

Abstract
Patients with congenital adrenal hyperplasia (CAH) with tenascin-X deficiency (CAH-X syndrome) have both endocrine imbalances and characteristic Ehlers Danlos syndrome phenotypes. Unlike other subtypes, tenascin-X-related Ehlers Danlos syndrome is caused by an extracellular matrix protein deficiency rather than a defect in fibrillar collagen or a collagen-modifying enzyme, and the understanding of the disease mechanisms is limited. We hypothesized that transforming growth factor-β pathway dysregulation may, in part, be responsible for connective tissue phenotypes observed in CAH-X, due to this pathway’s known role in connective tissue disorders.
Fibroblasts and direct tissue from human skin biopsies from CAH-X probands and age- and sex-matched controls were screened for transforming growth factor-β biomarkers known to be dysregulated in other hereditary disorders of connective tissue. In CAH-X fibroblast lines and dermal tissue, pSmad1/5/8 was significantly upregulated compared to controls, suggesting involvement of the bone morphogenetic protein pathway. Additionally, CAH-X samples compared to controls exhibited significant increases in fibroblast-secreted TGF-β3, a cytokine important in secondary palatal development, and in plasma TGF-β2, a cytokine involved in cardiac function and development, as well as palatogenesis. Finally, MMP-13, a matrix metalloproteinase important in secondary palate formation and tissue remodeling, had significantly increased mRNA and protein expression in CAH-X fibroblasts and direct tissue.
Collectively, these results demonstrate that patients with CAH-X syndrome exhibit increased expression of several transforming growth factor-β biomarkers and provide a novel link between this signaling pathway and the connective tissue dysplasia phenotypes associated with tenascin-X deficiency.
Keywords: CAH, tenascin-X deficiency, TGF-β pathway, cleft palate, cardiac abnormalities
Introduction
Congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency is an autosomal recessive disorder of the adrenal cortex characterized by cortisol deficiency, with or without aldosterone deficiency, and androgen excess.[1] Endocrine manifestations range in severity depending upon the degree of 21-hydroxylase impairment caused by defects in the CYP21A2 gene.[1] CYP21A2 is flanked by the TNXB gene that encodes tenascin-X (TNX), an extracellular matrix (ECM) glycoprotein that is highly expressed in connective tissue and functions in matrix maturation during wound healing.[2] TNX was the first essential protein identified for normal collagen fibril deposition independent of collagen synthesis and fibrillogenesis. Defects in normal collagen fibril deposition in connective tissue can impair collagenous matrix integrity and lead to Ehlers Danlos syndrome (EDS), a hereditary disorder of connective tissue.[3]
We recently described that approximately 7% of patients with CAH have an associated connective tissue phenotype due to TNXB haploinsufficiency, representing a contiguous gene syndrome termed CAH-X.[4] It is estimated that approximately 20 000 people in the US are living with CAH. Therefore, up to 1 400 people may be affected by CAH-X in the US alone. Using a conservative prevalence of CAH of 1 in 20 000 worldwide, about 350 000 people are at risk for CAH-X.
Complete TNX deficiency was first reported in a patient with CAH and EDS.[5] While autosomal recessive complete TNX deficiency is a cause of classical EDS,[6] TNXB haploinsufficiency is associated with the hypermobility type of EDS.[7] Previous investigations have been confined to TNX’s interactions with collagen and have suggested that the EDS phenotype in TNX deficiency may be predominantly related to its interactions with fibrillar collagens, particularly type V;[6] however, this hypothesis does not explain additional features such as clefting, cardiac developmental and midline defects, and myopathy found in CAH-X. The effects of TNX deficiency lead to an impaired ECM and connective tissue, which in turn lead to connective tissue dysplasia phenotypes. Interestingly, dysregulation in the transforming growth factor-beta (TGF-β) pathway has been found in other connective tissue dysplasias with similar outcomes,[4] such as Marfan syndrome (MFS), Loeys Dietz syndrome (LDS), Shprintzen-Goldberg syndrome (SGS), and a disorder in the LDS spectrum involving loss-of-function mutations in TGFB2 (Table 1).[8–11] In addition to EDS phenotypes such as joint hypermobility, piezogenic papules, soft tissue rheumatism, spondylosis, and functional bowel disorders, CAH-X patients exhibit structural cardiac valvular abnormalities such as quadricuspid aortic valve and congenital ventricular diverticulum. The presence of a bifid uvula, a forme fruste of cleft palate, has also been found in CAH-X.[4] Due to the phenotypic overlap of CAH-X with connective tissue dysplasias known to have aberrant TGF-β signaling, we hypothesized that abnormal expression of TGF-β pathway biomarkers may also be found in CAH-X (Table 1).
Table 1.
Involvement of the TGF-β pathway in disorders of connective tissue.
| Genetic Disorder | Causative Mutation | Dysregulated TGF-β Biomarker | Phenotype | Reference |
|---|---|---|---|---|
| Marfan Syndrome (MFS) | FBN1 | pSmad2 pERK1/2 Smad4 TGF-β1 BMP-2 p38 MAPK |
aortic aneurysm, skeletal features, lens dislocation, high palate, skin abnormalities | [32–35] |
| Loey’s Dietz Syndrome (LDS) |
TGFBRI TGFBRII TGFB2 SMAD3 |
TGF-βRI TGF-βRII TGF-β2 Smad3 |
cardiovascular, musculoskeletal, skin abnormalities, bifid uvula, hypertelorism, craniosynostosis | [9, 10, 36] |
| Shprintzen-Goldberg Syndrome (SGS) | SKI | SKI (Smad2/3 repressor) | MFS/LDS features, mental retardation, severe skeletal hypotonia | [11] |
| Case report: overlapping MFS/LDS features | TGFB3 | TGF-β3 | bifid uvula, distal contractures, myopathy, joint laxity | [23] |
| CAH-X Syndrome |
CYP21A2 TNXB |
pSmad1/5/8 TGF-β3 MMP-13 TGF-β2 |
bifid uvula, cardiac valvular abnormalities, joint hypermobility, osteoarthritis | [4] |
The objective of the current study, therefore, was to investigate the role of the TGF-β pathway in TNX deficiency within our CAH-X cohort. Though a TNXB knockout mouse model was shown to recapitulate the EDS phenotype,[3] a comparable TNXB knockout mouse with a CAH background is not currently available, thereby limiting mechanistic studies to available human tissue and cell lines. We therefore utilized patient skin tissue, fibroblasts, and EDTA-plasma to screen for TGF-β signaling biomarkers commonly associated with phenotypes found in other connective tissue disorders to identify a novel role for this signaling pathway in CAH-X.
Material and Methods
Ethics statement
Patients were enrolled in an ongoing prospective natural history study at the National Institutes of Health Clinical Center in Bethesda, MD (Clinical Trials # NCT00250159) and approval was obtained from the Eunice Kennedy Shriver National Institute of Child Health & Human Development Institutional Review Board. Written informed consent and assent were obtained for all participants. All clinical and molecular details of the CAH-X cohort have been recently described.[4]
Cell culture
Primary skin fibroblasts were initiated from explants of 4 mm-punch biopsies from 12 CAH-X probands (6 M/6 F) with TNXB haploinsufficiency and 19 age- and sex-matched CAH controls (age range ~5–25 yr) with a normal TNXB genotype. De-identified dermal fibroblast samples from apparently healthy age- and sex-matched controls were obtained from the Coriell repository (www.coriell.org). Fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA) with high glucose and supplemented with 10% fetal bovine serum (FBS, Invitrogen) in the presence of penicillin and streptomycin at 37°C in 5% CO2. Untreated CAH-X and control cells were grown to confluence before lysis. Treated CAH-X and controls cells were grown to ~80% confluence then serum starved for 18 hours. Cells were then treated for 1 hour with either 10 ng/mL TGF-β1, TGF-β2, or TGF-β3 or 50 ng/mL BMP-4 (R&D Systems, Minneapolis, MN, USA) as indicated before lysis. Cells were used between passages 2 and 8.
Western blot analysis
Protein expression was analyzed by SDS-PAGE electrophoresis and Western blot. Fibroblasts were lysed with RIPA buffer (Pierce, Rockford, IL, USA) containing protease and phosphatase inhibitors (Cocktail Sets I, II, and III, Calbiochem, Gibbstown, NJ, USA). Thirty μg of total cell protein (as determined by a BCA protein assay of whole cell protein extracts and using BSA as a standard, Pierce) was loaded onto a 4–20% Novex Tris-Glycine precast gel (Invitrogen). Proteins were then electrotransferred onto a PVDF membrane using Invitrogen’s iBlot dry blotting system and immunoblotting was performed with the appropriate human antibodies. Specifically, rabbit polyclonal anti-phospho-Smad1/5/8 (1:500, Cell Signaling Technology, Danvers, MA, USA), anti-Smad1 (1:500, Cell Signaling Technology), anti-phospho-Smad2 (1:500, Millipore, Billerica, MA, USA), anti-phospho-Erk1/2 (1:1000, Cell Signaling Technology), anti-p38 MAPK (1:500, Cell Signaling Technology), anti-MMP-13 (1:200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-β-tubulin (1:2,000, Cell Signaling Technology) or rabbit monoclonal anti-Smad2 (1:500, Cell Signaling Technology), anti-Erk1/2 (1:1000, Cell Signaling Technology), anti-phospho-p38 MAPK (1:500, Cell Signaling Technology), anti-β-actin (1:1000, Cell Signaling Technology) were used overnight at 4°C, followed by 1 hour incubation with a secondary donkey anti-rabbit IgG ECL-HRP linked antibody (1:5000, GE Healthcare, Piscataway, NJ, USA). Immunoreactive products were visualized by chemiluminescence using the ECL Plus kit (GE Healthcare). Quantification of immunoblots was performed using ImageJ software (NIH, Bethesda, MD, USA) and was done within the linear range of each antibody. All data were normalized to the loading control; however, the same results were seen when normalized to total protein. Results are from three independent experiments performed in triplicate.
Quantitative real-time PCR
An RNeasy Plus Mini kit (QIAGEN, Valencia, CA, USA) was used to extract RNA according to the manufacturer’s instructions. Synthesis of cDNA was done using 1 μg RNA and the qScript cDNA SuperMix (Quanta Biosciences, Gaithersburg, MD, USA). Gene expression was quantified using the SYBR green (Quanta Biosciences) method of real-time PCR with QuantumRNA Universal 18S primers (Ambion, Grand Island, NY, USA) or 200 nM of each gene specific primer in each reaction. The primers used for the human MMP13 gene were designed to cross intron-exon junctions and are as follows: forward (5′-ACTGAGAGGCTCCGAGAAATG-3′) and reverse (5′-GAACCCCGCATCTTGGCTT-3′) (Integrated DNA Technologies, Coralville, IA, USA). Quantitative PCR was performed on an ABI Prism 7300 sequence detection system (Applied Biosystems, Carlsbad, CA, USA) using standard conditions. Target gene mRNA levels were compared to standard curves and normalized to 18S mRNA. All PCR reactions were performed in triplicate from three independent experiments
Immunohistochemistry
Cryosections of human skin biopsies (10 μm) were immunostained for pSmad1/5/8 using a rabbit polyclonal anti-pSmad1/5/8 (1:50, Santa Cruz Biotechnology) and MMP-13 antibody using the antiserum (1:50) described above. As a negative control, the primary antibody was omitted and replaced with rabbit IgG (Santa Cruz Biotechnology) in lieu of a blocking peptide, which was not commercially available for these specific antibodies. Sections were treated with a biotinylated anti-rabbit IgG secondary antibody followed by an avidin peroxidase complex and 3,3′-diaminobenzidine (DAB) until a brown-colored immunostain developed (VECTASTAIN ABC kit, Vector Laboratories, Burlingame, CA, USA). Counterstaining was done with Gill’s hematoxylin #2 (Polysciences, Inc., Warrington, PA, USA) according to the manufacturer’s instructions. Slides were viewed at 10x magnification under an Olympus Ix51 light microscope.
Enzyme-linked immunosorbent assay
Total TGF-β1, -β2, -β3, and BMP-4 concentrations in secreted medium from human skin fibroblasts and total TGF-β1 and -β2 in platelet-poor EDTA-plasma were measured by enzyme-linked immunosorbent assay with the human TGF-β1, TGF-β2, and BMP-4 Quantikine ELISA kits and human TGF-β3 DuoSet ELISA kit (R&D Systems). TGF-β3 in plasma was measured using a ruthenium-based commercially available electrochemiluminescence platform according to the manufacturer’s instructions (Meso Scale Discovery, Gaithersburg, MD, USA). All samples were acid-activated prior to assaying according to the manufacturer’s instructions. Secretion data were normalized to protein concentration. All samples were run in duplicate.
Statistical analysis
Comparisons were made with the unpaired Student’s t-test to evaluate significance between groups. Sex was not a factor in statistical analyses. All p-values are two-tailed and considered significant when ≤ 0.05. Data are represented as mean ± standard error of the mean (SEM).
Results
The BMP pathway is upregulated in CAH-X syndrome
TGF-β biomarkers in human skin fibroblast whole cell lysate, both canonical (pSmad2 and pSmad1/5/8) and non-canonical (pERK/1/2 and p-p38 MAPK), were analyzed by Western blot in CAH-X patients versus CAH controls that did not harbor a TNXB mutation/deletion. Fibroblasts were treated with TGF-β1, TGF-β2, TGF-β3, and BMP-4 and probed for pSmad2, pSmad1/5/8, pERK1/2, and p-p38 MAPK. TGF-β1 and BMP-4 were used to induce pSmad2 and pSmad1/5/8, respectively, while endogenous levels of pERK1/2 and p-p38 MAPK were probed. While pSmad2, pERK1/2, and p-p38 MAPK levels did not change between groups, pSmad1/5/8 was significantly upregulated in CAH-X patients versus controls (p = 0.005), suggesting aberrant TGF-β signaling through the BMP pathway (Figure 1A, B). Only BMP-4-stimulated expression of pSmad1/5/8 was enhanced in CAH-X. No changes were seen with the other treatments (Figure 1C), suggesting a direct effect in the BMP pathway. Though other TGF-β pathway markers found in connective tissue dysplasias were screened by Western blot (Smad3, Smad4, TGF-βRI, TGF-βRII), only pSmad1/5/8 was different from controls (data not shown).
Figure 1. Western blot analysis of TGF-β signaling in human skin fibroblasts.
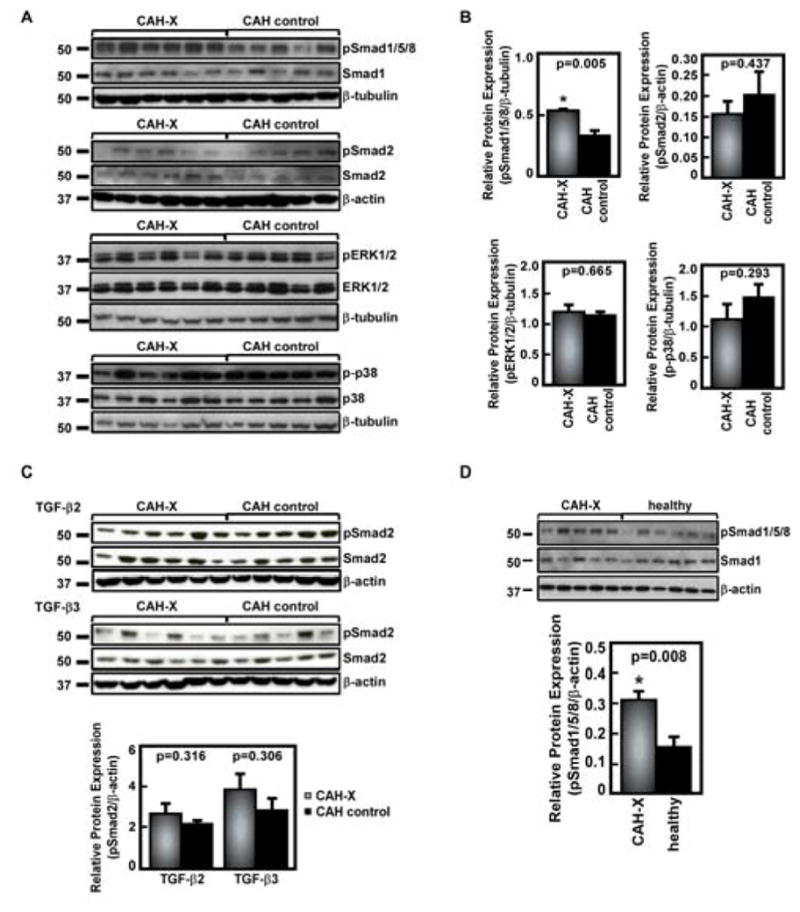
A. Representative Western blot analysis of TGF-β1-stimulated pSmad2, BMP-4-stimulated pSmad1/5/8, and endogenous pERK1/2 and p-p38 markers in CAH-X patients compared to CAH controls lacking a TNXB mutation/deletion. Significantly increased pSmad1/5/8 expression is seen in CAH-X patients. B. Western blot quantification graphs are shown from three independent experiments done in triplicate. All data were normalized to the loading control; however, the same results were seen when normalized to total protein (p ≤ 0.05 considered significant). C. Western blot analysis of TGF-β2 and TGF-β3 stimulation in fibroblasts. TGF-β2 and TGF-β3 did not induce changes in pSmad2 expression in fibroblasts. D. Western blot analysis of pSmad1/5/8 shows a similar pattern of increased pSmad1/5/8 expression in CAH-X patients compared to healthy controls. Western blot data were normalized to the loading control; however, the same results were seen when normalized to total protein (p ≤ 0.05 considered significant). The quantification graph is from three independent experiments done in triplicate.
The same markers were probed using CAH-X and age- and sex-matched apparently healthy controls in order to confirm the validity of using CAH patients without TNXB mutations/deletions as controls in this cohort. A similar pattern of elevated pSmad1/5/8 expression was seen when comparing to healthy controls (p = 0.008, Figure 1D); therefore, CAH patients without TNXB mutations/deletions were considered a valid control group in all further studies.
Immunoperoxidase staining of frozen human skin tissue sections with a pSmad1/5/8 antibody showed a marked increase in CAH-X patients versus CAH controls (Figure 2), validating the Western blot fibroblast results at the tissue level. Negative control slides where primary antibody was omitted and replaced by rabbit IgG showed no staining, confirming the pSmad1/5/8 antibody specificity.
Figure 2. Immunohistochemical analysis of pSmad1/5/8.
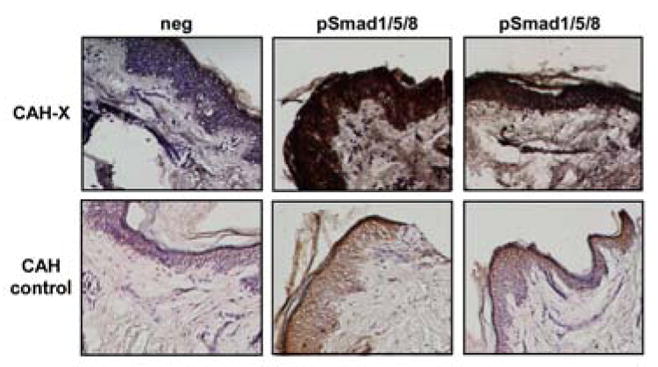
Immunoperoxidase staining using a human pSmad1/5/8 antibody shows markedly increased staining in representative skin sections from two CAH-X patients relative to CAH controls without a TNXB mutation/deletion. Neg refers to sections where rabbit IgG was used in place of the primary antibody, confirming antibody-specific staining. Hematoxylin was used to counterstain and sections were viewed at 10X magnification.
TGF-β3 secretion from CAH-X fibroblasts is increased
In addition to the intracellular findings, secretion into the medium from fibroblasts from CAH-X patients, CAH controls, and healthy controls revealed significantly elevated TGF-β3 in CAH-X patient samples by ELISA (p < 0.0001, Figure 3A). This result lends support to the Western blot findings, since it is known that excess TGF-β3 leads to increased pSmad1 expression.[12] However, secreted TGF-β1, TGF-β2, and BMP-4 were not different between groups (p > 0.05, data not shown). The possibility that the secreted TGF-β3 could potentiate an auto-feedback loop on the CAH-X fibroblasts was tested; however, no effect was seen in the fibroblasts (data not shown).
Figure 3. Analysis of TGF-β cytokines secreted from fibroblasts and in plasma.
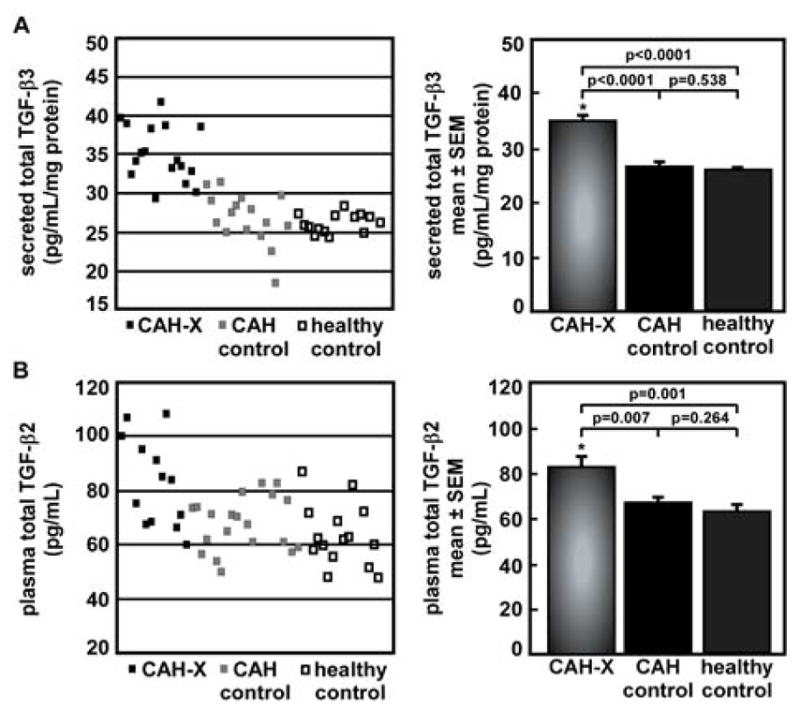
A. Secreted total TGF-β3 from fibroblasts from CAH-X patients compared to CAH controls without a TNXB mutation/deletion or healthy controls was determined by ELISA. CAH-X patient fibroblasts secrete significantly more TGF-β3 into the medium than controls. Secretion data were normalized to protein concentration (p ≤ 0.05 considered significant). Samples were run in duplicate. B. Levels of total TGF-β2 in platelet-poor EDTA-plasma from CAH-X patients compared to CAH controls without a TNXB mutation/deletion or healthy controls were determined by ELISA. CAH-X patients have significantly more TGF-β2 in circulation in plasma than controls (p ≤ 0.05 considered significant). Samples were run in duplicate.
Circulating TGF-β2 is increased in CAH-X plasma
Levels of TGF-β1, -β2, and -β3 in platelet-poor EDTA-plasma from CAH-X patients, CAH controls, and healthy controls were assayed, which revealed significantly elevated TGF-β2 in CAH-X patient samples by ELISA (p = 0.007 and 0.001, respectively, Figure 3B). Circulating TGF-β1 and TGF-β3 were not different between groups (p > 0.05, data not shown).
Matrix metalloproteinase-13 (MMP-13) is upregulated in CAH-X
Since MMP-13 is a downstream target of TGF-β3 during mammalian palatogenesis,[13] MMP-13 mRNA and protein expression was analyzed in fibroblasts following induction with TGF-β3 by real-time PCR and Western blot. Quantitative real-time PCR revealed that MMP13 was significantly upregulated in CAH-X patients versus CAH controls (p = 0.024, Figure 4A). Consistently, Western blot analysis showed significantly increased MMP-13 protein expression in CAH-X patients versus CAH controls (p = 0.006, Figure 4B). In order to determine the specificity of this response, MMP-13 expression was also tested in cells treated with TGF-β1, TGF-β2, and BMP-4 (Figure 4C). As expected, only TGF-β3 stimulation showed enhanced MMP-13 expression.
Figure 4. MMP-13 expression in CAH-X patients.

A. Relative MMP-13 mRNA expression in CAH-X patient fibroblasts compared to CAH controls without a TNXB mutation/deletion was tested by real-time PCR. CAH-X patients have increased expression of MMP13 relative to controls. MMP13 gene expression was normalized to 18S expression levels. Reactions from three independent experiments were run in triplicate. B. Representative Western blot analysis of TGF-β3-induced MMP-13 protein expression in skin fibroblasts shows increased MMP-13 in CAH-X patients compared to CAH controls without a TNXB mutation/deletion. Blot quantification data were normalized to β-actin. The quantification graph is from three independent experiments done in triplicate. C. Stimulation with TGF-β1 TGF-β2, and BMP-4 did not induce changes in MMP-13 expression in fibroblasts D. Immunoperoxidase staining using a human MMP-13 antibody shows markedly increased staining in representative skin sections from two CAH-X patients relative to CAH controls without a TNXB mutation/deletion. Neg refers to sections where rabbit IgG was used in place of the primary antibody, confirming antibody-specific staining. Hematoxylin was used to counterstain and sections were viewed at 10X magnification.
Immunoperoxidase staining of frozen human skin tissue sections with an MMP-13 antibody showed a marked increase in CAH-X patients versus CAH controls (Figure 4D), supporting the in vitro fibroblast results at the tissue level. Negative control slides where primary antibody was omitted and replaced by rabbit IgG showed no staining, confirming the MMP-13 antibody specificity.
Discussion
This study provides the first evidence of aberrant TGF-β expression in patients with CAH-X exhibiting features of a connective tissue dysplasia due to TNX deficiency. Although TNX deficiency was first associated with EDS over a decade ago,[5] little is known about how impairment of this ECM protein results in an EDS phenotype beyond its regulation of collagen deposition and crosslinking.[3, 14] Patients with CAH-X present with characteristic features of EDS, such as joint hypermobility, pain, and dislocations; soft tissue rheumatism, and other manifestations of tissue fragility. Midline defects, such as cardiac structural (quadricuspid aortic valve and congenital ventricular diverticulum) and palatal (bifid uvula) abnormalities, also have been described in a subset of affected CAH-X patients.[4] Our data suggest that alterations in TGF-β biomarkers play a role in the connective tissue dysplasia characteristics of CAH-X.
The TGF-β signaling pathway has been implicated in a number of connective tissue disorders[8–11] and is known to regulate collagen (Table 1).[15] Expression variability is often found in connective tissue disorders;[8–11] therefore, not all patients with the same underlying genetic defect necessarily display every phenotype associated with the disorder. However, it is apparent that the disruption of TGF-β signaling has a significant role in connective tissue dysplasias. The myriad of phenotypes associated with the fibrillin-TGF-β/BMP-Smad axes strongly support these pathways’ importance in connective tissue homeostasis.
Three TGF-β cytokines (TGF-β1, TGF-β2, and TGF-β3) are known to regulate diverse developmental processes and maintain tissue homeostasis. TGF-β1 is a potent stimulator of chemotaxis and is important in inflammation.[16] TGF-β2 has a vital role in embryonic development and disruption of the TGFB2 gene leads to a wide range of developmental defects, including cardiac and craniofacial deformities.[17] TGF-β3 is primarily responsible for proper secondary palatogenesis and palatal confluence.[18] In mouse models of cleft palate, it was shown that increased TGF-β3 in tissue and palatal fibroblasts led to increased signaling of pSmad1 and MMP-25 and MMP-13 (collagenase-3), which are also important in palate formation.[12, 13]
Upon screening known TGF-β pathway biomarkers found in connective tissue dysplasias, pSmad1/5/8 was found to be elevated in dermal fibroblasts and direct skin tissue, indicating abnormal BMP pathway function in CAH-X. BMP2 and BMP4 are expressed specifically within the epithelia and mesenchyme of the palatal shelves.[19] Overactive BMP signaling contributes to cleft palate formation by dysregulating cell proliferation and causing changes in gene expression.[20] In support of the BMP pathway findings, we found that CAH-X dermal fibroblasts secreted significantly more TGF-β3, which is known to induce pSmad1 expression.[12] TGF-β3 is essential for palatal confluence and mutations in TGFB3 are linked to cleft palate in mice.[18, 21] Rare variants in TGFB3 have been linked to nonsyndromic cleft palate in humans[22] and a mutation in TGFB3 has been shown to be disease causing in a human patient presenting with myopathy, joint laxity, distal contractures, bifid uvula, and other features overlapping with TGF-β spectrum disorders.[23] While these data may partially explain the palatal phenotype in CAH-X, a separate cohort of adult patients with TNXB deficiency was found to have myopathy and distal contractures, pointing to an overlap of the TGFB3 mutation phenotype with that of TNXB mutations.[24] In the current study, MMP-13, a downstream marker of TGF-β3 involved in embryogenesis and palatal fusion, was overexpressed in CAH-X dermal fibroblasts and direct skin tissue. Interestingly, it has been shown in a zebrafish model that exposure to glucocorticoids causes developmental abnormalities, including craniofacial, and leads to increased MMP-13 mRNA expression and activity.[25] Our patients were deficient in glucocorticoids during embryogenesis due to the enzymatic block in cortisol biosynthesis of CAH; the role of glucocorticoids in defining the phenotype is unknown.
CAH-X patient plasma had significantly higher amounts of circulating TGF-β2 than controls. Patients with CAH-X may present with cardiac structural valvular aberrations such as quadricuspid aortic valve and developmental defects such as congenital ventricular diverticulum. TGF-β2 appears to participate in mitral valve remodeling during development,[26, 27] and TGF-β2 mouse knockout studies suggested a specific role in cardiac cushion EMT.[28] Furthermore, recent studies showed that mutations that inactivate TGF-β2 led to aortic aneurysm syndrome, despite excess TGF-β2 being present in tissue.[29] Given that we have only cross-sectional data on circulating TGF-β2 from post-natal time points, the role of the elevated TGF-β2 in cardiac developmental defects in CAH-X remains speculative, albeit intriguing, and invites further exploration.
Screening known TGF-β biomarkers in connective tissue dysplasias in CAH-X revealed two potential disease-causing mechanistic axes: BMP/TGF-β3 and TGF-β2. Possibly the impact of TNX deficiency on TGF-β signaling stems from the role TNX plays in regulating collagen deposition and organization and TGF-β’s role in regulating collagen production. Therefore, the CAH-X phenotypes discussed appear to be related to TNX deficiency rather than the hormonal imbalances characteristic of CAH (Figure 5). Our cohort’s underlying CAH was consistent between groups, thus controlling for possible effects of the co-existing endocrine condition. Further studies are needed to describe this relationship.
Figure 5. Relationship Between TNX Deficiency and TGF-β Signaling in CAH-X.

CAH-X patients have two distinct genetic defects due to the proximity of CYP21A2, the gene encoding the 21-hydroxylase enzyme, and TNXB, the gene encoding TNX, an extracellular matrix protein. The 21-hydroxylase enzyme is essential in cortisol biosynthesis; 21-hydroxylase deficiency results in cortisol deficiency and androgen excess. TNX is associated with collagen reorganization; deficiency leads to a connective tissue dysplasia. A new link has been proposed in the current study between TNX deficiency and TGF-β signaling in CAH-X. Two possible mechanisms may be responsible for some CAH-X phenotypes, including BMP pathway dysregulation and abnormal TGF-β2 and TGF-β3 signaling, possibly through a connection between TNX’s collagen reorganization properties and TGF-β’s collagen production properties. The influence of the hormonal milieu on the connective tissue phenotype in CAH-X is unknown.
One weakness of our study is the lack of a haploinsufficient mouse model and animal data. Though a TNXB knockout mouse model that recapitulates an EDS phenotype is available, it lacks the CAH genetic background. Additionally, attempts have been made in other laboratories[30] to create a CAH mouse model with 21-hydroxylase deficiency through a homozygous deletion of CYP21A2. However, newborn homozygous mice are deficient in 21-hydroxylase activity and homozygosity results in death at an early postnatal stage. It is conceivable that protein expression patterns in the palatal, musculoskeletal, and cardiac defects described are different at embryonic and postnatal stages. Since palatal and vascular tissues were not accessible from our cohort, dermal fibroblasts were used as in studies of key pathologic features of other genetic disorders.[11, 31] Mouse models and human embryological studies have shown that TGF-β biomarkers are dysregulated and disease-causing in other related connective tissue diseases.[8–11] Therefore, it is likely that the TGF-β biomarkers found in CAH-X play a role in the connective tissue phenotypes.
Our study provides the first link between TGF-β pathway dysregulation and TNX deficiency in an endocrine disorder with connective tissue dysplasia features. The data suggest that abnormal BMP and TGF-β3 signaling may be involved in the palate abnormalities and myopathy, whereas TGF-β2 may be responsible for the cardiac phenotype. Given that other TGF-β pathway genes are associated with cardiovascular disease, including aneurysms, further mechanistic investigations of cardiac/vascular phenotypes and longitudinal cardiac and musculoskeletal follow-up in CAH-X patients is warranted. CAH-X is likely under recognized and underdiagnosed worldwide. Further discovery and understanding of the molecular mechanisms and the role of TGF-β signaling in the pathogenesis of CAH-X will provide novel insight into the role of tenascin-X deficiency as a cause of Ehlers Danlos syndrome.
Acknowledgments
We thank Dr. Justyna Fert-Bober and Dr. Jennifer van Eyk (Johns Hopkins Proteomics Center, Baltimore, MD) for technical support with the plasma TGF-β3 assay. R. Morissette and N. B. McDonnell declare no conflict of interest. D.P. Merke received research funds from Diurnal Limited, Ltd. This work was supported by the Intramural Research Programs of the National Institutes of Health, National Institute on Aging, the NIH Clinical Center, and the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Merke DP, Bornstein SR. Congenital adrenal hyperplasia. Lancet. 2005;365:2125–2136. doi: 10.1016/S0140-6736(05)66736-0. [DOI] [PubMed] [Google Scholar]
- 2.Egging D, van Vlijmen-Willems I, van Tongeren T, Schalkwijk J, Peeters A. Wound healing in tenascin-X deficient mice suggests that tenascin-X is involved in matrix maturation rather than matrix deposition. Connect Tissue Res. 2007;48:93–98. doi: 10.1080/03008200601166160. [DOI] [PubMed] [Google Scholar]
- 3.Mao JR, Taylor G, Dean WB, Wagner DR, Afzal V, Lotz JC, Rubin EM, Bristow J. Tenascin-X deficiency mimics Ehlers-Danlos syndrome in mice through alteration of collagen deposition. Nat Genet. 2002;30:421–425. doi: 10.1038/ng850. [DOI] [PubMed] [Google Scholar]
- 4.Merke DP, Chen W, Morissette R, Xu Z, Van Ryzin C, Sachdev V, Hannoush H, Shanbhag SM, Acevedo AT, Nishitani M, Arai AE, McDonnell NB. Tenascin-X Haploinsufficiency Associated with Ehlers-Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. 2013;98:E379–387. doi: 10.1210/jc.2012-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burch GH, Gong Y, Liu W, Dettman RW, Curry CJ, Smith L, Miller WL, Bristow J. Tenascin-X deficiency is associated with Ehlers-Danlos syndrome. Nat Genet. 1997;17:104–108. doi: 10.1038/ng0997-104. [DOI] [PubMed] [Google Scholar]
- 6.Schalkwijk J, Zweers MC, Steijlen PM, Dean WB, Taylor G, van Vlijmen IM, van Haren B, Miller WL, Bristow J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345:1167–1175. doi: 10.1056/NEJMoa002939. [DOI] [PubMed] [Google Scholar]
- 7.Zweers MC, Bristow J, Steijlen PM, Dean WB, Hamel BC, Otero M, Kucharekova M, Boezeman JB, Schalkwijk J. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73:214–217. doi: 10.1086/376564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 10.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doyle AJ, Doyle JJ, Bessling SL, Maragh S, Lindsay ME, Schepers D, Gillis E, Mortier G, Homfray T, Sauls K, Norris RA, Huso ND, Leahy D, Mohr DW, Caulfield MJ, Scott AF, Destree A, Hennekam RC, Arn PH, Curry CJ, Van Laer L, McCallion AS, Loeys BL, Dietz HC. Mutations in the TGF-beta repressor SKI cause Shprintzen-Goldberg syndrome with aortic aneurysm. Nat Genet. 2012;44:1249–1254. doi: 10.1038/ng.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown GD, Nazarali AJ. Matrix metalloproteinase-25 has a functional role in mouse secondary palate development and is a downstream target of TGF-beta3. BMC Dev Biol. 2010;10:93. doi: 10.1186/1471-213X-10-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zweers MC, Dean WB, van Kuppevelt TH, Bristow J, Schalkwijk J. Elastic fiber abnormalities in hypermobility type Ehlers-Danlos syndrome patients with tenascin-X mutations. Clin Genet. 2005;67:330–334. doi: 10.1111/j.1399-0004.2005.00401.x. [DOI] [PubMed] [Google Scholar]
- 15.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 16.Assoian RK, Komoriya A, Meyers CA, Miller DM, Sporn MB. Transforming growth factor-beta in human platelets. Identification of a major storage site, purification, and characterization. J Biol Chem. 1983;258:7155–7160. [PubMed] [Google Scholar]
- 17.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13(Spec No 1):R73–81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- 20.He F, Xiong W, Wang Y, Matsui M, Yu X, Chai Y, Klingensmith J, Chen Y. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev Biol. 2010;347:109–121. doi: 10.1016/j.ydbio.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang LT, Kaartinen V. Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev Biol. 2007;312:384–395. doi: 10.1016/j.ydbio.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lidral AC, Romitti PA, Basart AM, Doetschman T, Leysens NJ, Daack-Hirsch S, Semina EV, Johnson LR, Machida J, Burds A, Parnell TJ, Rubenstein JL, Murray JC. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet. 1998;63:557–568. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rienhoff HY, Jr, Yeo CY, Morissette R, Khrebtukova I, Melnick J, Luo S, Leng N, Kim YJ, Schroth G, Westwick J, Vogel H, McDonnell N, Hall JG, Whitman M. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlapping with marfan and loeys-dietz syndrome. Am J Med Genet A. 2013;161:2040–2046. doi: 10.1002/ajmg.a.36056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voermans NC, Jenniskens GJ, Hamel BC, Schalkwijk J, Guicheney P, van Engelen BG. Ehlers-Danlos syndrome due to tenascin-X deficiency: muscle weakness and contractures support overlap with collagen VI myopathies. Am J Med Genet A. 2007;143A:2215–2219. doi: 10.1002/ajmg.a.31899. [DOI] [PubMed] [Google Scholar]
- 25.Hillegass JM, Villano CM, Cooper KR, White LA. Matrix metalloproteinase-13 is required for zebra fish (Danio rerio) development and is a target for glucocorticoids. Toxicological sciences: an official journal of the Society of Toxicology. 2007;100:168–179. doi: 10.1093/toxsci/kfm192. [DOI] [PubMed] [Google Scholar]
- 26.Hulin A, Deroanne CF, Lambert CA, Dumont B, Castronovo V, Defraigne JO, Nusgens BV, Radermecker MA, Colige AC. Metallothionein-dependent up-regulation of TGF-beta2 participates in the remodelling of the myxomatous mitral valve. Cardiovasc Res. 2012;93:480–489. doi: 10.1093/cvr/cvr337. [DOI] [PubMed] [Google Scholar]
- 27.Azhar M, Brown K, Gard C, Chen H, Rajan S, Elliott DA, Stevens MV, Camenisch TD, Conway SJ, Doetschman T. Transforming growth factor Beta2 is required for valve remodeling during heart development. Dev Dyn. 2011;240:2127–2141. doi: 10.1002/dvdy.22702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Azhar M, Runyan RB, Gard C, Sanford LP, Miller ML, Andringa A, Pawlowski S, Rajan S, Doetschman T. Ligand-specific function of transforming growth factor beta in epithelial-mesenchymal transition in heart development. Dev Dyn. 2009;238:431–442. doi: 10.1002/dvdy.21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartler CS, Gouya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, Jondeau G, Milewicz DM. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gotoh H, Sagai T, Hata J, Shiroishi T, Moriwaki K. Steroid 21-hydroxylase deficiency in mice. Endocrinology. 1988;123:1923–1927. doi: 10.1210/endo-123-4-1923. [DOI] [PubMed] [Google Scholar]
- 31.Aoyama T, Francke U, Gasner C, Furthmayr H. Fibrillin abnormalities and prognosis in Marfan syndrome and related disorders. Am J Med Genet. 1995;58:169–176. doi: 10.1002/ajmg.1320580216. [DOI] [PubMed] [Google Scholar]
- 32.Holm TM, Habashi JP, Doyle JJ, Bedja D, Chen Y, van Erp C, Lindsay ME, Kim D, Schoenhoff F, Cohn RD, Loeys BL, Thomas CJ, Patnaik S, Marugan JJ, Judge DP, Dietz HC. Noncanonical TGFbeta signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332:358–361. doi: 10.1126/science.1192149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haskett D, Doyle JJ, Gard C, Chen H, Ball C, Estabrook MA, Encinas AC, Dietz HC, Utzinger U, Vande Geest JP, Azhar M. Altered tissue behavior of a non-aneurysmal descending thoracic aorta in the mouse model of Marfan syndrome. Cell and tissue research. 2012;347:267–277. doi: 10.1007/s00441-011-1270-y. [DOI] [PubMed] [Google Scholar]
- 34.Quarto N, Li S, Renda A, Longaker MT. Exogenous activation of BMP-2 signaling overcomes TGFbeta-mediated inhibition of osteogenesis in Marfan embryonic stem cells and Marfan patient-specific induced pluripotent stem cells. Stem cells. 2012;30:2709–2719. doi: 10.1002/stem.1250. [DOI] [PubMed] [Google Scholar]
- 35.Carta L, Smaldone S, Zilberberg L, Loch D, Dietz HC, Rifkin DB, Ramirez F. p38 MAPK is an early determinant of promiscuous Smad2/3 signaling in the aortas of fibrillin-1 (Fbn1)-null mice. J Biol Chem. 2009;284:5630–5636. doi: 10.1074/jbc.M806962200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collee M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]