

Abstract
The cAMP response element-binding protein (CREB) is a transcription factor that mediates the cellular response to metabolic and mitogenic signals. Whether CREB contributes to vascular function has received little attention, especially in relation to the processes associated with atherosclerotic disease progression and restenosis. This study examined the involvement of CREB in the mitogenic actions of angiotensin II (AngII), a growth factor that promotes neointimal hyperplasia in response to vascular injury. Treatments were performed on quiescent vascular smooth muscle cells (VSMCs) obtained from a porcine explant model. Organ culture was performed on porcine hearts subjected to angioplasty ex vivo. Stimulation of VSMCs with AngII resulted in transient CREB phosphorylation. Proliferation of smooth muscle cells in response to AngII was reduced by 90 % after infection with adenovirus expressing dominant-negative killer CREB (kCREB) mutant. Likewise, expression of kCREB prevented angioplasty-induced neointimal hyperplasia. AngII-induced CREB phosphorylation was independent of cAMP activation. Examination of putative CREB kinases revealed that MSK was responsible for phosphorylating CREB. In addition, inhibition of PKC revealed that this kinase operates upstream and activates MSK. These results indicate that activation of CREB via PKC and MSK is essential for SMC proliferation in response to AngII.
Keywords: Angiotensin II, CREB, Neointimal hyperplasia, Smooth muscle, Proliferation
Introduction
Humoral factors secreted in response to vascular injury are responsible for mediating the various processes that enable repair of the damaged tissue. However, production of these agents either in above normal amounts or for an increased length of time can trigger the onset of various pathophysiological conditions, including both atherosclerosis and restenosis. Among the factors commonly released following injury to vascular tissue is AngII (Newby and Zaltsman 2000). This peptide hormone operates by binding to the AT1 receptor (AT1R), which in turn activates specific intracellular signal transducers such as MAP kinase, phosphatidylinositol-3-kinase (PI3-kinase) and Rho that influence smooth muscle cell (SMC) function (Touyz and Schiffrin 2000). A consequence of these actions is the conversion of SMCs from their normal contractile state to the synthetic phenotype, thereby enabling these cells to proliferate and migrate, both essential processes for tissue repair but also key elements in vascular pathologies (reviewed in Owens et al. 2004).
Control of SMC phenotype is presumed to be mediated by transcription factors that are activated in response to various extracellular signals (Owens 2007). The cAMP response element-binding protein (CREB) is one of these transcription factors, and is an important nuclear target of cyclic nucleotide signaling. Klemm et al. (2001) were the first to report that CREB was a molecular “off-switch”, and that increased cellular content of CREB, along with its activation via phosphorylation, maintains SMCs in a differentiated state. Thus, de-activation of CREB would be required for the phenotypic switch that enables SMC proliferation and migration. In contrast, Funakoshi et al. (2002) subsequently reported that CREB phosphorylation occurred in response to AngII, and that there was an association between CREB modification and SMC hypertrophy. One of the conclusions reached in this study was that CREB phosphorylation was mediated by PKA. However, this contrasts with other reports showing that cAMP-induced stimulation of PKA results in suppression of SMC growth, rather than its promotion (Yau and Zahradka 2003). As the AT1R, the receptor primarily responsible for AngII effects on SMCs (Mehta and Griendling 2007), is a Gαi-coupled receptor that blocks cAMP production, it would be reasonable to expect that AngII activation would result in inhibition of cAMP-mediated activities, thereby leading to SMC growth. As the data implicating PKA were based on the actions of the pharmacological inhibitor H89, which is a compound also capable of blocking the actions of numerous other kinases (Lochner and Moolman 2006; Murray 2008), we sought to investigate whether the PKA-independent effects of H89 could be used to rationalize the contradicting results. Thus, the purpose of this study was to describe in greater detail the relationship between CREB, AngII and PKA in SMCs, specifically focusing on its role in SMC proliferation in relation to vascular injury, as well as to characterize the CREB kinase and other potential upstream regulators that mediate this event.
Materials and methods
Cell culture
Primary cultures of porcine coronary artery SMCs were generated from the left anterior descending (LAD) coronary artery of whole swine hearts obtained from a local abattoir, as previously described (Saward and Zahradka 1997b). Briefly, LAD arteries were flushed with PBS (consisting of 0.1 M Na3PO4 and 0.9 % NaCl) + 10× antibiotic-antimycotic, then dissected out and cleaned of adhering fat or cardiac tissue. Cells were obtained from migration of cells from cut segments onto culture plates and maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing 20 % FBS + 1× antibiotic-antimycotic. Only passages 3 to 5 were used to maintain consistency between cultures. Quiescence was induced by transferring cells at 80 % confluency into serum-free media consisting of DMEM supplemented with 0.1 mM pyruvate, 5 μg/mL transferrin, 1 nM selenium and 200 μM ascorbate (PTSA).
Western blotting
Extracts prepared by direct addition of 250 μl 2× SDS/gel loading buffer to cells in 6-well culture dishes were loaded onto either 10 % or 7.5 % polyacrylamide gel. Following electrophoresis, the proteins were transferred to PVDF membrane and stained with antibodies to CREB (Upstate & Cell Signaling), Ser-133 phospho-CREB (Upstate & Cell Signaling), phospho-PKD, phospho-MSK, MSK, phospho-p90RSK or β-tubulin (all from Cell Signaling). Band intensities were quantified as previously described (Yau et al. 2003).
Immunofluorescence
Epifluorescence microscopy of SMCs was conducted as previously described (Saward and Zahradka 1997a).
Adenovirus construction and infection
Dominant-negative (DN) killer CREB (kCREB) cDNA (RSVKCREB), generously provided by Richard H. Goodman (Oregon Health and Sciences University, Portland), was subcloned directly into the pSHUTTLE-CMV vector supplied in the AdEasy Vector System (Quantum Biotechnologies). The resulting adenoviral particles, designated as Ad-kC, were quantified by cytopathic titration as directed in the AdEasy Vector System manual. The Ad5.CMV-LacZ∆E1/∆E3 construct expressing β-galactosidase (β-gal) provided in the AdEasy kit, designated Ad-N, and an empty adenoviral vector, designated Ad-mt, were used as controls.
DNA synthesis assay
Cultures of quiescent SMCs were exposed to adenoviral particles 24 h prior to treatment with AngII. DNA synthesis was measured by incubating the cells with 1 μCi [3H]-thymidine, added 24 h after mitogen stimulation, for 48 h as previously described (Saward and Zahradka 1997a).
Coronary artery organ culture
Porcine hearts were subjected to angioplasty ex vivo as previously described (Wilson et al. 1999). A standard angioplasty catheter (20 × 3.5 mm) was inserted into the LAD coronary artery and passed along, distal to the first major bifurcation. The catheter was inflated to five atmospheres for 1 min. Coronary artery segments were extracted and maintained in culture for 14 days. Appropriate treatments (losartan or adenoviral particles) were added to the culture medium following each media change (every second day) over the 14-day incubation period. Vessel segments stained with Lee’s methylene blue were analyzed with StainPoint software (Lynx Graphics Ltd., www.lynxgl.com) to quantify the neointimal index as intimal area/medial area (Wilson et al. 1999).
cAMP assay
The cAMP levels of SMCs were measured with a cAMP EIA kit (Cayman Chemical) according to the manufacturer’s instructions.
Statistical analysis
Data are graphically represented as means ± SEM. All experiments were replicated at least 3 times, with each replicate employing independent cell or vessel isolations. Treatment means were compared using one-way ANOVA. Significance was set at the 5 % level (p < 0.05).
Results
AngII stimulates CREB phosphorylation via the AT1R
Western blot analysis with an antibody that recognizes CREB phosphorylation at Ser-133 was used to monitor the effects of AngII stimulation of SMCs. CREB phosphorylation was detectable within 2 min after treatment of quiescent coronary artery SMCs with 1 μM AngII, and returned to basal levels after 30 min (Fig. 1a). Immunofluorescence staining of SMCs with the same antibody revealed phosphorylated CREB was present in the nuclei of all AngII-treated cells (Fig. 1b). In order to determine which receptor mediated CREB activation in response to AngII, CREB phosphorylation was measured in the presence of either an AT1R (Losartan) or an AT2R (PD123319) blocker. This experiment (Fig. 1c) established that CREB activation by AngII is mediated exclusively by the AT1R.
Fig. 1.

Mitogens stimulate CREB phosphorylation. Panel a Quiescent SMCs were treated with 1 μM AngII for the indicated times. Cells were harvested and phosphorylation of CREB was monitored by Western blotting with a phospho-specific antibody. Protein loading was determined with an antibody to total CREB. Panel b Immunostaining for phosphorylated CREB was conducted with quiescent SMCs and with SMCs treated for 10 min with 1 μM AngII. Panel c Quiescent SMCs were treated with 1 μM AngII in the presence of either 10 μM losartan or 10 μM PD123319. Phosphorylation of CREB was assessed by Western blotting and intensities were quantified by scanning densitometry. Results are plotted as means ± SEM (n = 3); *Significantly different from untreated controls by one-way ANOVA, (p < 0.05)
Expression of dominant-negative CREB prevents SMC proliferation and neointimal hyperplasia
AngII is an SMC mitogen (Louis et al. 2011), which suggests CREB activation may be involved in the proliferation of these cells. To test this possibility, adenoviral particles capable of expressing DN-kCREB (Walton et al. 1992) were prepared and shown to increase the cellular content of CREB (Fig. 2a). Although kCREB expression could not be tracked directly in individual cells due to the presence of endogenous CREB, analogous experiments with a GFP reporter established that the typical adenovirus infection efficiency in our SMCs is ≥75 %. Cultures of quiescent SMCs were then exposed to adenoviral particles 24 h prior to treatment with AngII. Monitoring thymidine incorporation established that neither the empty adenoviral vector (Ad-mt), nor the vector expressing β-gal (Ad-φ), blocked the stimulation of SMC proliferation by AngII (Fig. 2b). In contrast, infection with Ad-kCREB prevented the increase in thymidine incorporation obtained with AngII (Fig. 2c), thus indicating that CREB activation is an essential event in SMC proliferation.
Fig. 2.

CREB is required for SMC proliferation. Panel a CREB protein (arrow) was measured by Western blotting after infection of quiescent SMCs with adenovirus expressing kCREB (Ad-kC, 100 moi for 90 min). Three separate viral preparations (V1, V2, V3) were compared with untreated cells (U) and cells infected with empty vector (V0, Ad-Mt). Panel b Quiescent SMCs were exposed to 100 moi adenovirus expressing β-gal (Ad-N) or empty vector (Ad-Mt) with and without AngII, and [3H]-thymidine incorporation was measured after 48 h. Panel c Quiescent SMCs were exposed to 100 moi adenovirus expressing kCREB or β-gal (Ad-N) with and without AngII, and [3H]-thymidine incorporation was measured after 48 h. Data are plotted as means ± SEM (n = 3). Each experiment was replicated three times. Significant differences (p < 0.05) were identified with one-way ANOVA; *, relative to no adenoviral exposure; and #, relative to AngII activation alone
Neointimal (NI) hyperplasia represents the proliferative response to vascular injury, and there is strong evidence supporting a role for AngII in this process (Moon et al. 2004; Wilson et al. 1999). To investigate the possibility that CREB is also required for neointimal formation, an organ culture model of balloon angioplasty-induced damage was employed. In this model, coronary vessels of isolated pig hearts are subjected to angioplasty and segments placed into culture for up to 14 days. Neointimal formation is then monitored by morphometric analysis of the intimal area in relation to the media (Wilson et al. 1999). This model system was shown to be appropriate by the fact that CREB phosphorylation was increased in vessels 2 days after undergoing angioplasty and that blockade of the AT1 receptor with losartan decreased the level of CREB phosphorylation (Fig. 3a). Morphometric examination of the stained vessel sections showed that the neointimal area obtained in vessels incubated with Ad-kC was significantly reduced in comparison with injured vessels that were either untreated or exposed to Ad-φ (Fig. 3b). Quantitative analysis of the data from all vessels indicated Ad-kC reduced the neointimal index, defined as the ratio between the intimal area and the medial area, by 53 % (Fig. 3b). These results establish that CREB activation is an important component of the vascular response to injury.
Fig. 3.
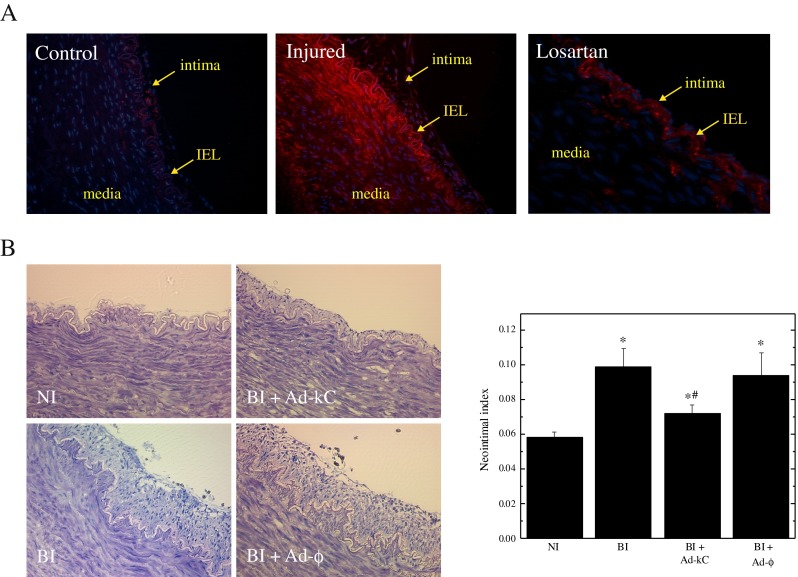
Effect of kCREB expression on neointimal formation after vascular injury. Porcine coronary artery was injured by balloon inflation and segments were cultured for 14 days. Panel a Representative depictions of vessel segments from non-injured and balloon-injured vessels stained for phospho-CREB. The media, intima and internal elastic lamina (IEL) are indicated by arrows. Panel b The neointimal index, representing the ratio between the intimal area to the medial area, was derived from morphological analysis of the vessel segments and is plotted as means ± SEM (n = 8). The experiment was replicated three times. Significant differences (p < 0.05) were identified with one-way ANOVA; *, significant increases relative to non-injured (NI); and #, reduction relative to balloon-injured (BI) vessels
PKA does not phosphorylate CREB in response to AngII
The inhibitor H89, previously used to report a connection between AngII and PKA-stimulated cAMP mediation of CREB (Funakoshi et al. 2002), is also a potent inhibitor of several other kinases known to affect cell proliferation (Lochner and Moolman 2006; Murray 2008). We therefore reexamined this issue with other inhibitors of PKA, namely KT5720 (Kase et al. 1987) and PKI (Cheng et al. 1986). When added to the cell culture medium, these compounds had no effect on the increase in CREB phosphorylation due to AngII treatment (Fig. 4a), whereas H89 was effective in blocking CREB phosphorylation (Fig. 4b). KT5720, like H89, has also been found to inhibit other kinases (Murray 2008) and thus cannot be used exclusively to distinguish the cellular actions of PKA. In contrast, the pseudosubstrate inhibitor PKI has been found to be highly selective. In support of these findings, it was shown that AngII treatment causes a decrease in cellular cAMP levels, thereby indicating that the AngII receptor is functional (Fig. 4c). These data establish that PKA is likely not involved in AngII-mediated CREB activation in SMCs, as previously proposed (Funakoshi et al. 2002).
Fig. 4.
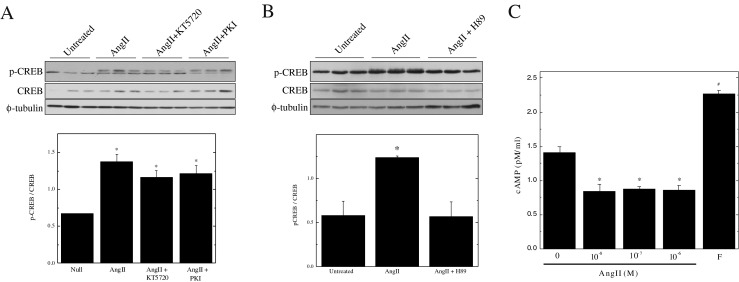
PKA is not required for AngII-mediated CREB activation. Panels a & b Quiescent SMCs were pre-treated with either KT5720 (10 μM), PKI (10 μM) or H89 (10 μM) and subsequently treated with for 10 min with 1 μM AngII. CREB phosphorylation was assessed by Western blotting of 5 μg of total starting protein. Band intensities were quantified by scanning densitometry. All results are plotted as means ± SEM (n = 3); *Significantly different from untreated controls by one-way ANOVA, (p < 0.05). Panel c Quiescent SMCs were treated with varying concentrations (0.01 – 1 μM) of AngII for 1 h or 10 μM forskolin. The cells were subsequently extracted and an EIA kit was used to measure the cAMP content. The results are presented as means ± SEM (n = 3). *Significantly decreased and #significantly increased from untreated controls by one-way ANOVA, (P < 0.05)
MSK meets the requirements of the CREB kinase and is likely downstream of PKC
To identify the CREB kinase activated by AngII, phosphorylation of likely CREB kinase candidates was monitored by Western blotting after stimulation of quiescent SMCs with 1 μM AngII. We found that PKD, p90RSK and MSK were all activated by AngII stimulation (Fig. 5a); however, only the MSK inhibitor SB747651 was able to prevent CREB phosphorylation (Fig. 5b). These results show that multiple pathways are activated by AngII, but that the CREB kinase that is responsive to AngII in SMCs is MSK.
Fig. 5.

MSK is the CREB kinase responsible for AngII-mediated signals. Panel a Quiescent VSMCs were treated with 1 μM AngII for 10 min. Kinase phosphorylation was assessed by Western blotting of 5 μg of total starting protein. Panel b Quiescent VSMCs were pre-treated for 15 min with the indicated inhibitors (CID: 10 μM; BI: 10 μM & SB: 5 μM) and subsequently treated for 10 min with 1 μM AngII. CREB phosphorylation was assessed by Western blotting of 5 μg of total starting protein. Panel c Quiescent VSMCs were pre-treated with Bisindol (10 μM) and subsequently treated for 10 min with 1 μM AngII. CREB and MSK phosphorylation was assessed by Western blotting of 5 μg of total starting protein. All results are plotted as means ± SEM (n = 3); *Significantly different from untreated controls by one-way ANOVA, (p < 0.05)
To further characterize the pathway by which AngII signals to CREB, we inhibited SMCs with the PKC inhibitor Bisindolylmaleimide (BIM) prior to stimulation with AngII. Results show that both CREB and MSK activation were blocked by PKC inhibition, indicating that PKC is likely upstream of these kinases (Fig. 5c). However, since BIM inhibits several PKC isoforms and other isoform-specific inhibitors are lacking, we are currently unable to distinguish which isoform of PKC mediates activation of MSK.
Discussion
Activation of CREB in SMCs by AngII was previously reported by Funakoshi et al. (Funakoshi et al. 2002). In this study, we have demonstrated that CREB activation in response to AngII stimulation promotes SMC proliferation and neointimal hyperplasia. Furthermore, we show that the kinase responsible for activating CREB in this instance is MSK, working downstream of PKC and independently of PKA or other cAMP-related signaling.
The cAMP-dependent kinase PKA stimulates Ser-133 phosphorylation of CREB in response to elevated intracellular levels of cAMP (Shaywitz and Greenberg 1999). The AngII receptor operates through both Gαq and Gαi (Higuchi et al. 2007); the latter G protein mediates a decrease in the levels of cAMP (Fig. 4c). Thus, under AngII-stimulated conditions, PKA activation should not occur, which contradicts the study by Funakoshi et al. (2002) who reported that PKA was activated in response to AngII stimulation. However, since the non-selective inhibitor H89 was used in their study, we sought to clarify this issue by comparing multiple inhibitors of PKA, including KT5720, PKI (Fig. 4a) and Rp-cAMPS (data not shown). The inhibition constant (Ki) of H89 for PKA is in the micromolar range (Chijiwa et al. 1990) and, although it does effectively inhibit PKA, it has also been shown to inhibit other kinases and to exhibit non-PKA-based activities (Murray 2008). Thus, alone, it is not the most suitable reagent for elucidating PKA function, despite its common use as such. The same may be said of KT5720, while PKI and Rp-cAMPS exhibit greater selectivity. Nevertheless, a comparative analysis of all of these compounds has confirmed that PKA is likely not involved in the pathways leading to AngII-induced CREB phosphorylation in SMCs (Fig. 4a). Consequently, inhibition of CREB activation by H89 is most likely explained by its non-PKA-selective effects. Interestingly, this includes the possibility of inhibition of the AT1R itself, since H89 has been shown to interfere with G protein receptor activation (Penn et al. 1999). It should be noted that our identification of mitogen- and stress-activated kinase (MSK) as the CREB kinase activated by AngII is also supported by the findings of Funakoshi et al. (2002), since H89 has been employed as an inhibitor of MSK as well as PKA (Khan et al. 2013).
A few studies have started to delineate the sequence of events leading from AngII to CREB. Previous studies have linked growth factor activation to MSK and CREB via ERK or p38MAPK (Arthur and Cohen 2000; Deak et al. 1998). However, these groups used tyrosine kinases and cellular stresses to stimulate CREB phosphorylation. With respect to AngII, much evidence suggests involvement of the epidermal growth factor receptor (EGFR) (Eguchi et al. 2001; Funakoshi et al. 2002; Mifune et al. 2005), and a review by Ichiki (2006) presented a model of transactivation of the EGFR by AT1R, eventually leading to MSK and CREB activation. Our study has confirmed using a highly selective inhibitor (Naqvi et al. 2012) that MSK is required for signal transduction from the AT1R to CREB and excludes PKA from this process. Likewise, the EGFR transactivation model of Ichiki (2006) also does not include PKA. However, in studies we have conducted, activation of CREB and MSK are not blocked by an EGFR inhibitor (data not shown), but by a selective Gαq inhibitor. This suggests that an entirely novel pathway is activated in this case (Fig. 6).
Fig. 6.

Schematic representation of the pathways leading to CREB activation by AngII and its contribution to vascular disease
The proposed pathway in the review of Ichiki (2006) does not include a role for PKC, which we have shown here. Rather, MSK activation was linked to ERK1/2 and p38 MAPK (Pearce et al. 2010). However, a role for PKC-dependent activation of MSK has been reported in the context of histone phosphorylation (Sutcliffe et al. 2011) and subsequent chromatin relaxation (Vermeulen et al. 2009). The group of Sutcliffe et al. (Sutcliffe et al. 2011) also found that PKC was associated with RNA polymerase II. Thus, PKC may directly associate with a chromatin-associated transcription complex that includes MSK and CREB for regulation of gene transcription.
The cellular and physiological consequences of CREB activation in SMCs remain highly contested. There are some convincing reports showing that CREB is down-regulated in response to signals of proliferation and migration, and in models of established vascular or metabolic disease (Herzig et al. 2001; Schauer et al. 2010; Watson et al. 2001), implying that CREB activity is important for normal vascular physiology. In contrast, there is much evidence that CREB is activated in vascular cells by agents associated with the development of vascular disease (Funakoshi et al. 2002; Norata et al. 2003; Ono et al. 2006; Schauer et al. 2010), especially in acute treatment settings where exposure to the agents ranged up to 60 min. While activation in these cases is temporary, there are also reports of elevated CREB after vascular injury, demonstrating an exacerbating effect of long-term, enhanced CREB activity (Tokunou et al. 2003). These authors observed that inhibiting CREB with a dominant-negative adenovirus decreased SMC proliferation, which agrees with the findings of the current study (Fig. 2c), although we obtained a 90 % inhibition of proliferation, as compared to the previously reported 40 % inhibition.
Our observations indicate that AngII is responsible for both short- and long-term CREB phosphorylation (Figs. 1 and 3, respectively), supporting the hypothesis that chronic CREB activation likely exacerbates vascular disease by promoting neointimal formation (Tokunou et al. 2003). Acute, transient activation (phosphorylation from 0 to 60 min) could be a factor relating to disease progression, where transient activation may modulate SMC phenotype, predisposing the cells to a different response in the case of subsequent chronic stimulation. Indeed, such a precedent has been set in a hypertensive cardiac model where the cellular responses to a combination of AngII and cAMP-elevating agent differed according to the activation state of cells (Sunga and Rabkin 1991).
While controversy remains as to the exact role of CREB, it has emerged as an important player in vascular disease, and its inhibition interferes with critical processes involved in vascular disease development, including inflammation and SMC luminal accumulation (Ono et al. 2006; Tokunou et al. 2003). The relevance of targeting CREB signaling for control of neointimal formation is further outlined in a study by Yang et al. (2010), who used lentiviral delivery of RNA interference against CREB binding protein (CBP) and showed inhibition of both neointimal hyperplasia and re-endothelialization in vivo. Thus, CREB signaling has the potential to be a legitimate pharmacological target, keeping in mind that short-term, localized inhibition may be the most appropriate strategy.
Smooth muscle proliferation is an essential element of the repair processes activated by mechanical injury of vascular tissue through an alteration in the SMC phenotype, and an aggressive hyperplastic response from a vessel can trigger the formation of a neointimal lesion. Whether the damage results from an intrinsic (eg. hypertension) or an extrinsic (eg. revascularization) insult, the factors that mediate this proliferative response are likely identical. We have shown here that one of the key agents responsible for neointimal hyperplasia is AngII (Moon et al. 2004), which initiates the vascular response to injury via activation of PKC, MSK and CREB.
Acknowledgments
We wish to thank Dr. Richard Goodman for generously supplying the kCREB cDNA. This investigation was supported by funds from the Canadian Institutes of Health Research.
Abbreviations
- CREB
cAMP response element-binding protein
- AngII
Angiotensin II
- VSMC/SMC
Vascular smooth muscle cells
- LAD
Left anterior descending
- DMEM
Dulbecco’s Modified Eagle Medium
- DN
Dominant negative
- kCREB
Killer CREB dominant negative mutant
- AT1R
Angiotensin type 1 receptor
- AT2R
Angiotensin type 2 receptor
- BIM
Bisindolylmaleimide
- ANOVA
Analysis of variance
- Ac-kC
pSHUTTLE-CMV construct expressing killer CREB
- β-gal
β-galactosidase
- Ad-N
pSHUTTLE-CMV control construct expressing β-galactosidase
- Ad-mt
Empty pSHUTTLE-CMV vector
- BrdU
Bromodeoxyuridine
- EGFR
Epidermal growth factor receptor
- MSK
Mitogen- and stress-activated kinase
- CBP
CREB binding protein
Footnotes
Summary
Past investigations have shown the implication of CREB in the development of vascular disease. Here, we show that AngII activation occurs upstream of transient CREB phosphorylation, thereby leading to a phenotypic switch involving MSK and PKC, but not PKA, and subsequently to smooth muscle proliferation and neointimal formation.
References
- Arthur JS, Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000;482:44–48. doi: 10.1016/S0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J Biol Chem. 1986;261:989–992. [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem. 2001;276:7957–7962. doi: 10.1074/jbc.M008570200. [DOI] [PubMed] [Google Scholar]
- Funakoshi Y, Ichiki T, Takeda K, Tokuno T, Iino N, Takeshita A. Critical role of cAMP-response element-binding protein for angiotensin II-induced hypertrophy of vascular smooth muscle cells. J Biol Chem. 2002;277:18710–18717. doi: 10.1074/jbc.M110430200. [DOI] [PubMed] [Google Scholar]
- Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S. Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 2007;112:417–428. doi: 10.1042/CS20060342. [DOI] [PubMed] [Google Scholar]
- Ichiki T. Role of cAMP response element binding protein in cardiovascular remodeling: good, bad, or both? Arterioscler Thromb Vasc Biol. 2006;26:449–455. doi: 10.1161/01.ATV.0000196747.79349.d1. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291X(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Khan P, Drobic B, Perez-Cadahia B, Healy S, He S, Davie JR. Mitogen- and stress-activated protein kinases 1 and 2 are required for maximal trefoil factor 1 induction. PLoS One. 2013;8:e63189. doi: 10.1371/journal.pone.0063189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm DJ, Watson PA, Frid MG, Dempsey EC, Schaack J, Colton LA, Nesterova A, Stenmark KR, Reusch JE. cAMP response element-binding protein content is a molecular determinant of smooth muscle cell proliferation and migration. J Biol Chem. 2001;276:46132–46141. doi: 10.1074/jbc.M104769200. [DOI] [PubMed] [Google Scholar]
- Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- Louis S, Saward L, Zahradka P. Both AT(1) and AT(2) receptors mediate proliferation and migration of porcine vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2011;301:H746–H756. doi: 10.1152/ajpheart.00431.2010. [DOI] [PubMed] [Google Scholar]
- Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- Mifune M, Ohtsu H, Suzuki H, Nakashima H, Brailoiu E, Dun NJ, Frank GD, Inagami T, Higashiyama S, Thomas WG, Eckhart AD, Dempsey PJ, Eguchi S. G protein coupling and second messenger generation are indispensable for metalloprotease-dependent, heparin-binding epidermal growth factor shedding through angiotensin II type-1 receptor. J Biol Chem. 2005;280:26592–26599. doi: 10.1074/jbc.M502906200. [DOI] [PubMed] [Google Scholar]
- Moon MC, Molnar K, Yau L, Zahradka P. Perivascular delivery of losartan with surgical fibrin glue prevents neointimal hyperplasia after arterial injury. J Vasc Surg. 2004;40:130–137. doi: 10.1016/j.jvs.2004.02.031. [DOI] [PubMed] [Google Scholar]
- Murray AJ. Pharmacological PKA inhibition: all may not be what it seems. Sci Signal. 2008;1:re4. doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- Naqvi S, Macdonald A, McCoy CE, Darragh J, Reith AD, Arthur JS. Characterization of the cellular action of the MSK inhibitor SB-747651A. Biochem J. 2012;441:347–357. doi: 10.1042/BJ20110970. [DOI] [PubMed] [Google Scholar]
- Newby AC, Zaltsman AB. Molecular mechanisms in intimal hyperplasia. J Pathol. 2000;190:300–309. doi: 10.1002/(SICI)1096-9896(200002)190:3<300::AID-PATH596>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Norata GD, Pirillo A, Callegari E, Hamsten A, Catapano AL, Eriksson P. Gene expression and intracellular pathways involved in endothelial dysfunction induced by VLDL and oxidised VLDL. Cardiovasc Res. 2003;59:169–180. doi: 10.1016/S0008-6363(03)00335-3. [DOI] [PubMed] [Google Scholar]
- Ono H, Ichiki T, Ohtsubo H, Fukuyama K, Imayama I, Iino N, Masuda S, Hashiguchi Y, Takeshita A, Sunagawa K. CAMP-response element-binding protein mediates tumor necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in endothelial cells. Hypertens Res. 2006;29:39–47. doi: 10.1291/hypres.29.39. [DOI] [PubMed] [Google Scholar]
- Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp. 2007;283:174–191. doi: 10.1002/9780470319413.ch14. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- Penn RB, Parent JL, Pronin AN, Panettieri RA, Jr, Benovic JL. Pharmacological inhibition of protein kinases in intact cells: antagonism of beta adrenergic receptor ligand binding by H-89 reveals limitations of usefulness. J Pharmacol Exp Ther. 1999;288:428–437. [PubMed] [Google Scholar]
- Saward L, Zahradka P. Angiotensin II activates phosphatidylinositol 3-kinase in vascular smooth muscle cells. Circ Res. 1997;81:249–257. doi: 10.1161/01.RES.81.2.249. [DOI] [PubMed] [Google Scholar]
- Saward L, Zahradka P. Coronary artery smooth muscle in culture: migration of heterogeneous cell populations from vessel wall. Mol Cell Biochem. 1997;176:53–59. doi: 10.1023/A:1006870827516. [DOI] [PubMed] [Google Scholar]
- Schauer IE, Knaub LA, Lloyd M, Watson PA, Gliwa C, Lewis KE, Chait A, Klemm DJ, Gunter JM, Bouchard R, McDonald TO, O’Brien KD, Reusch JE. CREB downregulation in vascular disease: a common response to cardiovascular risk. Arterioscler Thromb Vasc Biol. 2010;30:733–741. doi: 10.1161/ATVBAHA.109.199133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Sunga PS, Rabkin SW. Reversal of angiotensin II effect on the cyclic adenosine 3′,5′ monophosphate response to isoprenaline in cardiac hypertrophy. Cardiovasc Res. 1991;25:965–968. doi: 10.1093/cvr/25.11.965. [DOI] [PubMed] [Google Scholar]
- Sutcliffe EL, Bunting KL, He YQ, Li J, Phetsouphanh C, Seddiki N, Zafar A, Hindmarsh EJ, Parish CR, Kelleher AD, McInnes RL, Taya T, Milburn PJ, Rao S. Chromatin-associated protein kinase C-theta regulates an inducible gene expression program and microRNAs in human T lymphocytes. Mol Cell. 2011;41:704–719. doi: 10.1016/j.molcel.2011.02.030. [DOI] [PubMed] [Google Scholar]
- Tokunou T, Shibata R, Kai H, Ichiki T, Morisaki T, Fukuyama K, Ono H, Iino N, Masuda S, Shimokawa H, Egashira K, Imaizumi T, Takeshita A. Apoptosis induced by inhibition of cyclic AMP response element-binding protein in vascular smooth muscle cells. Circulation. 2003;108:1246–1252. doi: 10.1161/01.CIR.0000085164.13439.89. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52:639–672. [PubMed] [Google Scholar]
- Vermeulen L, Vanden Berghe W, Beck IM, De Bosscher K, Haegeman G. The versatile role of MSKs in transcriptional regulation. Trends Biochem Sci. 2009;34:311–318. doi: 10.1016/j.tibs.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Walton KM, Rehfuss RP, Chrivia JC, Lochner JE, Goodman RH. A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- Watson PA, Nesterova A, Burant CF, Klemm DJ, Reusch JE. Diabetes-related changes in cAMP response element-binding protein content enhance smooth muscle cell proliferation and migration. J Biol Chem. 2001;276:46142–46150. doi: 10.1074/jbc.M104770200. [DOI] [PubMed] [Google Scholar]
- Wilson DP, Saward L, Zahradka P, Cheung PK. Angiotensin II receptor antagonists prevent neointimal proliferation in a porcine coronary artery organ culture model. Cardiovasc Res. 1999;42:761–772. doi: 10.1016/S0008-6363(98)00340-X. [DOI] [PubMed] [Google Scholar]
- Yang J, Jiang H, Chen SS, Chen J, Li WQ, Xu SK, Wang JC. Lentivirus-mediated RNAi targeting CREB binding protein attenuates neointimal formation and promotes re-endothelialization in balloon injured rat carotid artery. Cell Physiol Biochem. 2010;26:441–448. doi: 10.1159/000320567. [DOI] [PubMed] [Google Scholar]
- Yau L, Zahradka P. PGE(2) stimulates vascular smooth muscle cell proliferation via the EP2 receptor. Mol Cell Endocrinol. 2003;203:77–90. doi: 10.1016/S0303-7207(03)00096-0. [DOI] [PubMed] [Google Scholar]
- Yau L, Litchie B, Thomas S, Storie B, Yurkova N, Zahradka P. Endogenous mono-ADP-ribosylation mediates smooth muscle cell proliferation and migration via protein kinase N-dependent induction of c-fos expression. Eur J Biochem. 2003;270:101–110. doi: 10.1046/j.1432-1033.2003.03366.x. [DOI] [PubMed] [Google Scholar]