

Abstract
Huntington’s disease (HD) is a fatal genetic disorder characterized by triad clinical symptoms of chorea, emotional distress, and cognitive decline. Genetic mutation in HD is identified by an expansion of CAG repeats coding for glutamine (Q) in exon 1 of the huntingtin (htt) gene. The exact mechanism on how mutant htt leads to the selective loss of medium spiny neurons (MSNs) in the striatum is still unknown. Recent studies suggest that nucleolar stress and dysfunction is linked to the pathogenesis of HD. Alterations of the nucleolar activity and integrity contributes to deregulation of ribosomal DNA (rDNA) transcription in HD pathogenesis. Furthermore, epigenetic modifications in the nucleolus are associated with neuronal damage in HD. In this review, we discuss about how post-translational modifications of upstream binding factor (UBF) are affected by histone acetyltransferase and histone methyltransferase and involved in the transcriptional regulation of rDNA in HD. The understanding of epigenetic modulation of UBF-dependent rDNA transcription in the nucleolus may lead to the identification of novel pathological markers and new therapeutic targets to treat HD.
Keywords: Huntington’s disease, nucleolus, upstream binding factor (UBF), rDNA transcription, acetylation and methylation, epigenetics
1. INTRODUCTION
1-1. From heritable dancing disorder (chorea) to Huntington’s disease (HD)
Huntington’s disease (HD) is an autosomal dominant genetic disorder featured by neurological symptoms with chorea (uncoordinated movement), psychiatric symptoms, and memory loss. The frequency of HD is reported as high as 10 cases per 100,000 and having a new mutation rate of 1–3% [1]. The occurrence of HD is not limited to a specific region or population but found throughout the globe in all races and ethnic groups [2]. The shores of Lake Maracaibo in Venezuela particularly have the highest HD frequency. The number of HD patients in United States alone reach 30,000 and those at genetic risk of HD is considered to be 200,000.
Before 17th century, Huntington’s disease (HD) was a mysterious dancing disorder (chorea) that shows jerking and twitching of muscles. Due to the lack of medical knowledge at that time, the people with involuntary movement as a feature of HD were being regarded as witches possessed by the devil [3]. In 1872, George Huntington, an American physician, provided the first clinical description of HD and its hereditary nature [4]. The initial understanding of HD as a chronic encephalitis has been replaced by Jergelsma in 1908, who described the characteristic neuropathological alterations affecting the basal ganglia that are now established as the imperative pathoanatomical feature of HD [5,6]. Gross atrophy of the neostriatal nuclei, the caudate nucleus, and putamen, combined by detectable neuronal loss and astrogliosis are the most salient neuropathological alterations of HD [7–9]. Within HD, not all striatal neurons are equally influenced, and there appears to be a selective pattern of neuronal vulnerability and topographic susceptibility. During the earliest stage of disease medium-sized spiny neurons are most severely affected while intrinsic locally arborizing aspiny striatal interneurons are comparably spared [9–15].
Most importantly, the Huntington’s disease Collaborative Research Group (HCRG) presented in 1993 that a previously unidentified ‘interesting transcript (IT) 15′ on human chromosome 4 was mutated in patients with HD and genetically linked to HD [16]. HCRG’s reported HD-related lethal gene was referred to as huntingtin (htt). The expansion of the wildtype htt allele that typically contains 15–35 CAG triplets in exon 1 to 36 or more repeats has been determined to be the mutation of htt gene. Furthermore, HD is considered to be related to other neurodegenerative diseases such as spinal and bulbar muscular atrophy (SBMA, also referred to as Kennedy’s disease (KD)) and spinocerebellar ataxias (SCAs) which are also caused by similar trinucleotide CAG repeat mutations. To be more specific, SBMA occurs when androgen receptor (AR) gene is mutated while the expansion of a polyglutamine tract within the SCA1 gene product ataxin-1 causes SCA1 [17, 18]. The significance of CAG expansion is observed in cases where affected individuals with greater number of CAG repeats show younger age of onset. In addition, there is a strong inverse relationship between age of onset and CAG repeat number in HD.
The cytoplasmic and vesicular protein, htt, is ubiquitously expressed and heterogeneously found in neurons throughout the brain. Numerous hypothetical pathologic mechanisms have been proposed since discovering htt gene but a direct pathway from the genetic mutation to neuronal degeneration has yet to be established. Even though the definitive functionality of htt remains unknown, it has been postulated that htt is involved in intracellular transport, autophagy, transcription, mitochondrial function, and signal transduction. On the other hand, mutant htt (mthtt) results in the inhibition of fast axonal transport and destabilization of microtubules within the cell [19, 20]. Within heterozygous HD, both normal and mutant alleles are expressed. As a “gain of function”, the expression of mutant htt protein is toxic and it triggers the pathologic cascades of HD. In contrast, the deletion of the normal htt gene is also lethal indicating that the function of normal htt is essential in survival [21–28]. The engagement in pathologic protein-protein interactions between mutant htt and its proteolytic fragments is well established and accepted. This engagement leads to alterations of cellular pathways that make neurons to be more vulnerable to generic stresses, ultimately resulting in neuronal damage and death [29]. Additionally, mutant htt interactomes involve transcriptional dysregulation, mitochondrial dysfunction, proapoptotic signaling, oxidative injury, excitotoxicity, inflammatory reactions, and malfunctioning proteolysis. Since the first clinical description of HD by George Huntington, there has been a strong progression towards the understanding of the disease mechanisms. However, a treatment to prevent the onset or to delay the insidious and unyielding course of HD is currently unavailable [30].
Epigenetic alterations in HD
A term “epigenetics” was described by Dr. Waddington to explain the biological events that are unexplainable by conventional genetic principals [31]. The field of epigenetics has evolved since its introduction, and is now defined as the study connecting genotype to phenotype in the absence of DNA sequence alteration [32]. In consideration to this, epigenetics is regarded as a highly beneficial field to investigate features and mechanisms responsible for the temporal and spatial control of gene activity regulated by processes beyond mutations in DNA sequence [33]. Epigenetic modifications explain for and encompass an array of molecular modifications to both chromatin and DNA, including ncRNAs regulation. For many genes in general, they contain DNA methylation sites (CpG islands) in their promoters [34]. This indicates that marked hypo- or hyper-DNA methylation may explain for significant components of the molecular and pathogenic complexity of human genomes. Expanding number of evidence suggests and point towards the alterations of epigenetic modifications to constitute a basic molecular mechanism contributing to HD pathogenesis. What the understanding of epigenetic mechanisms hold for is the opportunity to gain imperative insights leading to the identification of novel biological markers and therapeutic interventions to treat HD [35].
A growing body of evidence indicates that nucleolar stress and dysfunction is linked to the pathogenesis of HD. The nucleolus is a subnuclear compartment possessing the transcription machinery of ribosomal genes and the ribosomal DNA (rDNA) encoding ribosomal RNA (rRNA) [36]. The rDNA is organized as tandem repeats in the nucleolus and is transcribed into 47S precursor rRNA by a nucleolar transcription complex, consisting of RNA polymerase I and other co-regulatory factors [37]. Neurons have prominent nucleoli but the role of this structure and the regulatory mechanism of rDNA transcription are poorly understood [38]. RNA polymerase I and upstream binding factor (UBF) are major molecular components for the format ion of active nucleolar organizer regions and maintenance of rRNA transcriptional activity [39]. It has previously known that nucleolar accessory bodies (Cajal bodies) are associated with disorders caused by expansions of CAG repeats within genes, including HD [39]. Recently, our group and others have found that alteration of epigenetic components and deregulation of transcriptional machinery are directly involved in the down regulation of ribosomal RNA (rRNA) expression and neuronal damage in HD [40, 41] Together, in the present paper, we are going to address and discuss the epigenetic changes and mechanism that are associated with the nucleolar-dependent pathogenesis in HD.
2. Epigenetic Modifications in the nucleolus of HD
2-1. UBF acetylation in HD
UBF is a nucleolar transcription factor of the high mobility group (HMG) protein family and contains six HMG box DNA binding motifs. UBF consists of two polypeptides (UBF1 and UBF2, 97 and 94 kDa, respectively), which arise from alternative splicing of a single transcript [42]. UBF1 and 2 form hetero- and homodimers but UBF2 is five-fold less active than UBF1, and mechanisms by which UBF2 provides such poor transcriptional activity are not known [43]. UBF is essential for rDNA transcription by inducing remodeling of ribosomal gene chromatin and further playing a structural role by binding to other sequences across the entire rDNA [36]. UBF targeting to regions of heterochromatin is sufficient to induce large-scale chromatin decondensation [44]. UBF binding throughout the rDNA gene repeat might therefore contribute to the formation of the active chromatin state of rDNA genes [45]. Recently, we found that transcriptional modulation of rDNA is altered in HD, and both UBF and CREB binding protein (CBP) contribute to altered nucleolar chromatin remodeling and rRNA expression in HD. We found that protein levels UBF1, CBP, and UBF-mediated transcriptional activity of rDNA are impaired in cellular and animal model of HD as well as in HD patients [40]. Our study shows that UBF1 protein levels in the striatum were significantly decreased and the 45S was down re gulated in R6/2 mice in comparison with WT controls. These data indicate that abnormal protein level of UBF1 protein levels correlate with impaired ribosomal transcription in HD mice.
Post-translational modifications of UBF, such as acetylation and phosphorylation, have an important role in the control of rDNA transcription [46]. Transcription factor acetylation is responsible for both activation and subnuclear localization [47]. CBP functions as a HAT in acetylating histones that contributes to transcription by remodeling the chromatin structure and functions as a transcriptional cofactor [48]. A loss of CBP function interferes with transcription by altering the acetylation level of histones and chromatin structure in neurons and also by inhibiting recruitment of the basal transcription machinery to the promoter [49, 50]. Interestingly, the sequestration of CBP molecule by mthtt protein was found in intranuclear inclusions of striatal neurons and other neurons of HD [22, 51] (Figure 1). The polyQ repeats in mthtt interact physically with CBP and deregulate its intrinsic HAT activity and transcriptional coactivator function [52, 53]. Accordingly, the blocking of CBP activity by mthtt causes the hypoacetylation and hypermethylation of histone proteins and the subsequent transcriptional dysfunction of neurons in HD [54–58]. These specific interactions and epigenetic modifications are attributable to pathological transcriptional dysfunction [59]. Importantly, we determined that CBP interacts with UBF1, acetylates UBF1, and modulates rDNA transcription in HD. UBF acetylation is also important for cell cycle-dependent regulation of rRNA expression. It has been proposed that the transcription of rDNA is regulated upon the influence of two opposite processes through UBF acetylation by CBP and deacetylation by HDAC [26]. In this context, CBP-dependent acetylation of UBF provides a thread of mechanism on the transcription activation of rDNA [46]. Indeed, we found that the acetylation levels of UBF1 are significantly lowered due to deregulation of CBP function in both HD cells and HD mice. Our data shows that UBF1 and CBP interact in intact neurons. Reduced levels of acetylated UBF1 correlate with the reduced CBP level and activity in HD. We further demonstrated that the HAT activity of CBP directly controls UBF1 acetylation and this was found using a HAT do main deletion mutant of CBP that caused a marked reduction of UBF1 acetylation. Our findings indicate that the neurodegenerative process in HD is associated with changes in adaptive rDNA transcription caused by altered CBP acetyltransferase that affects the balance of acetylated and deacetylated UBF1 [40, 22] (Figure 1).
Figure 1. Mutant huntingtin (mthtt) sequestrates CBP and disrupts CBP-mediated UBF activation in the nucleolus of HD.
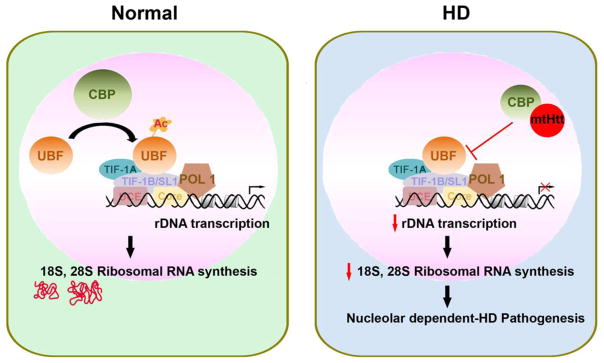
In normal conditions, CBP maintains the acetylation status of UBF through HAT activity and regulates the transcription of rDNA by interacting with transcriptional complexes including RNA polymerase I (Pol I). In HD, mthtt sequestrates CBP in nuclear inclusions (aggregate formation) and deregulates UBF-dependent nucleolar transcription. Consequently, imbalanced rDNA transcription leads to neuronal damage in HD.
The LC-MALDI-MS/MS analysis identified the specific acetylation site of UBF1 from in vitro-acetylated UBF protein by CBP [40]. CBP acetylates UBF1 at K352 that resides in the H MG3 domain. Mutations at K352 to alanine (A), glutamine (Q), and arginine (R) abrogates the acetylation of UBF1, as well as the transcriptional activation of UBF1 in response to CBP. This data confirms that the acetylation of UBF1 by CBP has a pivotal role in rDNA gene transcription. Consequently, due to the lack of CBP-dependent acetylation of UBF1, UBF1-induced rDNA expression is disrupted in HD models [46, 60]. It is unclear, however, whether mtHtt and HD-related cellular changes modulate the association of HDAC to the UBF1 complex. Future study remains to define precisely how UBF1 protein is deacetylated and what other factors are involved in this process. This will provide a better understanding of the rDNA transcription and ribosomal synthesis in the pathogenesis of HD.
2-2. UBF methylation in HD
The motor symptoms of HD are largely a consequence of profound neuronal damage in the GABAergic MSNs [3]. Deregulation of chromatin remodeling is regarded as one of the mechanisms by which mutant mthtt contributes to cell death of MSNs in the striatum [3]. Mthtt not only blocks the intrinsic HAT activity of CBP but also induces epigenetic enzymes such as histone methyltransferases (HMTs) [52, 61]. These specific epigenetic alterations show how mthtt modulates transcriptional signaling cascades that initiate a number of downstream pathophysiological mechanisms relevant to HD. ERG-associated protein with SET domain (ESET/SETDB1) is a histone H3K9-specific methyltransferase that contains both tudor and methyl-CpG binding domains that converge transcription and RNA processing factors [62]. It also acts as a signature motif for proteins regulating methylated DNA silencing [62]. SETDB1 is involved in neuronal dysfunction through its histone methyltransferase activity and the epigenetic silencing of neuronal genes [56]. However, the roles of SETDB1 on nucleolar chromatin landscaping and rDNA transcription have not been fully investigated yet [63]. We have recently found that elevations of SETDB1 expression and H3K9me3 level are correlated with transcriptional deregulation and neurodegeneration in HD [56, 64]. To examine the links between SETDB1 activity and nucleolar function, we evaluated whether SETDB1 interacts with UBF and contribute to the chromatin organization of the nucleolus and rDNA transcription in HD. First, we found that increased levels of methylated UBF are correlated with the increased SETDB1 activity, a pathological event found in HD (Figure 2). As we expected, the levels of SETDB1 protein and methylated UBF were significantly increased in the striatum of R6/2 transgenic HD mice and human HD patients (unpublished data). Second, we determined that SETDB1 physically interacts with UBF and methylates it in intact neurons. The methyltransferase activity of SETDB1 is directly responsible for UBF methylation. Accordingly, SET domain deletion mutants of SETDB1 resulted in a marked reduction of UBF methylation. In general, K (lysine) residues are mono-, di, or trimethylated, and the status of methylation contributes to different functional outcomes [65]. We confirmed that UBF is presented as a trimethylated form in intact cells. In addition, UBF is trimethylated by SETDB1 in vitro. We performed LC-MALDI-MS/MS analysis in vitro on UB-MG protein methylated by SETDB1 and identified the specific methylation site of UBF. We found that SETDB1 methylates UBF at the K232/254 residues of the HMG2 domain (unpublished data). Accordingly, mutations at K232/254 blocked UBF trimethylation and derepressed the transcriptional suppression of rDNA by methylated UBF in response to SETDB1. Furthermore, methylation site mutant UBF (K232/254A) restored the transcriptional level of intermediate (5′-ETS) and mature (18S and 28S) rRNA levels while SETDB1 decreased the expression of rRNA through the methylation of wild type UBF. We also determined that 45S levels were down regulated by SETDB1. Our data suggests that the increased level of methylated UBF is correlated with deregulation of ribosomal transcription in HD, and the SETDB1-dependent trimethylation of UBF plays a direct role in the regulation of nucleolar chromatin plasticity (Figure 2). Targeting of UBF trimethylation to regions of heterochromatin is sufficient to induce large-scale chromatin condensation in the nucleolus. In this paradigm, the binding of trimethylated UBF throughout the rDNA gene repeat might contribute to the formation of the inactive chromatin state of rDNA genes. It remains to be determined, however, how mthtt and HD-related cellular changes modulate the association of ESET to the UBF complex. It will also be important to precisely define the demethylation mechanism of UBF in order to more fully understand the dynamic role of UBF on nucleolar chromatin remodeling and rDNA transcription in HD.
Figure 2. A scheme represents an epigenetic mechanism that abnormal activity of histone H3K9-specific methyltransferase (ESET/SETDB1) leads to UBF trimethylation and impaired rDNA transcription in HD.
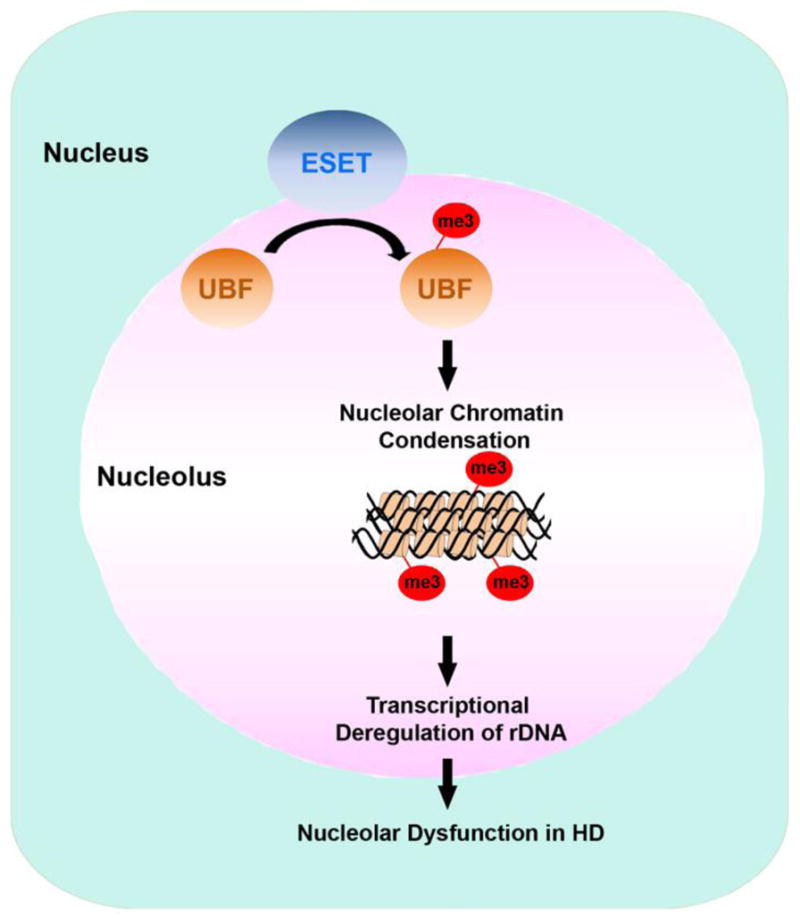
ESET methylates UBF and UBFme3-dependent nucleolar chromatin condensation results in the repression of rDNA transcription, and subsequent reduction of 18S and 28S RNA. Down regulation of rDNA transcription contributes to striatal dysfunction and neurodegeneration in HD.
3. Therapeutic approaches for improving the nucleolar function in HD
Genetic mutations are irreversible but epigenetic modifications are reversible processes. In this context, from a therapeutic perspective, epigenetic modifications are regarded as a useful therapeutic target. Small compounds that dynamically inhibit HDAC activity can modulate the status of rDNA transcription by post-translational modifications of histone and non-histone molecules, as well as remodeling of the chromatin structure in HD. Therefore, the development of such drug agents that realign the epigenetic balance and subsequently improve HD-related deficits in the nucleolus is necessary in HD-related researches. Because the acetylation on lysine residue in the UBF transforms nucleolar chromatin structure locally and results in increased DNA transcription, HDAC inhibitors can promote acetylation of UBF and transcription of rDNA. Indeed, trichostatin A, a deacetylase inhibitor, enhances UBF acetylation and rDNA gene expression [39, 26]. We also found that sodium butyrate elevates rDNA transcription in mutant HD Q111 cells and WT Q7 cells in the presence of UBF1 in a dose-dependent manner [40]. Our group has shown that the increased acetylation of transcription factors by three structurally distinct HDAC inhibitors improves neuronal survival in response to oxidative stress [66]. Collectively, these studies are consistent with a model in which altered rDNA transcription is an additional mechanism contributing to the protective effects of HDAC inhibitors in neurons. HDAC inhibitors have been preclinically tested in many neurodegenerative conditions, including animal models of HD, amyotrophic lateral sclerosis (ALS), and multiple sclerosis [49, 54–56, 67, 68]. Taken together, it is encouraging that HDAC inhibitors may improve phenotypes by upregulating rDNA transcription that is repressed in HD [40, 49, 54–56]. However, the underlying mechanisms whereby HDAC inhibitors modulate the function of nucleolus remain to be investigated. Thus, further studies will be needed to clarify the contribution of UBF1 acetylation and rDNA transcription to the neuroprotective effects of HDAC inhibitors.
4. Conclusions
Alterations of epigenetic modification in the nucleolus are closely associated with HD pathogenesis. In this context, posttranslational modifications of UBF play an important role in the regulation of nucleolar chromatin structure and transcription in striatal cells. Interestingly, an imbalance of transcriptional homeostasis through UBF hypoacethylation and hypermethylation provides another layer of pathological mechanisms that are relevant to HD. While the increased acetylation of UBF by CBP up regulates the transcription of rDNA under normal condition, the decreased acetylation of UBF due to the deficiency of CBP HAT activity le ads to the down regulation of rDNA transcription in HD. Since HDAC inhibitor increases the acetylation of UBF, the use of small compounds targeting HDAC in particular may be a successful therapeutic strategy for ameliorating the rDNA trascription in HD [40]. However, approaches using HDAC inhibitor-based therapeutic interventions need improved target specificity for UBF-dependent nucleolar transcription in HD. Otherwise, the trimethylation of UBF by HMT elevates the occupancy of UBF to the promoter region of rDNA, which condensates the structure of nucleolar chromatin, and leads to the down regulation of rDNA transcription. Prevention of UBP hypermethylation by inhibiting HMT activity may also be useful therapeutic targets for treating HD. It is evident that the status of UBF acetylation and methylation is an important marker directly or indirectly associated with transcriptional abnormality in the nucleolus of HD [40]. However, it remains to be further investigated whether epigenetic alterations are a fundamental aspect of HD pathogenesis or not. Accordingly, future studies are required to address whether epigenetic alterations in the nuclelous play a key role as a maker of disease progress in HD.
Highlights.
CBP is localized into the nucleus and acetylates UBF.
The decreased level of UBF acetylation is correlated with the reduction of rDNA transcription in HD.
Trimethyation of UBF leads to nucleolar chromatin condensation and impairs rDNA transcription in HD
Epigenetic modifications of UBF contribute to the nucleolus-dependent pathogenesis of HD.
Acknowledgments
This study was supported by NIH NS 067283-03 (H.R.), WCU Neurocytomics Program Grant (800-20080848) (H.R.) and SRC Grant (2010-0029-403) (H.R.) from National Research Foundation, and Flagship Grant (H.R.) from KIST.
Abbreviations
- HD
Huntington’s disease
- htt
huntingtin
- mthtt
mutant htt
- MSNs
medium spiny neurons
- rDNA
ribosomal DNA
- rRNA
ribosomal RNA
- UBF
upstream binding factor
- HAT
histone acetyltransferase
- Pol I
RNA polymerase I
- HMT
histone methyltransferase
- CBP
CREB binding protein
- HMG
high mobility group
- ESET
ERG-associated protein with SET domain
- HDAC
histone deacetylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Myers RH, MacDonald ME, Koroshetz WJ, Duyao MP, Ambrose CM, Taylor SA, Barnes G, Srinidhi J, Lin CS, Whaley WL, Lazzarini AM, Schwarz M, Wolff G, Bird ED, Vonsattel JPG, Gusella JF. De novo expansion of a (CAG)n repeat in sporadic Huntington’s disease. Nat Genet. 1993;5:168–173. doi: 10.1038/ng1093-168. [DOI] [PubMed] [Google Scholar]
- 2.Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E, Bird TD, Hayden MR. A worldwide study of the Huntington’s disease mutation the sensitivity and specificity of measuring CAG repeats. New Engl J Med. 1994;330:1401–1406. doi: 10.1056/NEJM199405193302001. [DOI] [PubMed] [Google Scholar]
- 3.Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 4.Huntington G. On chorea. Med Surg Rep. 1872;26:317–321. [Google Scholar]
- 5.Jergelsma G. Nue anatomische befunde bei paralysis agitans und bei chronischer progressive chorea. Neurol Centralbl. 1908;27:995–996. [Google Scholar]
- 6.Bruyn GW, Bots GTAM, Dom R. Huntinton’s chorea: Current neuropathological status. In: Chase TN, editor. Advances in Neurology. 23 . Raven Press; New York: 1979. pp. 83–93. [Google Scholar]
- 7.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington’s disease. J Neuropath Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Kowall NW, Ferrante RJ. Huntington’s disease. In: Markesbery WR, editor. Neuropathology of Dementing Disorders. Oxford University Press; New York: 1998. pp. 219–256. [Google Scholar]
- 9.Hersch SM, Rosas HD, Ferrante RJ. Neuropathology and pathophysiology of Huntington’s disease. In: Watts RL, Koller W, editors. Movement disorders: Neurologic principles and practice. Mcgraw-Hill; New York: 2004. pp. 503–526. [Google Scholar]
- 10.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 11.Ferrante RJ, Kowall NW, Richardson EP, Jr, Bird ED, Martin JB. Topography of encephalin, substance P and acetylcholinesterase staining in Huntington’s disease striatum. Neurosci Lett. 1986;71:283–288. doi: 10.1016/0304-3940(86)90634-8. [DOI] [PubMed] [Google Scholar]
- 12.Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP., Jr Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J Neuropathol Exp Neurol. 1987;1:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- 13.Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington’s disease: a combined study using the section-Golgi method and calbindin D28K immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 15.Kowall NW, Ferrante RJ, Martin JB. Patterns of cell loss in Huntington’s disease. TINS. 1987;10:24–29. [Google Scholar]
- 16.Huntington’s disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 17.Spada ARL, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 18.Orr HT, Chung MY, Banfi S, Kwiatkowski TJ, JR, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LP, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 19.Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- 20.Trushina E, Dyer RB, Badger JD, 2nd, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A, Chacko C, McMurray CT. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ross CA. Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron. 1997;19:1147–1150. doi: 10.1016/s0896-6273(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 22.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 23.Cattaneo E. Dysfunction of wild-type huntingtin in Huntington disease. News Physiol Sci. 2003;18:34–37. doi: 10.1152/nips.01410.2002. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Li M, Drozda M, Chen M, Ren S, Sanchez RO, Leavitt BR, Cattaneo E, Ferrante RJ, Hayden MR, Friedlander RM. Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem. 2003;87:101–106. doi: 10.1046/j.1471-4159.2003.01980.x. [DOI] [PubMed] [Google Scholar]
- 25.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med Suppl. 2004;10:S10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 26.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a corditional model of Huntington’s disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 28.Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, Wiener HW, Dure LS, 4th, Lindsey R, Hersch SM, Jope RS, Albin RL, Detloff PJ. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell. 1997;91:753–763. doi: 10.1016/s0092-8674(00)80464-x. [DOI] [PubMed] [Google Scholar]
- 29.Beal MF, Ferrante RJ. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat Rev Neurosci. 2004;5:373–384. doi: 10.1038/nrn1386. [DOI] [PubMed] [Google Scholar]
- 30.Ryu H, Rosas HD, Hersch SM, Ferrante RJ. The therapeutic role of creatine in Huntington’s disease. Pharmacol Ther. 2005;108:193–207. doi: 10.1016/j.pharmthera.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Waddington CH. The epigenotype. Endeavour. 1942;1:18. [Google Scholar]
- 32.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Sadri-Vakili G, Cha JH. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat Clin Pract Neurol. 2006;2:330–333. doi: 10.1038/ncpneuro0199. [DOI] [PubMed] [Google Scholar]
- 34.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 35.Ryu H, Ferrante RJ. Emerging chemotherapeutic strategies for Huntington’s disease. Expert Opin Emerg Drugs. 2005;10:345–363. doi: 10.1517/14728214.10.2.345. [DOI] [PubMed] [Google Scholar]
- 36.Casafont I, Bengoechea R, Navascués J, Pena E, Berciano MT, Lafarga M. The giant fibrillar center: a nucleolar structure enriched in upstream binding factor (UBF) that appears in transcriptionally more active sensory ganglia neurons. J Struct Biol. 2007;159:451–461. doi: 10.1016/j.jsb.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 37.Lafontaine DL, Tollervey D. The function and synthesis of ribosomes. Nat Rev Mol Cell Biol. 2001;2:514–520. doi: 10.1038/35080045. [DOI] [PubMed] [Google Scholar]
- 38.Akhmanova A, Verkerk T, Langeveld A, Grosveld F, Galjart N. Characterisation of transcriptionally active and inactive chromatin domains in neurons. J Cell Sci. 2000;113:4463–4474. doi: 10.1242/jcs.113.24.4463. [DOI] [PubMed] [Google Scholar]
- 39.McStay B, Grummt I. The epigenerics of rRNA genes: from molecular to chromosome biology. Annu Rev Cell Dev Biol. 2008;24:131–157. doi: 10.1146/annurev.cellbio.24.110707.175259. [DOI] [PubMed] [Google Scholar]
- 40.Lee J, Hwang YJ, Boo JH, Han D, Kwon OK, Todorova K, Kowall NW, Kim Y, Ryu H. Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ. 2011;18:1726–1735. doi: 10.1038/cdd.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kreiner G, Bierhoff H, Armentano M, Rodriguez-Parkitna J, Sowodniok K, Naranio JR, Bonfnati L, Liss B, Schütz G, Grummt I, Parlato R. A neuroprotective phase precedes striatal degeneration upon nucleolar stress. Cell Death Differ. 2013 doi: 10.1038/cdd.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stefanovsky VY, Moss T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008;36:5093–5101. doi: 10.1093/nar/gkn484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Mahony DJ, Rothblum LI. Identification of two forms of the RNA polymerase I transcription factor UBF. Proc Natl Acad Sci USA. 1991;88:3180–3184. doi: 10.1073/pnas.88.8.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hannan RD, Stefanovsky V, Taylor L, Moss T, Rothblum LI. Overexpression of the transcription factor UBF1 is sufficient to increase ribosomal DNA transcription in neonatal cardiomyocyte: implications for cardiac hypertrophy. Proc Natl Acad Sci USA. 1996;93:8750–8755. doi: 10.1073/pnas.93.16.8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, Belmont AS, Huang S. Upstream binding factor association induces large-scale chromatin decondensation. Proc Natl Acad Sci USA. 2004;101:15106–15111. doi: 10.1073/pnas.0404767101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanij E, Poortinga G, Sharkey K, Hung S, Holloway TP, Quin J, Robb E, Wong LH, Thomas WG, Stecanovsky V, Moss T, Rothblum L, Hannan KM, McArthur GA, Pearson RB, Hannan RD. UBF levels determine the number of active ribosomal RNA genes in mammals. J Cell Biol. 2008;183:1259–1274. doi: 10.1083/jcb.200805146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meraner J, Lechner M, Loidl A, Goralik-Schramel M, Voit R, Grummt I, Loidl P. Acetylation of UBF changes during the cell cycle and regulates the interaction of UBF with RNA polymerase I. Nucleic Acids Res. 2006;34:1798–1806. doi: 10.1093/nar/gkl101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- 49.Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 50.Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 52.Chouliaras L, Rutten BP, Kenis G, Peerbooms O, Visser PJ, Verhey F, van OS J, Steinbusch HW, van den Hove DL. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog Neurobiol. 2010;90:498–510. doi: 10.1016/j.pneurobio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 53.West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer’s disease patient. J Mol Neurosci. 1995;6:141–146. doi: 10.1007/BF02736773. [DOI] [PubMed] [Google Scholar]
- 54.Ferrante RJ, Ryu H, Kubilus JK, D’Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 56.Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3(K9) trimethylation in Huntington’s disease. Proc Natl Acad Sci USA. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- 58.McFarland KN, Das S, Sun TT, Leyfer D, Xia E, Sangrey GR, Kuhn A, Luthi-Carter R, Clark TW, Sadri-Vakili G, Cha JH. Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS One. 2012;7:e41423. doi: 10.1371/journal.pone.0041423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109:801–806. doi: 10.1016/s0092-8674(02)00798-5. [DOI] [PubMed] [Google Scholar]
- 60.Doetzlhofer A, Rotheneder H, Lagger G, Koranda M, Kurtev V, Brosch G, Winterberger E, Seiser C. Histone deacetylase 1 can repress transcription by binding to SP1. Mol Cell Biol. 1999;19:5504–5511. doi: 10.1128/mcb.19.8.5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee J, Hong YK, Jeon GS, Hwang YJ, Kim KY, Seong KH, Jung MK, Picketts DJ, Kowall NW, Cho KS, Ryu H. ATRX induction by mutant huntingtin via Cdx2 modulates heterochromatin condensation and pathology in Huntington’s disease. Cell Death Differ. 2012;19:1109–1116. doi: 10.1038/cdd.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21:148–152. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- 63.Feng W, Yonezawa M, Jenuwein T, Grummt I. PHF8 activates transcription of rRNA genes through H3K4me3 binding and H3K9me1/2 demethylation. Nat Struct Mol Biol. 2010;17:445–450. doi: 10.1038/nsmb.1778. [DOI] [PubMed] [Google Scholar]
- 64.Lee J, Hwang YJ, Shin JY, Lee WC, Wie J, Kim KY, Lee MY, Hwang D, Ratan RR, Pae AN, Kowall NW, So I, Kim JI, Ryu H. Epigenetic regulation of cholinergic receptor M1 (CHRM1) by histone H3K9me3 impairs Ca(2+) signaling in Huntington’s disease. Acta Neuropathol. 2013;125:727–39. doi: 10.1007/s00401-013-1103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–413. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 66.Stefanovsky VY, Moss T. Regulation of rRNA synthesis in human and mouse cells is not determined by changes in active gene count. Cell Cycle. 2006;5:735–739. doi: 10.4161/cc.5.7.2633. [DOI] [PubMed] [Google Scholar]
- 67.Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, Goto M, Sakoda S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20:3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x. [DOI] [PubMed] [Google Scholar]
- 68.Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]