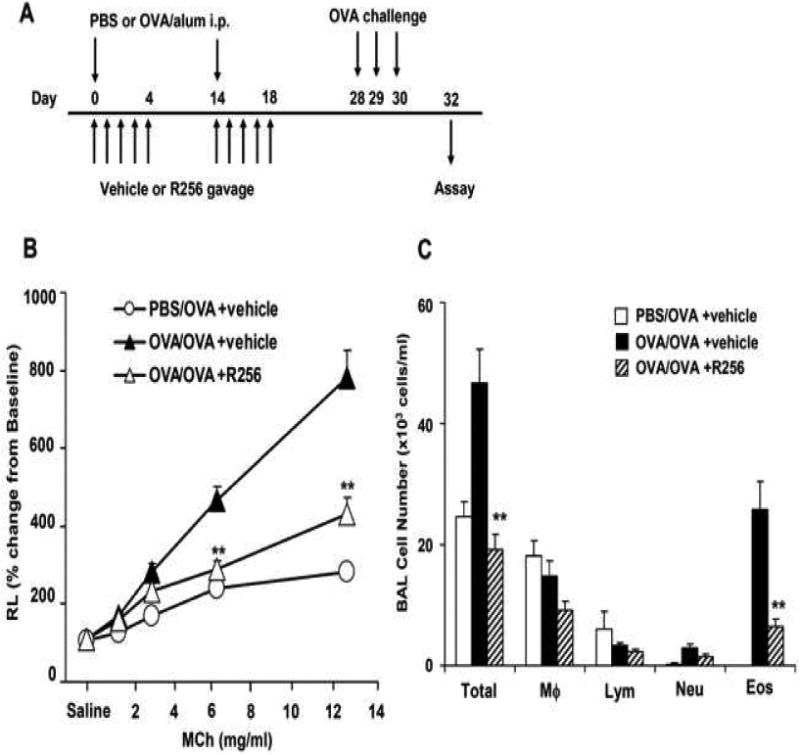
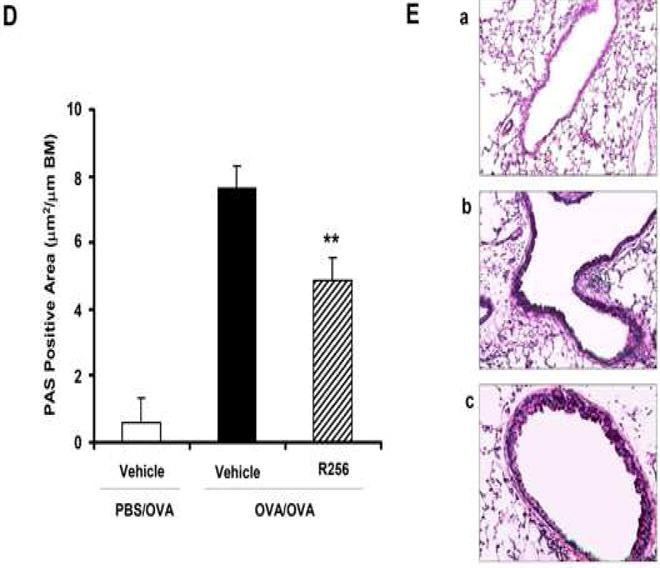
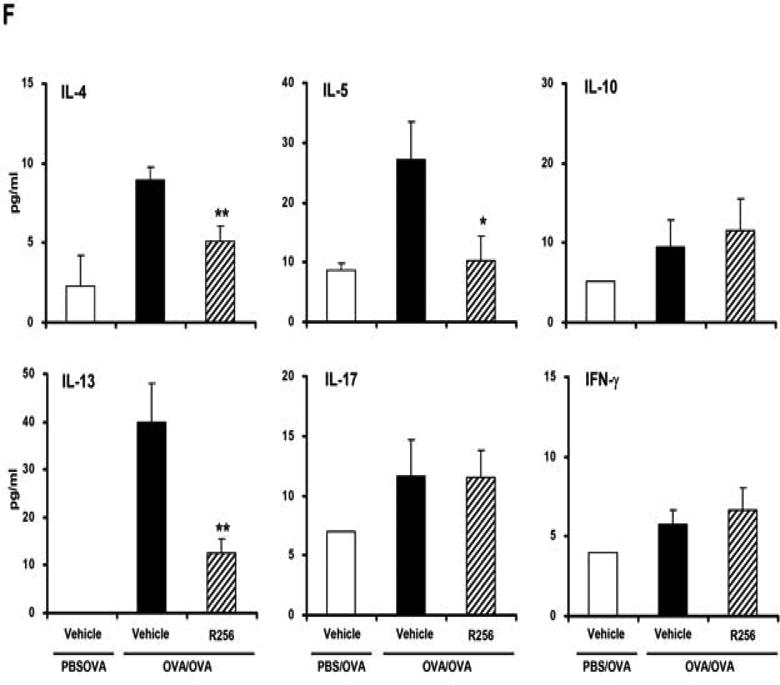
The effects of R256 treatment during allergen sensitization on the development of allergen-induced AHR and airway inflammation. (A) R256 was orally administered during the sensitization phase as described in Materials and Methods. Two weeks after the last allergen sensitization, mice were exposed to 3 consecutive days of allergen challenge followed by assessments of airway responsiveness to aerosolized MCh and BAL/lung tissue sampling 48 hrs after the last OVA challenge. (B) Lung resistance (RL). (C) BAL cell composition and (D) goblet cell metaplasia. (E) Representative pictures show (a) PBS/OVA+vehicle, (b) OVA/OVA+vehicle, and (c) OVA/OVA+R256. (F) Cytokine levels in BAL fluid. Mice were sham-sensitized and challenged to OVA (PBS/OVA) or sensitized and challenged to OVA (OVA/OVA). MCh; methacholine. Mϕ; macrophage. Lym; lymphocyte. Neu; neutrophil. Eos; eosinophil. The data for each group were expressed as means±SEM. The results are from 2 independent experiments with 4 mice per group, n=8. *p<0.05 and **p<0.01 compared to OVA/OVA+vehicle-treated group.
Figure 3.
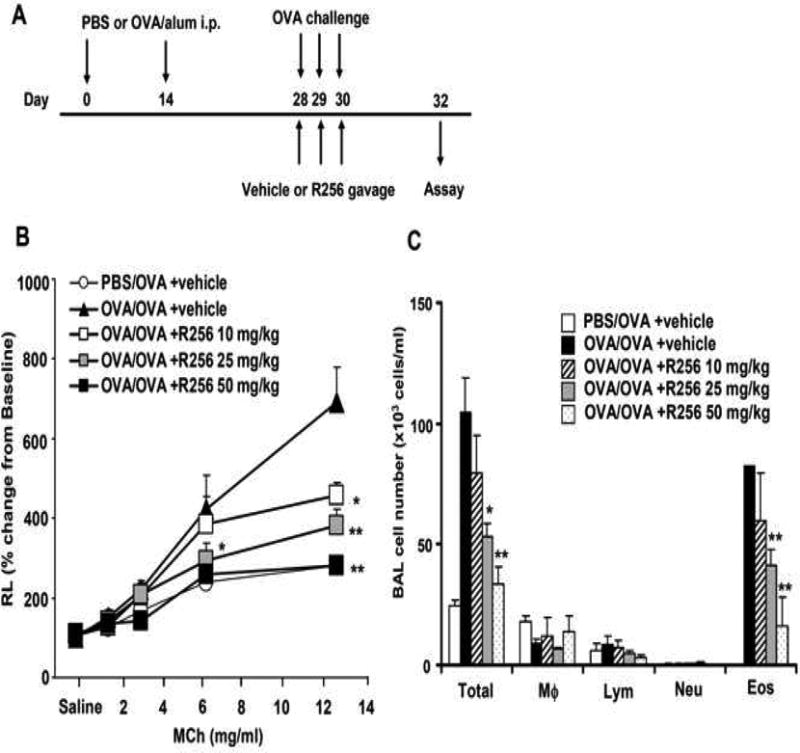
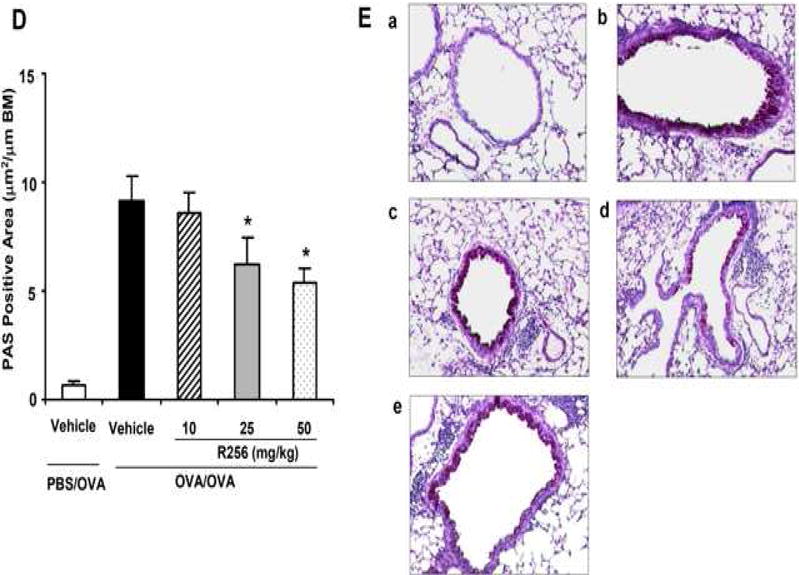
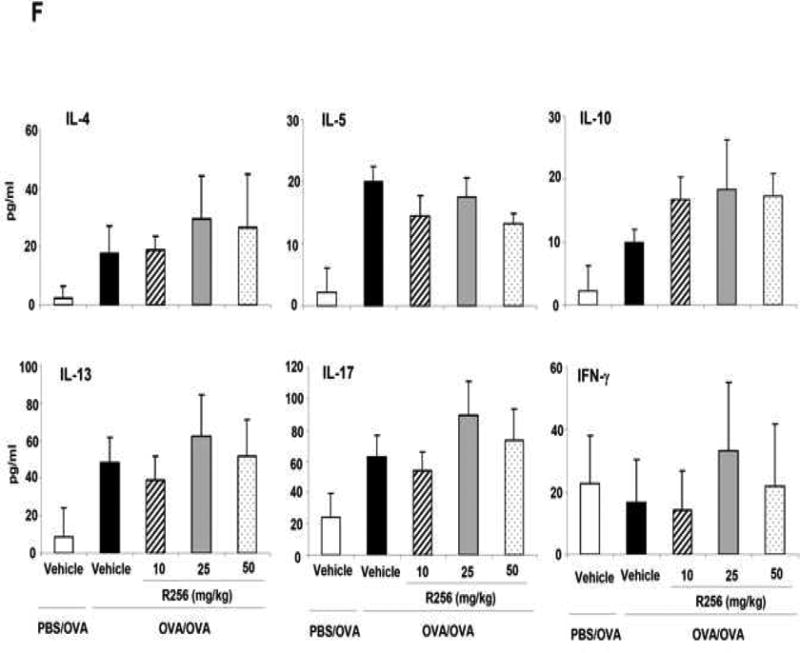
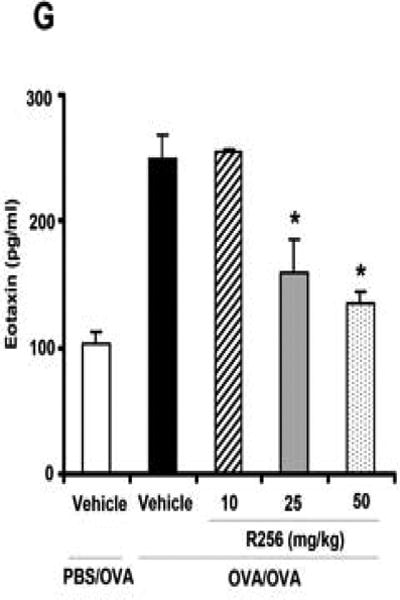
The effects of R256 treatment during primary allergen challenge on the development of allergen-induced AHR and airway inflammation. (A) The indicated doses of R256 were orally administered during the allergen challenge phase as described in Materials and Methods. Two weeks after the last allergen sensitization, mice were exposed to 3 consecutive days of allergen challenge followed by assessments of airway responsiveness and BAL/lung tissue sampling 48 hrs after the last OVA challenge. (B) Lung resistance (RL). (C) BAL cell composition, (D) goblet cell metaplasia. (E) Representative images show (a) PBS/OVA+vehicle, (b) OVA/OVA+vehicle, (c) OVA/OVA+R256 (10 mg/kg), (d) OVA/OVA+R256 (25 mg/kg), (e) OVA/OVA+R256 (50 mg/kg). (F) Cytokine levels, and (G) eotaxin levels in BAL fluid collected at 48 and 6 hrs after final OVA challenge. The data for each group were expressed as means±SEM. The results are from 3 independent experiments with 3 mice per group, n=9. *p<0.05 and **p<0.01 compared to OVA/OVA+vehicle-treated group.
Figure 4.
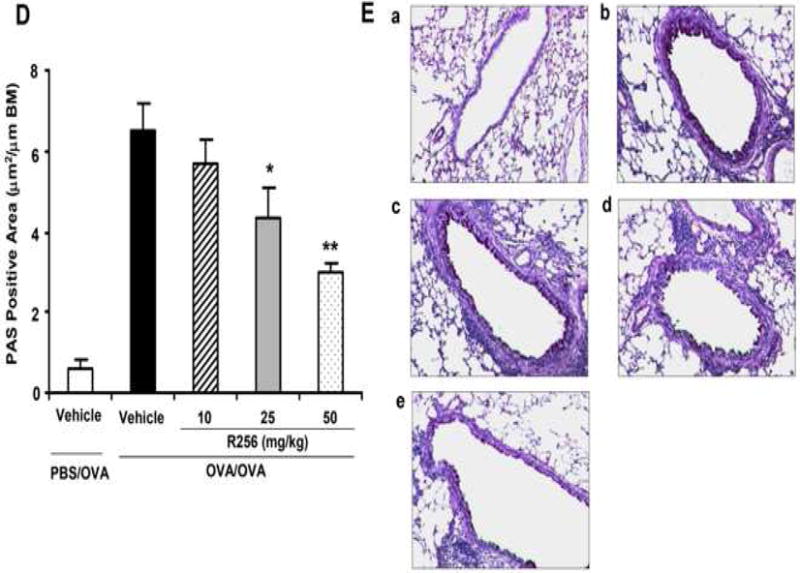
The effects of R256 treatment during the secondary allergen challenge on the development of allergen-induced AHR and airway inflammation. (A) The indicated doses of R256 were orally administered during and after the secondary allergen challenge as described in Materials and Methods. Four weeks after the primary allergen challenge, mice were exposed to a single allergen challenge followed by assessments of airway responsiveness and BAL/lung tissue sampling 48 hrs after the secondary OVA challenge. (B) Lung resistance (RL). (C) BAL cell composition, (D) goblet cell metaplasia. (E) Representative images show (a) PBS/OVA+vehicle, (b) OVA/OVA+vehicle, (c) OVA/OVA+R256 (10 mg/kg), (d) OVA/OVA+R256 (25 mg/kg), (e) OVA/OVA+R256 (50 mg/kg). The data for each group were expressed as means±SEM. The results are from 3 independent experiments with 4 mice per group, n=12. *p<0.05 and **p<0.01 compared to OVA/OVA+vehicle-treated group.
Assessment of airway function
Airway responsiveness was assessed as previously described by measuring changes in airway resistance in response to increasing doses of aerosolized methacholine (MCh; Sigma) (35). Data are expressed as the percent change from baseline values obtained after inhalation of saline.
Bronchoalveolar lavage
Immediately after assessment of AHR, lungs were lavaged with 1 mL of Hank's balanced salt solution through the trachea. Total leukocyte and cell differentials were performed as previously described (35).
Measurement of cytokines and chemokines
Cytokine levels in the BAL fluid and cell culture supernatants were measured by IL-4, IL-5, IL-10, IL-13, IL-17, and IFN-γ ELISAs (eBioscience, San Diego, CA) according to the manufacturer's directions. For measurement of eotaxin levels, BAL fluid was collected 6 hrs after the final OVA challenge, and assayed by ELISA (eBioscience).
Intracellular cytokine and Foxp3 staining of lung cells
Lung cells were isolated as previously described using collagenase digestion (36). Lung leukocytes were purified by 35% percoll gradient centrifugation. After 6 hrs of stimulation with plate-coated anti-CD3/anti-CD28 (eBioscience) in the presence of BFA, cells were examined for intracellular cytokine staining after cell surface staining with APC-conjugated anti-CD4 mAb (eBioscience) followed by fixation with 4% PFA and permealization with 0.1% saponin buffer. For detection of CD4+ Foxp3+ cells, the cells were stained with the anti-CD4 mAb followed by fixation and permeabilization with Foxp3 staining Buffer set (eBioscience), then stained with FITC-conjugated Foxp3 mAb (eBioscience).
T regulatory cell functional assay
CD4+CD25+ T regulatory cell (Treg) cells were isolated from spleens of naive mice using the Treg isolation kit (Miltenyi Biotec, Germany). Isolated Treg cells were treated with DMSO or R256 (50 nM) for 3 hrs. After washing out of drug, Treg cells were co-cultured with CFSE (eBioscience)-labeled CD4+CD25- cells which were isolated from spleens of naive mice. Following three days co-culture, division of CFSE-labeled CD4+ CD25- cells stimulated with anti-CD3/anti-CD28 was analyzed by flow cytometry.
Histologic studies
After lavage, the lungs were fixed in 10% formalin and processed into paraffin blocks. Tissue sections (5-μm) were stained with hematoxylin and eosin (H&E) and periodic acid Schiff (PAS) for identification of mucus-containing cells. The slides were transferred to pictures using a Nikon microscope (Melville, NY). PAS-positive cells in the tissues were quantified using NIH Image analysis system (version 1.63; NIH, Bethesda, MA) and counting 6-8 different fields per slide in a blinded manner.
Statistical analysis
Values for all measurements were expressed as means±SEM. For comparisons between multiple groups, the Tukey-Kramer test was used. Nonparametric analyses, using the Mann-Whitney U test or Kruskal-Wallis test, were also applied to confirm that statistical differences remained significant, even if the underlying distribution was uncertain. The p-value for significance was set at less than 0.05.
Results
R256 is a selective JAK1/JAK3 inhibitor identified through cell-based screening
R256 is a selective JAK1/3 inhibitor in cell-based assays (Table 1). R256 potently suppressed JAK1/JAK2/Tyk2-dependent STAT6 phosphorylation and eotaxin production in NHLF in response to IL-13 with EC50s of 37nM and 11 nM, respectively. R256 inhibited equally effectively JAK1/3-dependent STAT6 phosphorylation and eotaxin production in response to IL-4 in the same cells. Furthermore, R256 inhibited JAK1/JAK3-dependent T-cell proliferation and STAT5 phosphorylation in response to IL-2 with a EC50s below 50 nM. In contrast, R256 showed limited inhibition of JAK2-dependent primary erythroblast cell survival in response to EPO and JAK1/2-dependent ICAM-1 expression on the monocytic cell line, U937, in response to IFNγ.
Table 1. Activity of R256 in a cell-based assay panel assessing selectivity.
| Assay | Stimulation | Upstream signals | R256 (EC50 ± SD,μM) |
|---|---|---|---|
| NHLF eotaxin production | IL-13 | JAK1/JAK2/Tyk2 | 0.011 ± 0.003 |
| NHLF phospho-Stat6 | IL-13 | JAK1/JAK2/Tyk2 | 0.037 ± 0.021 |
| NHLF eotaxin production | IL-4 | JAK1/JAK3 | 0.020 ± 0.007 |
| NHLF phospho-Stat6 | IL-4 | JAK1/JAK3 | 0.037 ±0.016 |
| Primary T cell proliferation | IL-2 | JAK1/JAK3 | 0.017 ± 0.005 |
| Primary T cell phospho-STAT5 | IL-2 | JAK1/JAK3 | 0.044 ± 0.016 |
| U937 ICAM-1 | IFNγ | JAK1/JAK2 | 6.52 ± 0.788 |
| Primary erythroblast survival | EPO | JAK2 | 1.35 ± 0.493 |
| CHMC Tryptase | IgE | Syk | 0.409 ± 0.227 |
| Ramos phospho-ERK | IgM | Syk | 0.911 ± 0.015 |
| Primary T cell IL-2 production | TCR/CD28 | Lck/ZAP70 | > 4 |
| HUVEC phospho-VEGFR2 | VEGF | VEGFR2 | > 6 |
| HeLa phospho-Akt | EGF | EGFR | Inactive @ 10μM |
EC50 indicates the concentration of R256 at which the response is inhibited by 50%.
NHLF: normal human lung fibroblast, CHMC: cultured human mast cell, HUVEC: human umbilical vein endothelial cell
In a panel of cell-based assays to assess activity against a range of cytoplasmic and receptor tyrosine kinase-dependent pathways including Syk-dependent signaling, Lck/ZAP70-dependent signaling, VEGFR2 activation, R256 showed little effect.
Effects of R256 on differentiating Th cells
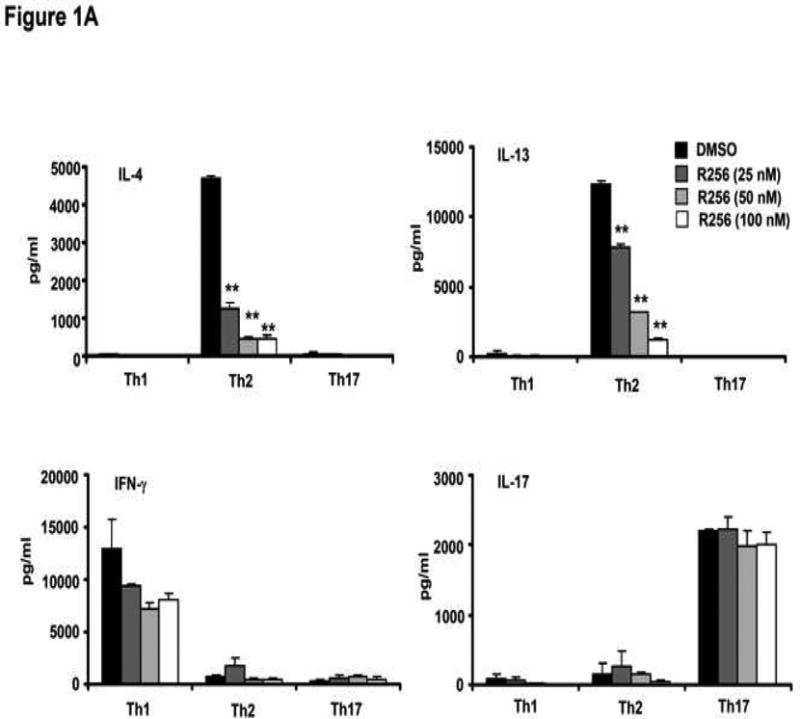
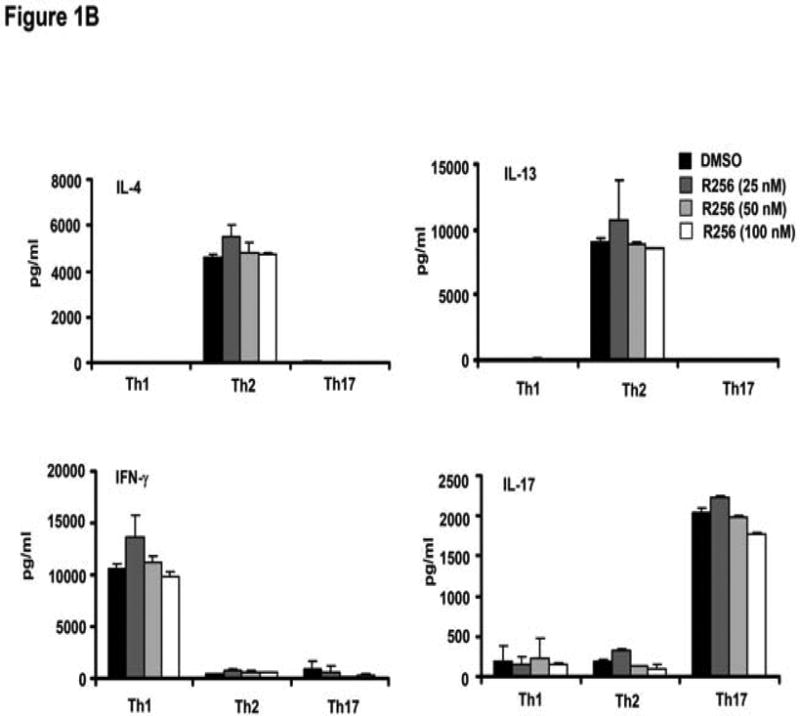
The effects of the selective JAK1/3 inhibitor on the differentiation of naive CD4 cells into Th1, Th2, and Th17 cells were determined. R256 was added to the culture medium from day 0 of the differentiation protocol followed at the end of the culture period (day 6) by analysis of cytokine production levels following TCR stimulation with anti-CD3/anti-CD28. As shown in Figure 1A, IL-4 and IL-13 production from cells cultured under the Th2 polarization protocol were reduced in a dose-dependent manner, in parallel to the decrease in their frequency (Fig. E1). In contrast, IFN-γ production from Th1 cells and IL-17 production from Th17 cells were not altered nor was the frequency of IFN-γ- or IL-17-producing cells. (Fig. E1). However, when cells were treated with R256 from days 6-8 of culture, after completion of Th cell differentiation, cytokine production levels from all of the Th subsets following TCR stimulation were not altered (Fig. 1B).
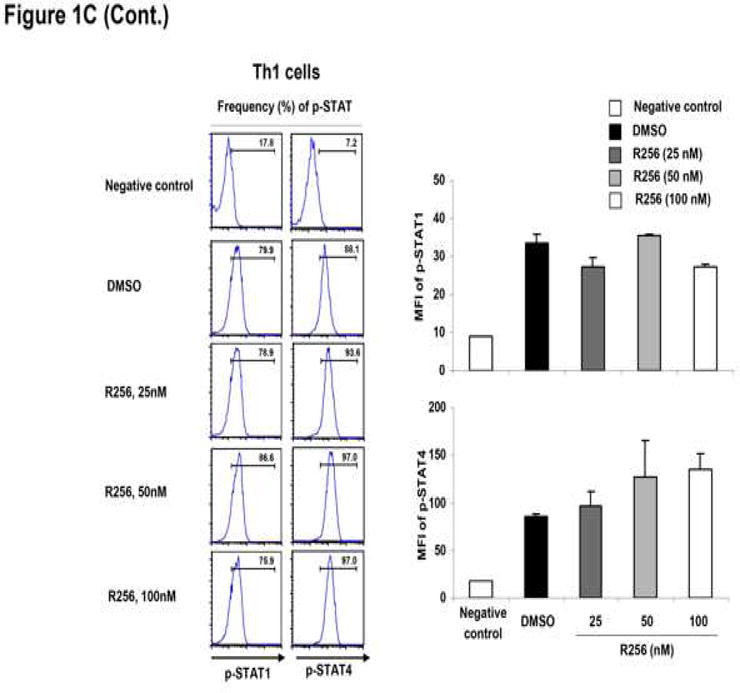
Figure 1.
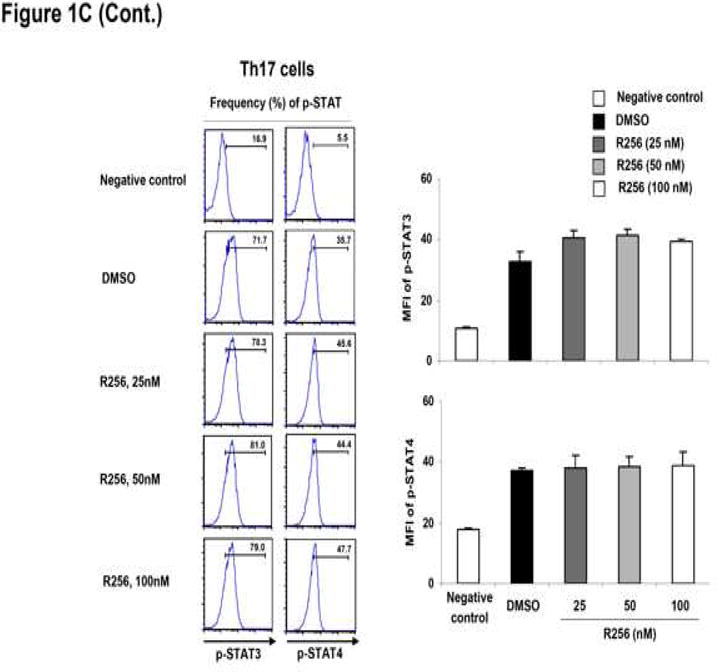
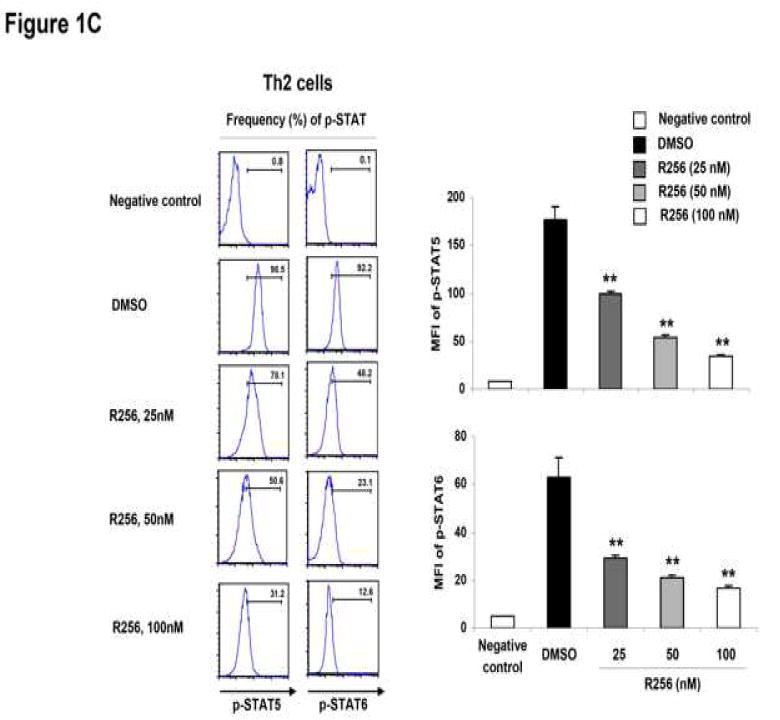
The effects of R256 on the in vitro differentiated Th subsets. (A) Cytokine levels in supernatants from Th cells. Cells were treated with R256 from day 0 to 6 and restimulated with plate-coated anti-CD3/anti-CD28 mAbs on day 6. (B) Cytokine levels in supernatants from differentiated Th cells. Cells were treated with R256 from days 6-8 and restimulated with anti-CD3/anti-CD28 mAbs on day 8. (C) On day 6 of cultures which included R256 from day 0 of Th subset differentiation, phosphorylated-STAT (p-STAT) 3, 4, 5, and 6 in the cells were stained using Alexa647-conjugated specific antibodies for flow cytometric analysis. Splenocyte CD4 cells from normal OT-2 mice were used as negative controls for p-STAT staining. The data for each group were expressed as means±SEM. The results are representative of 3 independent experiments carried out in duplicate. **p<0.01 compared to the DMSO-treated groups.
As Th2 differentiation requires IL-4-induced STAT6 and to some extent IL-2-induced STAT5 activation (37, 38), phosphorylation levels of STAT5 and STAT6 were determined by flow cytometry. As shown in Figure 1C, the JAK1/3 inhibitor blocked phosphorylation of both STAT6 and STAT5 in Th2 cells. By comparison, phosphorylation of STAT1 and STAT4 in Th1 cells and STAT3 and STAT4 in Th17 cells, which are essential in the polarization of Th1 cells in the presence of IFN-γ and IL-12 (39, 40) and Th17 cells in the presence of IL-6 and IL-23 (41, 42), respectively, were not altered by the drug. The phosphorylation levels of STAT5 in Th1 and Th17 cells were not altered by the drug (data not shown).
These data identify two important aspects related to JAK1/3 inhibition by R256. The first is that the pathway is essential for the differentiation of Th2 cells, but once Th2 cell differentiation has been completed, the inhibitor was no longer effective in blocking or inhibiting Th2 cytokine production. The second outcome of inhibiting JAK1/3 signaling by R256 was that the effects were restricted to Th2 differentiation without impacting Th1 or Th17 differentiation.
JAK1/3 inhibition during the sensitization phase prevents allergen-induced airway inflammation and AHR
In light of the in vitro findings, we determined the effects of R256 in vivo, in an animal model of allergen-induced airway inflammation and AHR during two distinct phases of the response. The initial, sensitization phase is thought to be IL-4-dependent and the latter or challenge phase was IL-13-dependent (43). In the first protocol, animals were treated with R256 only during the sensitization phase, followed two weeks later by allergen challenge (Fig. 2A). As shown in Figures 2B and 2C, R256 administered only during sensitization prevented the development of AHR and BAL eosinophilia. Histological analysis revealed marked reductions in airway inflammation and goblet cell metaplasia (Figs. 2D and 2E). In addition, Th2 cytokine levels (IL-4, IL-5, and IL-13) in BAL fluid following allergen challenge were also reduced following R256 administration during sensitization (Fig. 2F), paralleling the in vitro results when the inhibitor was added during Th2 induction (Fig. 1A). Under these in vivo conditions, no significant differences were observed in the levels of IFN-γ, IL-17, and IL-10 in the vehicle- and R256-treated groups.
JAK1/3 inhibition during the primary challenge phase prevents allergen-induced airway inflammation and AHR
We next determined the effects of R256 when administered after allergen sensitization but solely during the first allergen challenge phase (Fig. 3A). As shown in Figure 3B, when the inhibitor was given during the allergen challenge phase, the development of AHR was prevented and BAL eosinophilia (Fig. 3C) and goblet cell metaplasia (Figs. 3D and 3E) were significantly reduced. Surprisingly, unlike administration during the sensitization phase (Fig. 2F), levels of the Th2 cytokines, IL-4, IL-5, and IL-13 were not reduced (Fig. 3F); the levels of IFN-γ, IL-17, and IL-10 were unaltered similar to the in vitro results when the inhibitor was added to differentiated T cells (Fig. 1B). This was also confirmed by flow cytometry analyses which demonstrated that the frequencies of IFN-γ or IL-17 expressing CD4+ T cells in lung tissue were not altered by R256 treatment (Fig. E2). The frequencies of Treg cells in lung tissues were not affected by R256 treatment (Fig. E3). Moreover, R256 did not affect Treg suppressive activity when assayed against the proliferative activity of CD4+CD25- T cells (Fig. E4). When the inhibitor was administered during challenge, eotaxin levels in BAL fluid were significantly reduced when compared to non-treated mice (Fig. 3G).
JAK1/3 inhibition during the secondary challenge phase prevents allergen-induced airway inflammation and AHR
To determine the effects of R256 in the airway where allergen-induced airway inflammation and AHR were previously established, mice received the inhibitor during secondary allergen challenge. As shown in Figure 4, the development of AHR (Fig. 4B), airway eosinophilia (Fig. 4C), and goblet cell metaplasia (Figs. 4D and 4E) were significantly reduced by R256 in a dose-dependent manner, similar to the results observed in the primary allergen challenge model. As well, administration of the inhibitor during secondary challenge did not alter cytokine levels in BAL fluid (data not shown), despite changes in the other lung allergic response outcomes (Figs. 4A-E).
Discussion
JAK/STAT pathways play a critical role in the functional differentiation of immune cells (6). As a result, this pathway has been targeted in the treatment of various immune disorders (8). To date, there have been few studies investigating the effects of JAK pathway inhibition in asthma, where Th2 responses predominate. In this study, we first demonstrated in cell based assays that R256 activated JAK1 and JAK3 with 100-fold or greater selectivity over JAK2. R256 was shown to have limited activity on other kinase signaling pathways as shown in Table 1. We determined the effects of a selective JAK1/3 inhibitor on the differentiation of Th cells in vitro and the development of allergen-induced AHR, airway inflammation, and cytokine/chemokine production in the airways. Experiments in vitro demonstrated that selective inhibition of JAK1/3 targeted Th2 differentiation without altering Th1 and Th17 differentiation. In Th2 cells, p-STAT6 and p-STAT5 expression was reduced in a dose-dependent manner, without affecting STAT phosphorylation necessary for Th1 (p-STAT1 and p-STAT4) or Th17 (p-STAT3 and p-STAT4) differentiation. When the inhibitor was added to cultures after differentiation of the cells was completed, no changes in cytokine production were detected. These results suggested that the outcome of selective inhibition of JAK1/3 was restricted to Th2 cells and did not affect Th1 or Th17 cells. As well, the inhibitory effects of R256 were limited to the initiation of Th2 polarization. When administered in vivo at two different time points, the outcomes, although similar, appeared dictated by different events. When R256 was administered during the allergen sensitization phase, AHR and other lung allergic responses, including increases in BAL Th2 levels were markedly reduced, indicating that selective blockade of Th2 differentiation during allergen sensitization was critical. However, when administered during the primary challenge phase after sensitization was complete, although development of AHR, airway eosinophilia, and goblet cell metaplasia were prevented, this was not associated with any reduction in Th2 cytokine levels. In a confirmatory manner, administration of the inhibitor during secondary challenge, after sensitization and primary challenge were completed, also prevented development of AHR, eosinophilia, and goblet cell metaplasia without any alterations in Th2 cytokine levels, supporting the notion that during the challenge phases, the inhibitory effects were downstream of Th2 cytokine receptor signaling pathways.
Specific JAK/STAT pathways have been shown to be essential for the differentiation of distinct Th subsets: Th2 differentiation requires activation of the IL-2/STAT5 and IL-4/STAT6 signaling pathways (37, 38), Th1 differentiation depends on IFN-γ/STAT1 and IL-12/STAT4 signaling (39, 40) while differentiation of Th17 cells primarily requires STAT3 and STAT4 activated by IL-6 or IL-23 (41, 42). Our results confirmed that the selective inhibition of the JAK1/3 pathway by R256 was critical in the differentiation of Th2 cells through downregulation of STAT5 and STAT6 phosphorylation. Further, this was extended to observations in in vivo experiments; R256 given during the allergen sensitization phase but prior to allergen challenge prevented the development of allergen-induced AHR, eosinophilic airway inflammation, and mucus metaplasia and was associated with reduced Th2 cytokine levels in the airways. In contrast, it appeared that activation of JAK1/3 was not required after completion of Th2 differentiation in vivo as R256 did not alter the function of differentiated Th2 cells in vitro. This dichotomy was confirmed in vivo, as R256 given during the primary allergen challenge but after sensitization did not alter Th2 cytokine levels in BAL fluid, suggesting that sensitization initiated Th2 differentiation, subsequently rendering the cells resistant to the effects of the inhibitor. Regardless of sustained Th2 cytokine levels when administered during the primary challenge phase, development of AHR, eosinophilia, and goblet cell metaplasia were reduced on allergen challenge. These same outcomes were observed in the secondary allergen challenge model where allergic airway inflammation and altered airway function were previously established, and provoked by a single allergen challenge, perhaps similar to clinical asthma. The likely explanation was that these results were the consequence of the inhibition of downstream signaling pathways initiated by the unimpaired production of the Th2 cytokines. In previous studies, a critical role for JAK1 was identified as STAT6 was activated by IL-4 or IL-13 through JAK1 in rat alveolar type II cells and airway epithelial cells were shown to require IL-4 signaling after allergen exposure (44, 45). Although a predominant role for JAK3 over JAK1 was suggested from siRNA knockdown experiments (46), intracellular enzymatic levels of JAK1 were shown to be higher than levels of JAK3 (47). This dissociation might reflect differences in individual JAK kinase binding capabilities to substrates or ligands versus enzymatic activities. Activation of the JAK1-STAT6 pathway following IL-13 stimulation was shown in both rat and human bronchial smooth muscle cells, and this pathway was associated with activation of RhoA, which induces smooth muscle contraction (48, 49). When the inhibitor was administered in the primary challenge phase, a significant reduction in BAL eotaxin levels was demonstrated, similar to the inhibition of eotaxin triggered by IL-13 in lung fibroblasts (20, 22). Thus, in the challenge phase, despite abundant Th2 cytokine levels in the airways, where IL-13 and to some degree IL-4 effects are prominent (43), lower levels of eotaxin may be at the core of reduced airway eosinophilia, while the reduced mucus production and AHR are the result of R256-mediated impairment of IL-13 signaling in goblet cells and airway smooth muscle. Inhibition of IL-13 using a receptor antagonist resulted in similar effects (50). Taken together, the data indicate that the timing of inhibitor administration has a profound impact on effector responses which contribute to similar outcomes.
The selectivity of R256 for JAK1/3 and Th2 differentiation in allergen-driven diseases may have advantages over pan-JAK inhibitors. Despite reported efficacy in reducing airway eosinophilia and AHR to some extent, both tofacitinib and pyridone 6, unlike R256, may alter Treg, Th1, and Th17 responses (19, 51), potentially limiting efficacy in some diseases or increasing the likelihood of adverse events under some conditions. Selective targeting of JAK1/3 may provide important therapeutic advances in the treatment of Th2-mediated allergic diseases such as asthma.
Supplementary Material
Figure E1. The effects of R256 on the frequency of Th subsets in vitro. (A) On day 6, the Th subsets were restimulated with plate-coated anti-CD3/anti-CD28 mAbs for 6 hrs. The cells were collected and analyzed by intracellular staining. R256 or DMSO as a control was added from days 0-6. The results are from 2 independent experiments (n=6).
Figure E2. The effects of R256 treatment during primary allergen challenge on the frequency of cytokine expressing cells in the lungs. Cells were collected from lung tissues of mice following vehicle or R256 (50 mg/kg) treatment during primary OVA challenge. The cells were stimulated with anti-CD3 and anti-CD28 for 6 hrs in the presence of BFA, and then analyzed by intracellular staining as described in Material and Methods. A representative experiment is shown as well as the results from 2 independent experiments with 3 mice per group, n=6.
Figure E3. The effects of R256 treatment during primary allergen challenge on the frequency and numbers of Tregs cells in the lungs. Cells were collected from lung tissues of mice following vehicle or R256 (50 mg/kg) treatment during primary OVA challenge, and were stained with APC-conjugated anti-CD4 and FITC-conjugated Foxp3 mAb followed by flow cytometry analyses. Shown are a representative analysis and the results from 2 independent experiments with 3 mice per each group, n=6.
Figure E4. The effects of R256 on the function of Treg cells. CD4+CD25+ Treg cells isolated from naive mice were treated with DMSO or R256 (50 nM) for 3 hrs. The drug pre-treated Treg cells were co-cultured with anti-CD3/anti-CD28 pre-stimulated and CFSE-labeled CD4+CD25- cells. After 72 hrs of co-culture, CFSE fluorescence intensities were monitored by flow cytometry. A representative experiment is illustrated.
Acknowledgments
Grant Support: This work was supported by NIH grants HL-36577 and AI-77609. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NHLBI or the NIH.
Disclosures: Funding was received from Rigel Pharmaceuticals in support of this study and EWG was a consultant to Rigel.
We thank Diana Nabighian for her assistance in preparation of the manuscript.
Abbreviations
- AHR
Airway hyperresponsiveness
- BFA
Brefeldin A
- CHEP
Cultured human erythroid progenitor cells
- EPO
Erythropoietin
- H&E
Hematoxyline and eosin
- JAK
Janus kinases
- MCh
Methacholine
- OVA
Ovalbumin
- PAS
Periodic acid Schiff
- STAT
Signal transducers and activator of transcription
- Th
T helper
- Treg
T regulatory cell
- WT
Wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Casanova JL, Holland SM, Notarangelo LD. Inborn errors of human JAKs and STATs. Immunity. 2012;36:515–28. doi: 10.1016/j.immuni.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 4.Shuai K, Liu B. Regulation of JAK-STAT signaling in the immune system. Nat Rev Immunol. 2003;3:900–11. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 5.Schindler CW. JAK-STAT signaling in human disease. J Clin Invest. 2002;109:1133–37. doi: 10.1172/JCI15644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–87. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O'Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223:132–42. doi: 10.1111/j.1600-065X.2008.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Shea JJ, Plenge R. JAK and STAT signaling molecules in immune regulation and immune-mediated disease. Immunity. 2012;36:542–50. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kontzias A, Kotlyar A, Laurence A, Changelian P, O'Shea JJ. Jakinibs: a new class of kinase inhibitors in cancer and autoimmune disease. Curr Opin Pharmacol. 2012;12:464–70. doi: 10.1016/j.coph.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heine A, Held SA, Daecke SN, Wallner S, Yajnanarayana SP, Kurts C, et al. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122:1192–202. doi: 10.1182/blood-2013-03-484642. [DOI] [PubMed] [Google Scholar]
- 11.Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. doi: 10.1056/NEJMoa1109071. [DOI] [PubMed] [Google Scholar]
- 12.van Vollenhoven RF, Fleischmann R, Cohen S, Lee EB, García Meijide JA, Wagner S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–19. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]
- 13.Ghoreschi K, Laurence A, O'Shea JJ. Selectivity and therapeutic inhibition of kinases: to be or not to be? Nat Immunol. 2009;10:356–60. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fox DA. Kinase inhibition-a new approach to the treatment of rheumatoid arthritis. N Engl J Med. 2012;367:565–7. doi: 10.1056/NEJMe1206315. [DOI] [PubMed] [Google Scholar]
- 15.Bosnjak B, Stelzmueller B, Erb KJ, Epstein MM. Treatment of allergic asthma: modulation of Th2 cells and their responses. Respir Res. 2011;12:114. doi: 10.1186/1465-9921-12-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szefler SJ, Zeiger RS, Haselkorn T, Mink DR, Kamath TV, Fish JE, et al. Economic burden of impairment in children with severe or difficult-to-treat asthma. Ann Allergy Asthma Immunol. 2011;107:110–9.e1. doi: 10.1016/j.anai.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Walsh GM. An update on emerging drugs for asthma. Expert Opin Emerg Drugs. 2012;17:37–42. doi: 10.1517/14728214.2012.657625. [DOI] [PubMed] [Google Scholar]
- 18.Kudlacz E, Conklyn M, Andresen C, Whitney-Pickett C, Changelian P. The JAK-3 inhibitor CP-690550 is a potent anti-inflammatory agent in a murine model of pulmonary eosinophilia. Eur J Pharmacol. 2008;582:154–61. doi: 10.1016/j.ejphar.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 19.Matsunaga Y, Inoue H, Fukuyama S, Yoshida H, Moriwaki A, Matsumoto T, et al. Effects of a Janus kinase inhibitor, pyridone 6, on airway responses in a murine model of asthma. Biochem Biophys Res Commun. 2011;404:261–7. doi: 10.1016/j.bbrc.2010.11.104. [DOI] [PubMed] [Google Scholar]
- 20.May RD, Monk PD, Cohen ES, Manuel D, Dempsey F, Davis NHE, et al. Preclinical development of CAT-354, an IL-13 antibody for the treatment of severe uncontrolled asthma. Brit J Pharmacol. 2012;166:177–93. doi: 10.1111/j.1476-5381.2011.01659.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakano T, Inoue H, Fukuyama S, Matsumoto K, Matsumura M, Tsuda T, et al. Niflumic acid suppresses interleukin-13-induced asthma phenotypes. Am J Respir Crit Care Med. 2006;173:1216–1221. doi: 10.1164/rccm.200410-1420OC. [DOI] [PubMed] [Google Scholar]
- 22.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–90. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 23.Wenzel SE, Trudeau JB, Barnes S, Zhou X, Cundall M, Westcott JY, et al. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J Immunol. 2002;169:4613–9. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- 24.Liao W, Lin JX, Leonard WJ. IL-2 family cytokines: new insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr Opin Immunol. 2011;23:598–604. doi: 10.1016/j.coi.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oda A, Sawada K, Druker BJ, Ozaki K, Takano H, Koizumi K, et al. Erythropoietin induces tyrosine phosphorylation of Jak2, STAT5A, and STAT5B in primary cultured human erythroid precursors. Blood. 1998;92:443–51. [PubMed] [Google Scholar]
- 26.Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood. 2005;105:4604–12. doi: 10.1182/blood-2004-10-4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang YJ, Holtzman MJ, Chen CC. Interferon-gamma-induced epithelial ICAM-1 expression and monocyte adhesion. Involvement of protein kinase C-dependent c-Src tyrosine kinase activation pathway. J Biol Chem. 2002;277:7118–26. doi: 10.1074/jbc.M109924200. [DOI] [PubMed] [Google Scholar]
- 28.Scicchitano MS, McFarland DC, Tierney LA, Narayanan PK, Schwartz LW. In vitro expansion of human cord blood CD36+ erythroid progenitors: temporal changes in gene and protein expression. Exp Hematol. 2003;31:760–9. doi: 10.1016/s0301-472x(03)00185-1. [DOI] [PubMed] [Google Scholar]
- 29.Rossi AB, Herlaar E, Braselmann S, Huynh S, Taylor V, Frances R, et al. Identification of the Syk kinase inhibitor R112 by a human mast cell screen. J Allergy Clin Immunol. 2006;118:749–55. doi: 10.1016/j.jaci.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 30.Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastic breast cancer. Cancer Res. 2010;70:1544–54. doi: 10.1158/0008-5472.CAN-09-2997. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi M, Ashino S, Shiohama Y, Wakita D, Kitamura H, Nishimura T. IFN-γ elevates airway hyper-responsiveness via up-regulation of neurokinin A/neurokinin-2 receptor signaling in a severe asthma model. Eur J Immunol. 2012;42:393–402. doi: 10.1002/eji.201141845. [DOI] [PubMed] [Google Scholar]
- 32.Ashino S, Wakita D, Zhang Y, Chamoto K, Kitamura H, Nishimura T. CpG-ODN inhibits airway inflammation at effector phase through down-regulation of antigen-specific Th2-cell migration into lung. Int Immunol. 2008;20:259–66. doi: 10.1093/intimm/dxm138. [DOI] [PubMed] [Google Scholar]
- 33.Ashino S, Wakita D, Shiohama Y, Iwakura Y, Chamoto K, Ohkuri T, et al. A T(h)17-polarized cell population that has infiltrated the lung requires cells that convert to IFN-{gamma} production in order to induce airway hyperresponsiveness. Int Immunol. 2010;22:503–13. doi: 10.1093/intimm/dxq034. [DOI] [PubMed] [Google Scholar]
- 34.Kanehiro A, Ikemura T, Makela MJ, Lahn M, Joetham A, et al. Inhibition of phosphodiesterase 4 attenuates airway hyperresponsiveness and airway inflammation in a model of secondary allergen challenge. Am J Respir Crit Care Med. 2001;163:173–84. doi: 10.1164/ajrccm.163.1.2001118. [DOI] [PubMed] [Google Scholar]
- 35.Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. 1997;186:449–54. doi: 10.1084/jem.186.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshiba A, Hamelmann E, Takeda K, Bradley KL, Loader JE, Larsen GL, et al. Passive transfer of immediate hypersensitivity and airway hyperresponsiveness by allergen-specific immunoglobulin (Ig) E and IgG1 in mice. J Clin Invest. 1996;97:1398–408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu J, Guo L, Watson CJ, Hu-Li J, Paul WE. Stat6 is necessary and sufficient for IL-4's role in Th2 differentiation and cell expansion. J Immunol. 2001;166:7276–81. doi: 10.4049/jimmunol.166.12.7276. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–48. doi: 10.1016/s1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 39.Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, et al. T-bet is rapidly induced by interferon-gamma in lymphoid and myeloid cells. Proc Natl Acad Sci USA. 2001;98:15137–15142. doi: 10.1073/pnas.261570598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 41.Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- 42.Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, et al. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol. 2007;178:4901–4907. doi: 10.4049/jimmunol.178.8.4901. [DOI] [PubMed] [Google Scholar]
- 43.Takeda K, Dakhama A, Gelfand EW. Allergic asthma: what have we learned from the mouse model? Allergology Int. 2005;54:263–71. [Google Scholar]
- 44.Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–31. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 45.Kuperman DA, Huang X, Nguyenvu L, Hölscher C, Brombacher F, Erle DJ. IL-4 receptor signaling in Clara cells is required for allergen-induced mucus production. J Immunol. 2005;175:3746–52. doi: 10.4049/jimmunol.175.6.3746. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Valades AG, Llamas M, Blanch S, Perales JC, Roman J, Gomez-Casajus, et al. Specific Jak3 downregulation in lymphocytes impairs γc cytokine signal transduction and alleviates antigen-driven inflammation in vivo. Mol Ther-Nuc Acids. 2012;1:e42. doi: 10.1038/mtna.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haan C, Rolvering C, Raulf F, Kapp M, Drückes P, Thoma G, et al. Jak1 has a dominant role over Jak3 in signal transduction through γc-containing cytokine receptors. Chem Biol. 2011;18:314–2. doi: 10.1016/j.chembiol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Chiba Y, Goto K, Matsusue K, Kimura S, Misawa M. Identification and characterization of rat RhoA gene promoter. J Pharmacol Sci. 2010;112:467–72. doi: 10.1254/jphs.09346sc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chiba Y, Goto K, Misawa M. Interleukin-13-induced activation of signal transducer and activator of transcription 6 is mediated by an activation of Janus kinase 1 in cultured human bronchial smooth muscle cells. Pharmacol Rep. 2012;64:454–8. doi: 10.1016/s1734-1140(12)70788-0. [DOI] [PubMed] [Google Scholar]
- 50.Taube C, Duez C, Chi ZH, Takeda K, Rha YH, Park JW, et al. The role of IL-13 in established allergic airway disease. J Immunol. 2002;169:6482–9. doi: 10.4049/jimmunol.169.11.6482. [DOI] [PubMed] [Google Scholar]
- 51.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–43. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure E1. The effects of R256 on the frequency of Th subsets in vitro. (A) On day 6, the Th subsets were restimulated with plate-coated anti-CD3/anti-CD28 mAbs for 6 hrs. The cells were collected and analyzed by intracellular staining. R256 or DMSO as a control was added from days 0-6. The results are from 2 independent experiments (n=6).
Figure E2. The effects of R256 treatment during primary allergen challenge on the frequency of cytokine expressing cells in the lungs. Cells were collected from lung tissues of mice following vehicle or R256 (50 mg/kg) treatment during primary OVA challenge. The cells were stimulated with anti-CD3 and anti-CD28 for 6 hrs in the presence of BFA, and then analyzed by intracellular staining as described in Material and Methods. A representative experiment is shown as well as the results from 2 independent experiments with 3 mice per group, n=6.
Figure E3. The effects of R256 treatment during primary allergen challenge on the frequency and numbers of Tregs cells in the lungs. Cells were collected from lung tissues of mice following vehicle or R256 (50 mg/kg) treatment during primary OVA challenge, and were stained with APC-conjugated anti-CD4 and FITC-conjugated Foxp3 mAb followed by flow cytometry analyses. Shown are a representative analysis and the results from 2 independent experiments with 3 mice per each group, n=6.
Figure E4. The effects of R256 on the function of Treg cells. CD4+CD25+ Treg cells isolated from naive mice were treated with DMSO or R256 (50 nM) for 3 hrs. The drug pre-treated Treg cells were co-cultured with anti-CD3/anti-CD28 pre-stimulated and CFSE-labeled CD4+CD25- cells. After 72 hrs of co-culture, CFSE fluorescence intensities were monitored by flow cytometry. A representative experiment is illustrated.