

Abstract
14-3-3 is a family of regulatory proteins highly expressed in the brain. Previous invertebrate studies have demonstrated the importance of 14-3-3 in the regulation of synaptic functions and learning and memory. However, the in vivo role of 14-3-3 in these processes has not been determined using mammalian animal models. Here, we report the behavioral and electrophysiological characterization of a new animal model of 14-3-3 proteins. These transgenic mice, considered to be a 14-3-3 functional knock-out, express a known 14-3-3 inhibitor in various brain regions of different founder lines. We identify a founder-specific impairment in hippocampal-dependent learning and memory tasks, as well as a correlated suppression in long-term synaptic plasticity of the hippocampal synapses. Moreover, hippocampal synaptic NMDA receptor levels are selectively reduced in the transgenic founder line that exhibits both behavioral and synaptic plasticity deficits. Collectively, our findings provide evidence that 14-3-3 is a positive regulator of associative learning and memory at both the behavioral and cellular level.
Keywords: 14-3-3 proteins, fear conditioning, long-term potentiation, NMDA receptors, passive avoidance
Introduction
14-3-3 refers to a family of conserved acidic proteins consisting of seven homologous mammalian isoforms denoted β, γ, ε, η, ζ, σ, and τ. They exist as homo- and heterodimers which bind to target proteins via specific phosphoserine/phosphothreonine-containing motifs (Muslin et al., 1996; Obsil and Obsilova, 2011). To date, 14-3-3 proteins are known to interact with hundreds of proteins and participate in the regulation of a wide range of cellular processes, including signal transduction (Fantl et al., 1994; Hausser et al., 1999), cell cycle (Peng et al., 1997), transcription (Brunet et al., 1999), apoptosis, and neuronal development (Zha et al., 1996; Skoulakis et al., 1998).
14-3-3 proteins are abundantly expressed in the brain, comprising ∼1% of its total soluble proteins (Moore, 1967). Certain 14-3-3 isoforms are enriched at the synapse and implicated in the regulation of synaptic transmission and plasticity (Martin et al., 1994; Zhou et al., 1999). The first in vivo evidence was provided by studies of the mutant leonardo (leo), a gene encoding one (ζ) of the two 14-3-3 isoforms in Drosophila. These leo flies display olfactory learning deficits (Skoulakis and Davis, 1996), as well as exhibit a reduction in basal synaptic transmission and a disruption in short-term plasticity at the neuromuscular junction (Broadie et al., 1997). In the vertebrate nervous system, Simsek-Duran et al. (2004) reported that 14-3-3 is required for a presynaptic form of long-term potentiation (LTP) in the mouse cerebellum. We have also recently identified a regulatory protein complex comprised of 14-3-3 and Cav2.2 calcium channels, which may play a modulatory role in hippocampal synaptic plasticity (Li et al., 2006). As these results were obtained from in vitro studies, the importance of 14-3-3 proteins in synaptic and cognitive functions has yet to be demonstrated using appropriate mammalian animal models.
Here, we developed a new mouse model to examine the involvement of 14-3-3 proteins in learning, memory and synaptic plasticity. These mice were generated through transgenic expression of a fusion protein that inhibits the functions of the 14-3-3 family of proteins in an isoform-independent manner (Wang et al., 1999; Masters and Fu, 2001). Transgene expression is driven by the neuronal-specific Thy-1 promoter, which produces variable expression patterns in the brains of different founder lines. This approach allows us to assess the behavioral and synaptic alterations associated with expression of the 14-3-3 inhibitor in certain brain regions. Our results establish a critical role of 14-3-3 proteins in long-term synaptic plasticity and associative learning and memory.
Materials and Methods
Generation of transgenic mice.
All animal procedures were performed in accordance with the guidelines for the Care and Use of Laboratory Animals of the Florida State University (FSU), and approved by the FSU Animal Care and Use Committee. Transgenic 14-3-3 functional knock-out (FKO) mice were generated by expressing the YFP fused difopein (dimeric 14-3-3 peptide inhibitor) using the Thy-1 promoter (Fig. 1A). Transgenic founders were backcrossed to C57BL/6 mice for at least eight generations before being subjected to behavioral and electrophysiological analyses. Heterozygous transgenic mice and their WT littermates were identified by PCR genotyping using the following primers: Thy1F (AAGGGGATAAAGAGAGGGGCTGAG) from the Thy1 sequence, and difopeinR (CTCGCCGGACACGCTGAACTTG) from the difopein sequence.
Figure 1.

Transgene expressions in the brains of two different founder lines of 14-3-3 FKO mice. A, Schematic representation of the Thy1-YFP-difopein expression cassette used in this study. The gray bars represent the untranslated exons of the Thy-1.2 gene. B, C, Detection of YFP-difopein fusion proteins by fluorescence microscopy in sagittal brain sections of 132 and 142 founder line mice. D–G, Confocal images showing YFP-difopein signals in the hippocampal CA1 (132, D; 142, F) and CA3 (132, E; 142, G) subregions of 132 and 142 founder line mice. Brain sections were immunostained with a NeuN antibody to visualize neuronal nuclei (D′, E′, F′, G′). Transgene expression in NeuN positive neurons is shown by merged images (132, D” and E”; 142, F” and G”). Scale bar, 40 μm.
Histology.
Mice were anesthetized and transcardially perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4 (PBS). After an overnight postfixation in the same fixative at 4°C, the brains were cut into 40 μm sections on a vibratome (Leica Microsystems). Brain sections were mounted with Vectashield to retard fluorescence fading, and imaged on a fluorescence microscope (Eclipse FN1, Nikon) using a 4× objective. The pictures of complete sagittal brain sections were acquired as stitched images using the NIS-Elements AR software (Nikon). To evaluate the transgene expression in neurons of hippocampal subregions, brain sections were immunostained with a monoclonal anti-NeuN antibody (Millipore), followed by incubation with AlexaFluor 647 donkey anti-mouse secondary antibodies (Invitrogen). The images were attained on a Leica TCS SP2 SE laser scanning confocal microscope using a 20× objective.
PSD preparation and Western blot.
The postsynaptic density (PSD) fraction of mouse hippocampi was prepared as described previously (Hallett et al., 2008). Briefly, mouse hippocampi (100 mg) were homogenized in TEVP buffer + 320 mm sucrose solution (10 mm Tris base, 5 mm NaF, 1 mm Na3VO4, 1 mm EDTA, 1 mm EGTA, pH 7.4). Homogenates were centrifuged (800 g) to remove nuclei and large tissue debris (brain lysate fraction). The supernatant was centrifuged at 9200 × g to yield the crude synaptosomal membrane fraction (P2 pellet) and the soluble fraction. The P2 pellet was subjected to hypo-osmotic shock (TEVP buffer + 35.6 mm sucrose) and centrifuged at 25,000 × g to yield the lysed synaptosomal membrane fraction (LP pellet). The LP pellet was then resuspended in 60 μl of TEVP, separated by SDS-PAGE and probed with specific antibodies. The relative amount of GAPDH was used as a loading control for quantification. Other primary antibodies used in this experiment include monoclonal antibodies against GluK2/3 (GluR6/7, Millipore), GluN1 (NR1, Millipore), and PSD95 (Abcam), as well as polyclonal antibodies against GluA1 (GluR1, Abcam), GluA2 (GluR2, Abcam), GluN2A (NR2A, Millipore), and GluN2B (NR2B, Millipore). The Western blot signals were generated by incubating the membranes with fluorescently labeled secondary antibodies (LI-COR Biosciences), and acquired using the LI-COR Odyssey Infrared Fluorescent scanner. Band intensities on Western blots were then analyzed and quantified with ImageJ software. Protein levels were determined by normalizing the band intensities to that of the corresponding GAPDH bands.
Evaluation of cognitive behaviors.
All behavioral tests were performed using adult male heterozygous transgenic mice and their WT littermates. Mice were allowed to habituate to the dimly lit testing room for 1 h before all behavioral tests. To evaluate hippocampal-dependent learning and memory, animals were subjected to the contextual fear conditioning and passive avoidance tests.
The contextual fear conditioning test was performed over two consecutive days using the Med Associates' Contextual NIR Video Fear Conditioning System for Mouse and Video Freeze Software. For training on day 1, mice were placed in the testing chamber and their freezing behavior was recorded before (3 min), during, and after (2 min) three footshocks (2 s, 0.50 pA, separated by a 2 min intershock interval). For testing on day 2, mice were placed back into the same chamber and their freezing behavior in the first 3 min of exposure was recorded and compared.
The passive avoidance task was performed over two consecutive days using the Med Associates' Shuttle Box Avoidance Chamber for Mouse. For training on day 1, the test mouse was placed in the light chamber and their latency to the dark chamber was recorded. Upon entering the dark chamber, the automatic guillotine door was closed and mice were given a brief 2 s footshock (0.70 pA) and remained in the chamber for 1 min. For testing on day 2, mice were placed back into the light chamber and their latency to enter the dark chamber was recorded.
Slice preparation.
Acute hippocampal slices (400 μm for fEPSP recording and 250 μm for EPSC recording) were prepared from male heterozygous transgenic mice and their WT littermates (4–6 weeks old). Mice were killed after being deeply anesthetized with ketamine (100 mg/kg) and xyzaline (10 mg/kg). The whole brain was removed and placed in ice-cold and oxygenated (95% O2/5% CO2) cutting medium including the following (in mm): 230 sucrose, 2.5 KCl, 10 MgSO4, 1.25 Na2HPO4, 26 NaHCO3, 0.5 CaCl2, and 10 d-glucose. The sagittal slices were cut with VT1200S vibratome (Leica) and moved to the artificial CSF (ACSF) saturated with 95% O2/5% CO2. The ACSF contains the following (in mm): 124 NaCl, 5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1.2 KH2PO4, 26 NaHCO3, and 10 Glucose. The slices were incubated at 37°C for at least 45 min in oxygenated ACSF before use.
Field potential recording.
The mouse brain slice (400 μm) was transferred to the recording chamber and continuously perfused with oxygenated ACSF (2 ml/min) at room temperature (22 ± 1°C). fEPSPs were evoked through a concentrical bipolar stimulating electrode which was placed in the Schaffer collaterals (Sch) and recorded by the Axon MultiClamp 700B (Molecular Devices) amplifier through a glass electrode which was back-filled with 4 m NaCl and placed in the CA1 stratum radiatum. Test stimuli consisted of monophasic 100 μs pulses of constant currents with intensity adjusted to produce 50–60% of the maximum response at a frequency of 0.033 Hz. The strength of synaptic transmission was determined by measuring the initial (10–60% rising phase) slope of fEPSPs. LTP was induced by applying two trains of 50 pulses (100 Hz) separated by a 10 s interval.
Whole-cell recording.
A single slice (250 μm) in the recording chamber was kept in place with a slice anchor (Warner Instruments) under a fluorescence microscope (ECLIPSE FN1, Nikon) equipped with infrared differential interference contrast and water-immersion objective for visualization of neurons in live tissue. During recordings, the chamber was continuously superfused with ACSF (2 ml/min) and saturated with 95% O2/ 5% CO2 at room temperature. The patch electrodes (DC resistance: 5–7 MΩ) were filled with a standard intracellular solution containing the following (in mm): 145 K-gluconate, 7.5 KCl, 9 NaCl, 1 MgSO4, 10 HEPES, 0.2 EGTA, 2 Na-ATP, and 0.25 Na-GTP (290–300 mOsm, pH 7.4 with KOH). EPSCs were evoked by electrical stimulation of axons in the stratum radiatum with 100 μs pulse of constant currents with intensity adjusted to produce 50–60% of the maximum response at a frequency of 0.033 Hz. LTP was induced by two bursts of 100 Hz stimuli (10 s interval), each composed of 50 pulses. Currents were filtered at 2 kHz with a low-pass filter. Data were digitized at 10 kHz and stored online using the pClamp 10 software. After high-frequency stimulation, 360 sweeps were recorded per experiment. The neuron was rejected from statistical analysis if the membrane resistance changed >20% relative to the initial 3 min period of recordings. EPSCs were recorded at a holding potential of −70 mV. Each EPSC was normalized by the average synaptic response before high-frequency stimulation and each three consecutive traces were averaged for statistical analysis and plot.
NMDA/AMPA ratios were determined following published protocols (Schmeisser et al., 2012; Won et al., 2012). In these experiments, whole-cell recording was done in the presence of 50 μm picrotoxin. To stabilize the preparation, the ACSF contained a high concentration of divalent cations, including 4 mm CaCl2 and 4 mm MgCl2. Here, we used a cesium-based intracellular solution containing the following (in mm): 145 CsCl, 10 HEPES, 0.2 EGTA, 2 MgCl2, 2 NaATP, 0.5 NaGTP, and 5 phosphocreatine (305 mOsm, pH 7.2 with CsOH). Compound EPSCs were recorded at holding potentials of −70 and +40 mV by stimulation that was adjusted to produce a single-peaked short-latency response. Ten consecutive EPSCs for each holding potential were averaged. The AMPA receptor-mediated component of the EPSC was determined by measuring the peak amplitude of the averaged EPSC at −70 mV, whereas the NMDAR-mediated component was estimated by measuring the amplitude of the averaged EPSC with a holding potential of +40 mV at 75 ms after stimulation. To generate the input-output curves of the NMDAR component of EPSCs, whole-cell currents at +40 mV were recorded from CA1 pyramidal neurons with varying stimulus intensities. Recordings were performed in a Mg2+-free ACSF containing 50 μm CNQX and 10 μm bicuculline.
Statistical analyses.
Data are expressed as mean ± SEM with statistical significance assessed by one-way ANOVA tests for comparisons using Origin 7. The value of *p < 0.05 was considered to be a statistically significant difference.
Results
Generation of the 14-3-3 functional knock-out mice
To examine in vivo functions of 14-3-3 proteins in the nervous system, we generated a line of transgenic mice expressing the YFP-fused difopein that binds to all 14-3-3 isoforms with a very high affinity, disrupting their interactions with endogenous binding partners (Wang et al., 1999; Masters and Fu, 2001). This strategy can be considered a FKO of 14-3-3 proteins, and has been successfully applied to various cell-based assays (Cao et al., 2010). To circumvent potential lethality associated with 14-3-3 inhibition during embryonic development (Toyo-oka et al., 2003), the YFP-difopein transgene was expressed under the control of a neuronal-specific promoter, Thy-1, that normally initiates transgene expression in the perinatal period (Fig. 1A; Caroni, 1997). In addition, the Thy-1 promoter produces a variety of transgene expression patterns that are variable between different founder lines but preserved among descendants of a single founder (Feng et al., 2000). This approach may allow the assessment of 14-3-3 functions in specific brain regions.
We identified a total of nine positive transgenic founders by PCR genotyping. Western blot analyses confirmed a preferential expression of the transgene in neuronal tissues, with expression levels varying between different founders (data not shown). Further examination by fluorescence microscopy revealed that the YFP-difopein transgene was uniquely and discretely expressed in various brain regions of each founder mouse. This study focused on two of the founder lines (132 and 142) with a relatively higher level of transgene expression in their hippocampi (Fig. 1B,C). In the CA1 subregion, the 132 mice have transgene expression in a large population of pyramidal neurons, whereas the transgene expression was minimal in the 142 mice (Fig. 1D,F). However, for both 132 and 142 founder lines, the transgene is expressed only in a fraction of the CA3 neurons (Figs. 1E,G).
14-3-3 FKO mice exhibit founder-specific impairments in associative learning and memory behaviors
Previously, the Drosophila 14-3-3ζ mutants were shown to have a defect in associative learning and memory. To determine whether the 14-3-3 FKO mice exhibit associative cognitive deficits, we first conducted the contextual fear conditioning test that is functionally dependent upon the hippocampus and other forebrain structures (Kim and Jung, 2006). In this test, freezing behavior is measured as an index of learned association between a context (test chamber) and an aversive stimulus (foot shock, FS). Both 132 and 142 founder line mice displayed similar baseline freezing behavior compared with their WT littermates (Fig. 2A, Pre-FS). After training, both WT and 142 mice exhibited enhanced freezing responses, which were maintained 24 h later (Fig. 2A, Test). However, the 132 mice showed significantly less freezing behavior than WT when recorded either immediately after the footshock or 24 h later in the same context (Fig. 2A, Post-FS and Test). Because 132 and 142 mice were not different from the WT in either pain sensitivity or threshold for eliciting foot shock (data not shown), the reduction in freezing responses of 132 mice was likely due to deficits in associative learning and memory.
Figure 2.
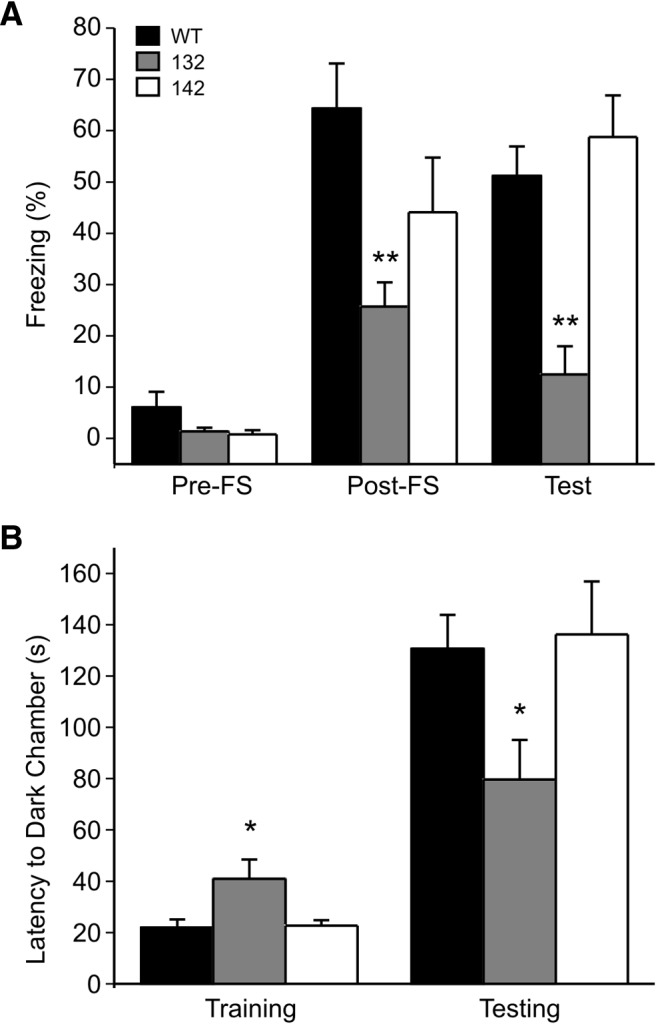
Founder-specific deficits of learning and memory behaviors in 14-3-3 FKO mice. Hippocampal-dependent associative learning and memory were assessed using contextual fear conditioning (A) and passive avoidance testing (B). A, Compared with WT, the 132 founder line mice showed significantly less freezing immediately post-FS and 24 h later when re-exposed to the same context (Test). 142 founder line mice showed no difference from the WT in the freezing responses. (WT, n = 7; 132, n = 10; 142, n = 5). B, Compared with WT, the 132 mice had a shorter latency to enter the dark chamber when tested 24 h (Testing) after footshock (Training). There was no difference between 142 mice and the WT in their latency to the dark side (WT, n = 21; 132, n = 18; 142, n = 10). All data are represented as mean ± SEM, with *p < 0.05, **p < 0.01, one-way ANOVA.
Next, we used the passive avoidance test to further characterize the cognitive abilities of the mice (Pittenger et al., 2006). This behavioral assay is another hippocampal-dependent learning and memory paradigm, which tests the ability of mice to form a negative association between the dark chamber and a footshock. Compared with WT or 142 mice, the 132 mice had a significantly shorter latency to the dark chamber when tested 24 h after training (Fig. 2B, Testing). However, the 132 mice had a longer latency to enter the dark chamber before footshock-pairing (Fig. 2B, Training), which may be indicative of neuropsychiatric changes that have also been observed in this founder line (unpublished data). Nevertheless, these results provide additional evidence that the 14-3-3 FKO mice have founder-specific impairments in associative learning and memory.
14-3-3 FKO mice display founder-specific defects in hippocampal long-term synaptic plasticity
One of the crucial cellular mechanisms for associative learning and memory is a use-dependent change in hippocampal synaptic strength, including LTP that can be induced by high-frequency stimulation. To test whether the founder-specific behavioral impairments are associated with alterations in hippocampal LTP, we performed electrophysiological recordings on acute hippocampal slices prepared from the two founder lines of the 14-3-3 FKO mice. LTP was induced by two trains of high-frequency stimulation (HFS) on the Sch-commissural projection to CA1 pyramidal cells. We first recorded field EPSPs (fEPSPs) in the stratum radium of the hippocampus and observed robust and sustained LTP in slices from both WT and 142 mice (Fig. 3A). In contrast, HFS-induced enhancement of fEPSP was markedly attenuated in the 132 mice. At 60 min post-HFS, LTP was completely eliminated in this founder line (Fig. 3A). Moreover, we measured LTP by recording whole-cell EPSCs from CA1 pyramidal neurons on mouse brain slices. Consistently, LTP of EPSCs was abolished when recorded from YFP-difopein-positive CA1 pyramidal neurons (identified by fluorescence, referred to as “132 green”) in the 132 brain slices, whereas no difference in EPSC LTP was observed between WT and 142 mice in which the transgene was minimally expressed (Figs. 1F, 3B). Together, these results reveal a founder-specific deficit in LTP of the hippocampal synapses in the 14-3-3 FKO mice. Furthermore, we found that EPSC LTP was preserved in YFP-difopein negative neurons (referred to as “132 non-green”) of the 132 mice, directly demonstrating that transgene expression in the CA1 neurons results in LTP deficit (Fig. 3B).
Figure 3.

Founder-specific impairment of hippocampal LTP in 14-3-3 FKO mice. A, HFS-induced LTP at Sch-CA1 synapses was measured by field recording in WT, 132, or 142 founder line mice. Representative fEPSP traces before (at position 1) and after (at position 2) the induction of LTP are shown on the top (WT, n = 15; 132, n = 12; 142, n = 10). B, Hippocampal LTP was assessed by whole-cell recording in CA1 pyramidal cells from WT and the two founder lines of 14-3-3 FKO mice. For 132 founder line mice, LTP of EPSCs was evaluated separately in neurons with (132 green) and without (132 non-green) fluorescence. Shown on the top are representative EPSC traces recorded from CA1 pyramidal neurons before and after LTP induction. (WT, n = 16; 132 green, n = 15; 132 non-green, n = 6; 142, n = 10).
Synaptic NMDA receptors are reduced in the CA1-expressing founder line of 14-3-3 FKO mice
We assessed basal properties of the Sch-CA1 synapses by measuring the relationship of fiber volley amplitudes (input) and fEPSP slopes (output). The input–output curve of 132 mice was not different from that of either WT or 142 mice (Fig. 4A), indicating that the LTP impairment of 132 mice does not result from an alteration in basal synaptic transmissions. After the induction of LTP, the input–output curve of WT mice showed a typical increase in fEPSP for a given stimulus intensity (Fig. 4B). Such a leftward shift of input–output curve was not observed in 132 mice (Fig. 4B), further signifying their impairments in LTP. Moreover, there was no significant difference between 132, 142 and WT mice in paired-pulse facilitation (PPF; Fig. 4C), a measure of presynaptic functions associated with the probability of neurotransmitter release. This observation is in line with the results of whole-cell recordings (Fig. 3B). It further supports transgene-induced postsynaptic changes as a potential mechanism for the founder-specific LTP impairment.
Figure 4.

Normal synaptic efficacy and paired-pulse facilitation in 14-3-3 FKO mice. A, Compared with WT, 132 and 142 founder line mice show no difference in input–output function of fEPSPs recorded in the CA1 region. (WT, black circles, n = 18; 132, gray circles, n = 12; 142, open circles, n = 9). Representative fEPSP traces with varying stimulus intensities are shown on the top. B, Assessed by input–output relationships, synaptic response was enhanced in WT, but not in 132 founder line mice after LTP induction. (WT n = 5; 132, n = 5). C, The averaged paired-pulse ratios at three interstimulus intervals are not different between WT, 132, and 142 mice. (WT, n = 17; 132, n = 9; 142, n = 7). Shown on the top are representative EPSC traces recorded from CA1 pyramidal neurons.
At the postsynaptic site, NMDA receptors (NMDARs) are one of the key mediators of LTP in the Sch-CA1 synapses (Bannerman et al., 2012). Thus, we recorded EPSCs from CA1 neurons of hippocampal slices and assessed the fractions of currents mediated by either NMDA or AMPA receptors. The NMDA/AMPA ratio in 142 mice was no different from that of the WT mice (Fig. 5A). However, 132 mice had significantly reduced NMDA/AMPA ratio in the CA1 pyramidal neurons (Fig. 5A). Considering that the AMPAR-mediated transmissions appear to be normal in 132 mice (Fig. 4A), the observed reduction in NMDA/AMPA ratio may reflect a selective decrease in NMDAR-mediated synaptic transmissions. Furthermore, we directly measured evoked NMDAR synaptic currents after blocking AMPA receptors. As illustrated by the input–output curves, the NMDAR-mediated currents are significantly smaller in transgene expressing CA1 neurons of 132 mice than that of WT controls (Fig. 5B).
Figure 5.

Founder-specific reduction of NMDA/AMPA ratio in 14-3-3 FKO mice. A, Representative traces of averaged compound EPSCs evoked at −70 and +40 mV from CA1 neurons of WT, 132, or 142 founder line mice. The ratio of synaptic NMDA to AMPA receptors is significantly less in 132 founder line than that of either WT or 142 mice. (WT, n = 11; 132, n = 14; 142, n = 6) All data are represented as mean ± SEM, with **p < 0.01, one-way ANOVA. B, Input–output curves of NMDAR-mediated EPSCs in CA1 neurons of WT and 132 founder line mice. (WT, n = 4, 132, n = 6).
To explore the molecular basis for the founder-specific deficit in NMDAR-mediated synaptic currents, we used Western blot analysis to assess the protein levels of various glutamate receptor subunits in hippocampal PSD fractions. Compared with their WT littermates, 132 mice had significantly lower levels of GluN1 and GluN2A proteins (Fig. 6A,B), which are two of the major NMDAR subunits in the adult brain. This reduction appears to be specific for NMDARs, as neither AMPA nor kainate receptors were altered in the PSDs of this founder line (Fig. 6A,B). On the other hand, 142 mice showed no difference from their WT littermates in the PSD levels of NMDA, AMPA or kainate receptor subunits (Fig. 6C,D). These results are consistent with our electrophysiological data (Fig. 5), and reveal a founder-specific reduction of synaptic NMDARs in the 14-3-3 FKO mice.
Figure 6.

Founder-specific reduction of synaptic NMDA receptors in 14-3-3 FKO mice. A, Representative Western blots of hippocampal PSD fractions from WT and 132 founder line mice. B, Protein levels of GluN1 and GluN2A subunits in hippocampal PSD fractions of 132 mice are reduced compared with that of the WT. C, Representative Western blots of hippocampal PSD fractions from 142 and WT mice. D, 142 mice show no difference in the level of examined postsynaptic proteins compared with WT. Blots were probed with GAPDH as loading control and protein levels were normalized to that of WT; *p < 0.05, **p < 0.01, one-way ANOVA; all data are presented as means ± SEM; for 132 group n = 5, for 142 group n = 4. Each sample is a combined pool of hippocampal tissue from 2–3 animals in order for the total tissue weight to equal 100 mg.
Discussion
This study provides in vivo evidence that the 14-3-3 family of proteins is a positive regulator of synaptic plasticity as well as learning and memory in mammals. We demonstrate that neuronal expression of a peptide inhibitor of 14-3-3 proteins, particularly in major subregions of the hippocampus, impairs associative learning and memory behaviors and suppresses LTP at the Sch-CA1 synapses. Interestingly, these cognitive and LTP deficits are not observed in another transgenic founder line that has more restricted transgene expressions in the hippocampus. Through comparative analyses of these two different founder lines, we further identify that postsynaptic inhibition of 14-3-3 proteins may contribute to the impairments in LTP and cognitive functions.
The importance of 14-3-3 proteins in learning, memory and synaptic functions was initially demonstrated by behavioral and electrophysiological characterizations of the 14-3-3ζ mutant fly, leonardo (Skoulakis and Davis, 1996; Broadie et al., 1997). More recently, several mammalian models of individual 14-3-3 isoforms have been created using standard genetic approaches, including knock-out mice with deletion of the 14-3-3γ, -ζ, or -ε gene (Toyo-oka et al., 2003; Steinacker et al., 2005; Cheah et al., 2012). Due to the presence of multiple functionally redundant 14-3-3 isoforms in the brain, knock-out or deficiency of a single isoform may not be sufficient to reveal a potentially critical role for the 14-3-3 family of proteins in regulating synaptic plasticity and cognitive behaviors. By transgenically expressing the isoform-nonspecific 14-3-3 inhibitor, we have generated a new mouse model that genetically manipulates the function of 14-3-3 proteins as a whole. The utility of this animal model is demonstrated by the observed behavioral and electrophysiological alterations in a certain founder line. In the future, this strategy may be combined with more advanced genetic techniques to further define the role of 14-3-3-dependent regulatory pathways in a spatially and temporally controlled manner.
14-3-3 proteins have been previously implicated as important regulators of presynaptic functions. Based on a study conducted in cultured mouse cerebellar neurons, 14-3-3 is required for a presynaptic form of LTP in parallel fiber synapses through its binding to phosphorylated RIM1α, a scaffolding protein at the presynaptic active zone (Simsek-Duran et al., 2004). However, the in vivo role of this molecular pathway has not been supported by recent genetic studies from two independent groups. Using either a knock-in or an acute genetic rescue approach, they demonstrate that phosphorylation of RIM1α at the putative 14-3-3 binding motif (serine-413) is not essential for presynaptic LTP or learning in mice (Kaeser et al., 2008; Yang and Calakos, 2010). In this report, we find that PPF in hippocampal Sch-CA1 synapses is not altered in the two different 14-3-3 FKO founder lines. However, there is limited colocalization of the YFP-difopein transgene with a presynaptic marker synaptophysin in the CA1 stratum radiatum (data not shown), which is consistent with the observation that the 14-3-3 peptide inhibitor is expressed only in a small population of hippocampal CA3 neurons for both 132 and 142 founder line mice (Fig. 1E,G). Thus, it is possible that the transgene expression within the presynaptic neurons is not extensive enough to exert a significant effect in the CA3-CA1 synapses of the 14-3-3 FKO mice. Nevertheless, we identify a founder-specific deficit in hippocampal LTP, which correlates with the expression of the 14-3-3 inhibitor in postsynaptic (CA1) neurons (Fig. 3B). These observations thus reveal a novel postsynaptic function for 14-3-3 proteins in regulating long-term synaptic plasticity in the hippocampus.
Consistent with the proposed function of 14-3-3 at the postsynaptic site, we find a reduction of NMDA receptor subunits in the PSD fraction of the 14-3-3 FKO founder line mice with transgene expression in CA1 neurons. This finding is in line with the established role of NMDA receptors in long-term synaptic plasticity and cognitive behaviors (Morris et al., 1986; Tsien et al., 1996; Martin et al., 2000; Bannerman et al., 2012), and provides a potential molecular mechanism for 14-3-3-dependent regulation of postsynaptic functions. Previously, Chen and Roche, (2009) demonstrated that 14-3-3-ε promotes surface expression of NMDA receptors in cerebellar neurons through its binding to PKB-phosphorylated GluN2C subunits. Thus, future studies should be directed to examine whether 14-3-3 proteins have similar effects on the surface expression of GluN1 and GluN2A subunits. Because 14-3-3 interacts with and modulates numerous target proteins which contain specific phosphoserine/phosphothreonine motifs, it is also possible that 14-3-3 might enhance synaptic targeting of NMDA receptors at other critical steps, especially those known to be regulated by protein phosphorylation (Shirao and Sekino, 2001; Groc and Choquet, 2006; Lau and Zukin, 2007). Interestingly, a recent study showed that synaptic levels of 14-3-3-ε are reduced in GluN1 knockdown mice, but not by subchronic administration of an NMDAR antagonist in WT mice (Ramsey et al., 2011). Together with our finding that inhibition of 14-3-3 decreases synaptic NMDAR levels, there is likely a reciprocal regulation between 14-3-3 and NMDARs at the postsynaptic site.
As a family of brain-rich regulatory proteins, dysfunction of 14-3-3 has been linked to a variety of neurological and psychiatric disorders with cognitive impairments (Berg et al., 2003; Foote and Zhou, 2012). Particularly, genetic and postmortem analyses have identified certain 14-3-3 isoforms as candidate risk genes for schizophrenia (Toyooka et al., 1999; Vawter et al., 2001; Wong et al., 2003, 2005; Ikeda et al., 2008). Additionally, a reduction of synaptic 14-3-3-τ in the hippocampus has been observed in mice with age-related cognitive decline (VanGuilder et al., 2011). These findings are consistent with our results that 14-3-3 plays a positive role in learning, memory and synaptic plasticity. Therefore, it is important to further elucidate the molecular mechanisms of 14-3-3-dependent regulation of synaptic functions, which may shed light on the underlying pathological processes of cognitive deficits associated with aging and neuropsychiatric diseases.
Footnotes
This work was supported by National Institute of Health Grant NS50355 to Y.Z. and National Natural Science Foundation of China grant 81273831 to H.Q. We thank Ruth Didier of the Confocal Microscopy Core laboratory at the FSU Biomedical Sciences Department for her assistance with imaging.
The authors declare no competing financial interests.
References
- Bannerman DM, Bus T, Taylor A, Sanderson DJ, Schwarz I, Jensen V, Hvalby Ø, Rawlins JN, Seeburg PH, Sprengel R. Dissecting spatial knowledge from spatial choice by hippocampal NMDA receptor deletion. Nat Neurosci. 2012;15:1153–1159. doi: 10.1038/nn.3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- Broadie K, Rushton E, Skoulakis EM, Davis RL. Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron. 1997;19:391–402. doi: 10.1016/S0896-6273(00)80948-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/S0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Cao W, Yang X, Zhou J, Teng Z, Cao L, Zhang X, Fei Z. Targeting 14-3-3 protein, difopein induces apoptosis of human glioma cells and suppresses tumor growth in mice. Apoptosis. 2010;15:230–241. doi: 10.1007/s10495-009-0437-4. [DOI] [PubMed] [Google Scholar]
- Caroni P. Overexpression of growth-associated proteins in the neurons of adult transgenic mice. J Neurosci Methods. 1997;71:3–9. doi: 10.1016/S0165-0270(96)00121-5. [DOI] [PubMed] [Google Scholar]
- Cheah PS, Ramshaw HS, Thomas PQ, Toyo-Oka K, Xu X, Martin S, Coyle P, Guthridge MA, Stomski F, van den Buuse M, Wynshaw-Boris A, Lopez AF, Schwarz QP. Neurodevelopmental and neuropsychiatric behaviour defects arise from 14-3-3zeta deficiency. Mol Psychiatry. 2012;17:451–466. doi: 10.1038/mp.2011.158. [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW. Growth factor-dependent trafficking of cerebellar NMDA receptors via protein kinase B/Akt phosphorylation of NR2C. Neuron. 2009;62:471–478. doi: 10.1016/j.neuron.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantl WJ, Muslin AJ, Kikuchi A, Martin JA, MacNicol AM, Gross RW, Williams LT. Activation of Raf-1 by 14-3-3 proteins. Nature. 1994;371:612–614. doi: 10.1038/371612a0. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Foote M, Zhou Y. 14-3-3 proteins in neurological disorders. Int J Biochem Mol Biol. 2012;3:152–164. [PMC free article] [PubMed] [Google Scholar]
- Groc L, Choquet D. AMPA and NMDA glutamate receptor trafficking: multiple roads for reaching and leaving the synapse. Cell Tissue Res. 2006;326:423–438. doi: 10.1007/s00441-006-0254-9. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Collins TL, Standaert DG, Dunah AW. Biochemical fractionation of brain tissue for studies of receptor distribution and trafficking. Curr Protoc Neurosci. 2008;42:1.16l1–1.16.16. doi: 10.1002/0471142301.ns0116s42. [DOI] [PubMed] [Google Scholar]
- Hausser A, Storz P, Link G, Stoll H, Liu YC, Altman A, Pfizenmaier K, Johannes FJ. Protein kinase C mu is negatively regulated by 14-3-3 signal transduction proteins. J Biol Chem. 1999;274:9258–9264. doi: 10.1074/jbc.274.14.9258. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Hikita T, Taya S, Uraguchi-Asaki J, Toyo-oka K, Wynshaw-Boris A, Ujike H, Inada T, Takao K, Miyakawa T, Ozaki N, Kaibuchi K, Iwata N. Identification of YWHAE, a gene encoding 14-3-3epsilon, as a possible susceptibility gene for schizophrenia. Hum Mol Genet. 2008;17:3212–3222. doi: 10.1093/hmg/ddn217. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Kwon HB, Blundell J, Chevaleyre V, Morishita W, Malenka RC, Powell CM, Castillo PE, Südhof TC. RIM1alpha phosphorylation at serine-413 by protein kinase A is not required for presynaptic long-term plasticity or learning. Proc Natl Acad Sci U S A. 2008;105:14680–14685. doi: 10.1073/pnas.0806679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev. 2006;30:188–202. doi: 10.1016/j.neubiorev.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Li Y, Wu Y, Zhou Y. Modulation of inactivation properties of CaV2.2 channels by 14-3-3 proteins. Neuron. 2006;51:755–771. doi: 10.1016/j.neuron.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Martin H, Rostas J, Patel Y, Aitken A. Subcellular localisation of 14-3-3 isoforms in rat brain using specific antibodies. J Neurochem. 1994;63:2259–2265. doi: 10.1046/j.1471-4159.1994.63062259.x. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Ann Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Masters SC, Fu H. 14-3-3 proteins mediate an essential anti-apoptotic signal. J Biol Chem. 2001;276:45193–45200. doi: 10.1074/jbc.M105971200. [DOI] [PubMed] [Google Scholar]
- Moore BEP, VJ . Specific acidic proteins of the nervous system. In: Carlson FD, editor. Physiological and biochemical aspects of nervous integration. Englewood Cliffs, NJ: Prentice Hall; 1967. pp. 343–359. [Google Scholar]
- Morris RG, Anderson E, Lynch GS, Baudry M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature. 1986;319:774–776. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Tanner JW, Allen PM, Shaw AS. Interaction of 14-3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell. 1996;84:889–897. doi: 10.1016/S0092-8674(00)81067-3. [DOI] [PubMed] [Google Scholar]
- Obsil T, Obsilova V. Structural basis of 14-3-3 protein functions. Semin Cell Dev Biol. 2011;22:663–672. doi: 10.1016/j.semcdb.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science. 1997;277:1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Fasano S, Mazzocchi-Jones D, Dunnett SB, Kandel ER, Brambilla R. Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J Neurosci. 2006;26:2808–2813. doi: 10.1523/JNEUROSCI.5406-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey AJ, Milenkovic M, Oliveira AF, Escobedo-Lozoya Y, Seshadri S, Salahpour A, Sawa A, Yasuda R, Caron MG. Impaired NMDA receptor transmission alters striatal synapses and DISC1 protein in an age-dependent manner. Proc Natl Acad Sci U S A. 2011;108:5795–5800. doi: 10.1073/pnas.1012621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ, Ey E, Wegener S, Bockmann J, Stempel AV, Kuebler A, Janssen AL, Udvardi PT, Shiban E, Spilker C, Balschun D, Skryabin BV, Dieck St, Smalla KH, Montag D, Leblond CS, Faure P, Torquet N, Le Sourd AM, Toro R, et al. Autistic-like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature. 2012;486:256–260. doi: 10.1038/nature11015. [DOI] [PubMed] [Google Scholar]
- Shirao T, Sekino Y. Clustering and anchoring mechanisms of molecular constituents of postsynaptic scaffolds in dendritic spines. Neurosci Res. 2001;40:1–7. doi: 10.1016/S0168-0102(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Simsek-Duran F, Linden DJ, Lonart G. Adapter protein 14-3-3 is required for a presynaptic form of LTP in the cerebellum. Nat Neurosci. 2004;7:1296–1298. doi: 10.1038/nn1348. [DOI] [PubMed] [Google Scholar]
- Skoulakis EM, Davis RL. Olfactory learning deficits in mutants for leonardo, a Drosophila gene encoding a 14-3-3 protein. Neuron. 1996;17:931–944. doi: 10.1016/S0896-6273(00)80224-X. [DOI] [PubMed] [Google Scholar]
- Skoulakis EM, Davis RL. 14-3-3 proteins in neuronal development and function. Molecular neurobiology. 1998;16:269–284. doi: 10.1007/BF02741386. [DOI] [PubMed] [Google Scholar]
- Steinacker P, Schwarz P, Reim K, Brechlin P, Jahn O, Kratzin H, Aitken A, Wiltfang J, Aguzzi A, Bahn E, Baxter HC, Brose N, Otto M. Unchanged survival rates of 14-3-3gamma knock-out mice after inoculation with pathological prion protein. Mol Cell Biol. 2005;25:1339–1346. doi: 10.1128/MCB.25.4.1339-1346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka K, Muratake T, Tanaka T, Igarashi S, Watanabe H, Takeuchi H, Hayashi S, Maeda M, Takahashi M, Tsuji S, Kumanishi T, Takahashi Y. 14-3-3 protein η chain gene (YWHAH) polymorphism and its genetic association with schizophrenia. Am J Med Genet. 1999;88:164–167. doi: 10.1002/(SICI)1096-8628(19990416)88:2<164::AID-AJMG13>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3ϵ is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller–Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/S0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- VanGuilder HD, Farley JA, Yan H, Van Kirk CA, Mitschelen M, Sonntag WE, Freeman WM. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol Dis. 2011;43:201–212. doi: 10.1016/j.nbd.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood WH, 3rd, Donovan DM, Webster M, Freed WJ, Becker KG. Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res Bull. 2001;55:641–650. doi: 10.1016/S0361-9230(01)00522-6. [DOI] [PubMed] [Google Scholar]
- Wang B, Yang H, Liu YC, Jelinek T, Zhang L, Ruoslahti E, Fu H. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry. 1999;38:12499–12504. doi: 10.1021/bi991353h. [DOI] [PubMed] [Google Scholar]
- Won H, Lee HR, Gee HY, Mah W, Kim JI, Lee J, Ha S, Chung C, Jung ES, Cho YS, Park SG, Lee JS, Lee K, Kim D, Bae YC, Kaang BK, Lee MG, Kim E. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature. 2012;486:261–265. doi: 10.1038/nature11208. [DOI] [PubMed] [Google Scholar]
- Wong AH, Macciardi F, Klempan T, Kawczynski W, Barr CL, Lakatoo S, Wong M, Buckle C, Trakalo J, Boffa E, Oak J, Azevedo MH, Dourado A, Coelho I, Macedo A, Vicente A, Valente J, Ferreira CP, Pato MT, Pato CN, et al. Identification of candidate genes for psychosis in rat models, and possible association between schizophrenia and the 14-3-3eta gene. Mol Psychiatry. 2003;8:156–166. doi: 10.1038/sj.mp.4001237. [DOI] [PubMed] [Google Scholar]
- Wong AH, Likhodi O, Trakalo J, Yusuf M, Sinha A, Pato CN, Pato MT, Van Tol HH, Kennedy JL. Genetic and post-mortem mRNA analysis of the 14-3-3 genes that encode phosphoserine/threonine-binding regulatory proteins in schizophrenia and bipolar disorder. Schizophr Res. 2005;78:137–146. doi: 10.1016/j.schres.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Acute in vivo genetic rescue demonstrates that phosphorylation of RIM1alpha serine 413 is not required for mossy fiber long-term potentiation. J Neurosci. 2010;30:2542–2546. doi: 10.1523/JNEUROSCI.4285-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/S0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Schopperle WM, Murrey H, Jaramillo A, Dagan D, Griffith LC, Levitan IB. A dynamically regulated 14-3-3, slob, and slowpoke potassium channel complex in Drosophila presynaptic nerve terminals. Neuron. 1999;22:809–818. doi: 10.1016/S0896-6273(00)80739-4. [DOI] [PubMed] [Google Scholar]