

Abstract
Blood flow increases to exercising skeletal muscle, and this increase is driven primarily by vasodilation in the contracting muscles. When oxygen delivery to the contracting muscles is altered by changes in arterial oxygen content, the magnitude of the vasodilator response to exercise changes. It is augmented during hypoxia and blunted during hyperoxia. Because the magnitude of the increased vasodilation during hypoxic exercise tends to keep oxygen delivery to the contracting muscles constant, we have termed this phenomenon “compensatory vasodilation.” In a series of studies, we have explored metabolic, endothelial, and neural mechanisms that might contribute to compensatory vasodilation. These include the contribution of vasodilating substances like nitric oxide (NO) and adenosine, along with altered interactions between sympathetic vasoconstriction and metabolic vasodilation. We have also compared the compensatory vasodilator responses to hypoxic exercise with those seen when oxygen delivery to contracting muscles is altered by acute reductions in perfusion pressure. A synthesis of our findings indicate that NO contributes to the compensatory dilator responses during both hypoxia and hypoperfusion, while adenosine appears to contribute only during hypoperfusion. During hypoxia, the NO-mediated component is linked to a β-adrenergic receptor mechanism during lower intensity exercise, while another source of NO is engaged at higher exercise intensities. There are also subtle interactions between α-adrenergic vasoconstriction and metabolic vasodilation that influence the responses to hypoxia, hyperoxia, and hypoperfusion. Together our findings emphasize both the tight linkage of oxygen demand and supply during exercise and the redundant nature of the vasomotor responses to contraction.
Keywords: hypoxia, vasodilation, exercise, oxygen
this review is broadly about the phenomenon we have termed “compensatory vasodilation” and is based on a presentation given at the 18th International Hypoxia Symposia held in early 2013. This phenomenon describes the tendency of muscle blood flow to increase (or decrease), depending on changes in arterial oxygen content. This is especially true during exercise, when the ability to maintain oxygen consumption by changes in extraction alone is somewhat limited, because extraction is already high during exercise (1). The basic idea is that, when arterial oxygen content is reduced by hypoxia, anemia, or other reductions of functional oxygen-carrying capacity (carbon monoxide, for example), there is an increase in blood flow to active muscles that “compensates” for the reduced arterial oxygen content and keeps oxygen delivery to the active muscles relatively constant. It should also be noted that the opposite occurs during hyperoxia, when there are increases in arterial oxygen content and vasodilation is reduced. In this paper, we briefly review our data and data from others and discuss potential mechanisms responsible for these observations. Figure 1 is a classic slide adapted from Rowell and colleagues (39) showing this phenomenon.
Fig. 1.

Muscle vascular conductance (i.e., vasodilation) responses to dynamic single-leg knee-extensor exercise during either normoxic or hypoxic (∼80% saturation) conditions in young men. Arrows indicate the magnitude of compensatory vasodilation during hypoxic exercise relative to normoxic conditions at a given workload. FiO2, fraction of inspired O2. [Adapted from Fig. 7 in Ref. 34.]
Our interest in this topic stems from our general interest in exercise hyperemia. In this context, there appears to be redundant mechanisms that contribute to exercise hyperemia, including various “metabolic” vasodilators (28). The idea is that vasodilating substances released by the contracting muscles or other tissues in proportion to the level of contractile activity are responsible for the well-known matching of blood flow and metabolism. However, identifying the dominant metabolic dilating substance (if there is one) or substances has proven challenging, and we hoped that, by altering oxygen delivery to the active muscle, we might either amplify or suppress one or more of the major contributors to this response.
HOW HAVE WE STUDIED THIS IN HUMANS?
Our basic strategy to study compensatory vasodilation in humans has used the isolated forearm handgripping model, which involves a handgrip device that allows subjects to perform rhythmic forearm exercise by lifting a given weight 4–5 cm over a pulley 20 times/min. This model has a number of advantages: the small muscle mass involved means that we can study vasodilator responses and give drugs in a way that does not affect the systemic circulation, and it is possible to either blunt or eliminate the effects of various reflex changes in autonomic outflow on the responses we seek to study. We can also use high doses of drugs to block vasodilating or vasoconstricting pathways. However, the handgripping model can also be criticized because clearly the forearm is not a major locomotor muscle, and cardiovascular reflexes and competition between the demands of skeletal muscle for blood flow, blood pressure regulation, and blood flow to other organs are clearly present during whole body exercise in real-life situations.
HOW HAVE WE ALTERED ARTERIAL OXYGEN CONTENT?
We have used three basic strategies to alter arterial oxygen content to the exercising muscles we have studied. First is simple systemic hypoxia. For these series of experiments, we used a self-regulating partial rebreathe system to lower arterial oxygen saturation to ∼80%, while clamping end-tidal CO2 at baseline levels, despite large changes in minute ventilation that occur with hypoxia (3, 11–13, 45, 46). This is accomplished by titrating the level of inspired oxygen to achieve the desired arterial oxygen saturation. The amount of inspired oxygen provided in the inspiratory gas was controlled by mixing nitrogen with medical air via an anesthesia gas blender.
Another strategy we have used to alter arterial oxygen content is systemic hyperbaric hyperoxia (9, 10). Traditionally, studies that have examined the impact of increased levels of oxygen on blood flow during dynamic exercise have been limited to small increases in arterial oxygen content (∼5–10%) using 100% inspired O2 under normobaric conditions (16, 18, 23, 44). Since our hypoxia studies have typically reduced arterial oxygen content by ∼20%, we have used several atmospheres of pressure to increase arterial oxygen content by ∼20%. This has permitted us to study the relationship between arterial oxygen content and blood flow “in both directions.”
Our final strategy to manipulate arterial oxygen content has been local hypoperfusion. In this technique, we place a balloon in the brachial artery and inflate the balloon to partially occlude it (5–8). Using this technique, we can acutely reduce perfusion pressure to the contracting skeletal muscles of the forearm by 15–20% and forearm blood flow by 40–50%. We have not used anemia or carbon monoxide in our studies.
HYPOXIA IS SYMPATHOEXCITATORY
The first thing to remember about hypoxia is that it is sympathoexcitatory. This sympathetic activation is due to stimulation of the carotid body chemoreceptors, and there is a 50–60% rise in muscle sympathetic nerve activity during systemic hypoxia (25). Normally, this increase in sympathetic traffic would potentially compete with or limit any local vasodilation caused by the hypoxia, and at rest this is clearly the case (43, 45). In other words, the vasodilation seen with hypoxia is greater after α-adrenergic blockade of the forearm than with hypoxia alone. As demonstrated in Fig. 2, there also appears to be enhanced sympathetic vasoconstrictor restraint of the skeletal muscle vasculature during hypoxic exercise, as α-adrenergic blockade reveals a greater vasodilation during hypoxic exercise compared with control hypoxic exercise conditions (no pharmacological blockade) (45). Thus while the ability of norepinephrine released from sympathetic nerves to cause vasoconstriction in active skeletal muscle is blunted during exercise, the additional sympathetic outflow evoked by systemic hypoxia can still cause vasoconstriction in the active muscles.
Fig. 2.

Blockade of α-adrenergic receptors via phentolamine reveals a substantial hypoxic (H) vasodilation [change (Δ) in forearm vascular conductance (FVC)] relative to respective normoxic (N) condition at rest and during rhythmic forearm contractions performed at a duty cycle of 1-s contraction and 2-s relaxation (20 contractions/min). Nos. within each bar indicate the magnitude of ΔFVC during α-adrenergic blockade compared with control (saline) conditions. MVC, maximal voluntary contraction.
So our first question was to determine whether functional sympatholysis was augmented during exercise under hypoxic conditions. In this study, we used tyramine to cause local release of norepinephrine from the sympathetic nerves in the forearm. We showed that exercise clearly caused functional sympatholysis, and that functional sympatholysis (compared with rest) was greater during rhythmic handgripping at 10 and 20% of maximum voluntary contraction (46). However, compared with what is observed during normoxic exercise, hypoxia did not augment sympatholysis. So the classic compensatory vasodilation seen during hypoxia could not be ascribed simply to augmented sympatholysis during hypoxia: clearly something else, perhaps related to a “metabolic signal” from the active muscles or circulating factor, drives the compensatory dilator response.
CANDIDATE DILATORS AND COMPENSATORY VASODILATION
Along these lines, in our hypoxia studies, we have focused on the role of β-adrenergic receptors, adenosine, and nitric oxide (NO) acting alone or in combination as potential candidate dilators, which might contribute to compensatory vasodilation during hypoxia. During mild rhythmic handgripping (10% of maximum voluntary contraction), there is evidence that β-adrenergic receptor stimulation plays a role in compensatory vasodilation. However, this role is not as prominent during heavier rhythmic handgripping (20% maximum voluntary contraction) (45). It is also likely that some or all of this β2-mediated vasodilation is due to stimulation of NO release from the vascular endothelium (3).
We have also studied the interactions of adenosine and NO in a complex series of experiments that have blocked both pathways alone and in combination. Based on our data, we have concluded that NO plays a major role (∼50% or more) in compensatory vasodilation, and that adenosine is not obligatory for this response (11, 12). Our findings that adenosine is not obligatory for the compensatory vasodilation in the forearm is in agreement with what is observed in exercising human quadriceps femoris muscle during hypoxia (27). However, it should be noted that, under resting conditions, adenosine plays a major role in skeletal muscle vasodilation in experimental animals when oxygen is reduced by acute systemic hypoxia, and a substantial amount of the response is mediated by NO (19, 33, 34). Recent evidence from others has demonstrated that the combined blockade of NO and prostaglandin pathways also reduces the vasodilator response to hypoxic exercise by ∼50% (17). However, single inhibition trials of NO and prostaglandins were not performed in the aforementioned study, and, therefore, it is difficult to discern the relative contribution of prostaglandins and its interaction with NO in the compensatory vasodilation during hypoxic exercise. As is the case for both β2-adrenergic receptor- and adenosine-mediated vasodilation, at least some of the vasodilator response to prostaglandins is NO mediated (37).
There are a variety of other pathways that might contribute to compensatory vasodilation, including direct effects of hypoxia on vascular smooth muscle and ATP release from red blood cells as they desaturate. This latter pathway has emerged as an exciting new mechanism to help match blood flow, metabolism, and oxygen delivery in contracting skeletal muscles (20). However, in some models, ATP-mediated dilation is due in part to stimulation of NO release from the vascular endothelium (14, 35, 36). There has also been speculation about NO release from hemoglobin as a contributor to vasodilation during hypoxia (42), but at least in our model the dramatic effects of a local blockade of NO synthesis by selective infusions of NO synthase inhibitors into the forearm would tend to argue against a systemic mechanism centered on NO release from circulating hemoglobin as predominant. In both the case of ATP and NO release from desaturating hemoglobin, there are parallels to historic ideas about either oxygen or carbon dioxide per se playing a role in exercise hyperemia (41). Additionally, neuronal NO synthase (NOS)-derived NO plays a significant role in the regulation of skeletal muscle vascular tone at rest in humans (40) and in low-oxidative muscles of rats during high-intensity treadmill running (15). Therefore, it is possible that neuronal NOS-derived NO might be another contributor to compensatory vasodilation during hypoxic exercise. Lastly, accumulating evidence suggests that nitrite reduction (i.e., nitrate-nitrite-NO pathway) represents an alternative and differentially regulated system for NO generation that operates in parallel to the classic l-arginine-NOS pathway (32). Interestingly, the nitrate-nitrite-NO pathway is greatly enhanced under hypoxic conditions (29). Moreover, recent evidence in experimental animals suggests that dietary nitrate supplementation increases skeletal muscle blood flow and vasodilation during exercise (21). Taken together, these findings might suggest that the nitrate-nitrite NO pathway potentially can contribute to compensatory vasodilation. Lastly, other hypotheses and models have been proposed to explain the regulation of local blood flow under various levels of oxygen availability. For a provocative review of an alternative mechanism that may contribute to hyperemia within skeletal muscle and the compensatory vasodilation during hypoxic exercise, please see Ref. 22.
Based on our previous data demonstrating that β-adrenergic receptor activation is responsible for a portion of hypoxic vasodilation (45) and evidence supporting the idea that β-adrenergic receptors are either more sensitive or upregulated in young women compared with men (26, 31), we recently examined whether hypoxic vasodilation is influenced by sex. Our retrospective analysis of the blood flow and vasodilator responses during hypoxic exercise revealed that young women demonstrated a greater compensatory vasodilation compared with young men (12a). The mechanisms responsible for the discrepancies between sexes are currently unclear. Compensatory dilation is also blunted with aging (13). In this context, a loss of NO-mediated endothelial dilator responses with aging likely explains these observations. Additionally, the ATP mechanisms mentioned above are also blunted by aging (30), and, unlike younger subjects, there do not appear to be major sex differences in the compensatory dilator responses seen in older men and women.
WHAT ABOUT HYPEROXIA?
We have used hyperbaric hyperoxia (100% oxygen at 2.82 ATA) and also hyperbaric normoxia (7.4% oxygen at 2.82 ATA) to study the effects of increased oxygen content on blood flow responses to contracting muscle (9). The studies conducted at 7.4% fraction of inspired O2 served as an atmospheric pressure control trial and are done to control for the effects of chamber pressurization on any observed responses. The fundamental observations with this approach are that, when arterial oxygen content is increased by an estimated 25%, the blood flow responses are reduced by a similar amount. In other words, the opposite directional changes are seen compared with hypoxia. Additionally, this reduction in flow during hyperoxia is not due to a loss of functional sympatholysis and an augmented sympathetic vasoconstrictor response in the active skeletal muscles (10). The extent to which hyperbaric hyperoxia affects other potential vasodilator pathways, such as NO, adenine nucleotides, or other factors, is currently under investigation.
HYPOPERFUSION
When we use our balloon catheter system to reduce perfusion pressure in the contracting forearm, we see a brief reduction in flow, followed by local vasodilation, which tends to return flow to normal (8). Interestingly, the compensatory response appears to be “less than perfect” (4–8). That is, in the majority of the studies we have conducted, flow does not return completely (100% recovery) to preinflation levels. Figure 3 is an individual record of such a response. During a series of studies aimed at identifying the potential vasodilators involved in the partial restoration of blood flow in the hypoperfused muscles, we have shown that most of the compensatory vasodilation is due to either NO or adenosine acting alone or in combination with NO (5, 6). In this context, 1) the magnitude of compensatory vasodilation is reduced by both single inhibition of NOS or adenosine receptor antagonism; and 2) the combined inhibition of the two pathways results in additive reduction in the compensatory response (5). At some level, these responses are an important confirmation of the so-called “adenosine hypothesis” postulated in the 1960s. This hypothesis, primarily directed at coronary circulation, suggested that, when there was a mismatch between flow and metabolism, which caused local tissue hypoxia, adenosine released from hypoperfused muscle as a metabolic “error signal” might be a critical mediator of vasodilator responses (2). It is of interest to note that there was discussion of adenosine, ADP, and ATP as potential candidate dilators during the initial wave of interest in these mechanisms as part of the adenosine hypothesis (41).
Fig. 3.

Representative responses to forearm exercise with balloon-induced hypoperfusion under control (no drug; ●) and nitric oxide synthase (NOS) inhibition [NG-monomethyl-l-arginine (l-NMMA); ○] conditions. Circles at baseline (rest) represent 5-s FVC averages, whereas circles during exercise (20% MVC) represent FVC averages per duty cycle (1-s contraction and 2-s relaxation period). The vasodilation and related recovery of flow following balloon inflation were blunted by NOS inhibition.
In this context, it is interesting to note that adenosine does not appear to be an important contributor to responses seen during systemic hypoxia in humans (12, 27), but is an important contributor seen during hypoperfusion. We should also note that prostaglandins do not appear to play a major role during the local responses to hypoperfusion (7). Lastly, similar to systemic hypoxia, α-adrenergic-mediated vasoconstriction limits the amount of compensatory vasodilation during exercise with hypoperfusion (4). Along these lines, α-adrenergic blockade via phentolamine enhances the recovery of flow and vasodilation in hypoperfused contracting muscle.
SUMMARY AND FUTURE DIRECTIONS
Blood flow to contracting skeletal muscles has a remarkable ability to adapt to changes in arterial oxygen content. Our studies have shown this both with systemic hypoxia and hyperoxia, along with skeletal muscle hypoperfusion. Others have shown similar responses during anemia and systemic carbon monoxide administration (24, 38).
During both hypoxia and hyperoxia, alterations in the way that the sympathetic nervous system interacts with local vasodilator mechanisms (functional sympatholysis) probably do not play a major role in modulating these responses. During systemic hypoxia, there can be release of epinephrine, and this epinephrine release and increase in circulating epinephrine could in fact contribute to the compensatory vasodilator response.
At this time, NO appears to be a major contributor to the compensatory dilator responses seen during both hypoxia and hypoperfusion, but it is unclear whether this NO release is due to direct stimulation of the vascular endothelium, NO release from the active skeletal muscles, potentiation of a non-NOS NO pathway, release of some mediator (ATP?) that then evokes NO release from the vascular endothelium, or perhaps a direct effect of hypoxia on vascular smooth muscle that then stimulates flow-induced NO release. The contribution and interactions between potential vasodilator substances are illustrated in Figure 4. These responses appear to be augmented in healthy young women. Our studies in healthy older humans demonstrate that compensatory vasodilation during hypoxic exercise is blunted with aging in both sexes and likely due to a loss of NO-mediated endothelial vasodilator function. In this context, there may be further reductions in compensatory vasodilator responses with pathophysiological conditions such as hypertension and diabetes. If these responses also occurred in other vascular beds, then the consequences of tissue hypoxia or hypoperfusion might be even more problematic in disease states. As always for both exercise hyperemia and NO, it could be “some of the above” or “all of the above,” when thinking about vasodilator mechanisms and exercise. The fact that differences in dilator mechanisms are seen when different approaches are used to alter oxygen delivery to contracting muscles is not surprising to us, given the highly redundant regulatory control mechanisms associated with exercise hyperemia.
Fig. 4.
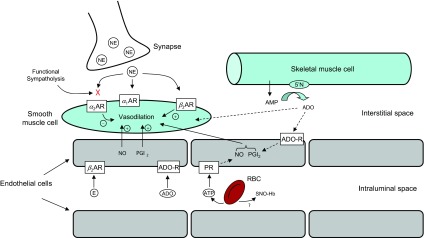
Proposed mechanisms for hypoxia-induced vasodilation at rest and during exercise. During hypoxic exercise, nitric oxide (NO) is the final common pathway for the compensatory dilator response. Systemic epinephrine (E) release, acting via β-adrenergic receptors, contributes to the NO-mediated vasodilation at lower exercise intensities, but this β-adrenergic contribution decreases with increasing exercise intensity. ATP released from the red blood cell (RBC) and/or endothelial-derived prostacyclin (PGI2) remain attractive candidates for stimulating NO during higher intensity hypoxic exercise. Neuronal NOS and nitrate are also potential sources of NO generation and might be another contributor to compensatory vasodilation during hypoxic exercise. Adenosine receptor (ADO-R) activation does not appear to be a major source of NO production during hypoxic exercise. α1AR, α2AR, and β2AR: α1-, α2-, and β2-adrenergic receptors, respectively; NE, norepinephrine; PR, purinergic receptor; SNO-Hb, S-nitrosohemoglobin.
GRANTS
This work was supported, in part, by National Heart, Lung, and Blood Institute grant HL-46493 and by UL1TR000135-07 (Mayo Clinic Center for Translational Science Activities).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.J.J. conception and design of research; M.J.J. and D.P.C. interpreted results of experiments; M.J.J. and D.P.C. drafted manuscript; M.J.J. and D.P.C. approved final version of manuscript; D.P.C. performed experiments; D.P.C. analyzed data; D.P.C. prepared figures; D.P.C. edited and revised manuscript.
REFERENCES
- 1.Barcroft H, Greenwood B, Whelan RF. Blood flow and venous oxygen saturation during sustained contraction of the forearm muscles. J Physiol 168: 848–856, 1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berne RM. Cardiac nucleotides in hypoxia: possible role in regulation of coronary blood flow. Am J Physiol 204: 317–322, 1963 [DOI] [PubMed] [Google Scholar]
- 3.Casey DP, Curry TB, Wilkins BW, Joyner MJ. Nitric oxide-mediated vasodilation becomes independent of beta-adrenergic receptor activation with increased intensity of hypoxic exercise. J Appl Physiol 110: 687–694, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Casey DP, Joyner MJ. α-Adrenergic blockade unmasks a greater compensatory vasodilation in hypoperfused contracting muscle. Front Physiol 3: 271, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casey DP, Joyner MJ. Contribution of adenosine to compensatory dilation in hypoperfused contracting human muscles is independent of nitric oxide. J Appl Physiol 110: 1181–1189, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casey DP, Joyner MJ. NOS inhibition blunts and delays the compensatory dilation in hypoperfused contracting human muscles. J Appl Physiol 107: 1685–1692, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casey DP, Joyner MJ. Prostaglandins do not contribute to the nitric oxide-mediated compensatory vasodilation in hypoperfused exercising muscle. Am J Physiol Heart Circ Physiol 301: H261–H268, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casey DP, Joyner MJ. Skeletal muscle blood flow responses to hypoperfusion at rest and during rhythmic exercise in humans. J Appl Physiol 107: 429–437, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Casey DP, Joyner MJ, Claus PL, Curry TB. Hyperbaric hyperoxia reduces exercising forearm blood flow in humans. Am J Physiol Heart Circ Physiol 300: H1892–H1897, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Casey DP, Joyner MJ, Claus PL, Curry TB. Vasoconstrictor responsiveness during hyperbaric hyperoxia in contracting human muscle. J Appl Physiol 114: 217–224, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casey DP, Madery BD, Curry TB, Eisenach JH, Wilkins BW, Joyner MJ. Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J Physiol 588: 373–385, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casey DP, Madery BD, Pike TL, Eisenach JH, Dietz NM, Joyner MJ, Wilkins BW. Adenosine receptor antagonist and augmented vasodilation during hypoxic exercise. J Appl Physiol 107: 1128–1137, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Casey DP, Shepherd JR, Joyner MJ. Sex and vasodilator responses to hypoxia at rest and during exercise. J Appl Physiol 10.1152/japplphysiol.00409.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casey DP, Walker BG, Curry TB, Joyner MJ. Ageing reduces the compensatory vasodilatation during hypoxic exercise: the role of nitric oxide. J Physiol 589: 1477–1488, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins DM, McCullough WT, Ellsworth ML. Conducted vascular responses: communication across the capillary bed. Microvasc Res 56: 43–53, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Copp SW, Holdsworth CT, Ferguson SK, Hirai DM, Poole DC, Musch TI. Muscle fibre-type dependence of neuronal nitric oxide synthase-mediated vascular control in the rat during high speed treadmill running. J Physiol 591: 2885–2896, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corcondilas A, Koroxenidis GT, Shepherd JT. Effect of a brief contraction of forearm muscles on forearm blood flow. J Appl Physiol 19: 142–146, 1964 [DOI] [PubMed] [Google Scholar]
- 17.Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Augmented skeletal muscle hyperaemia during hypoxic exercise in humans is blunted by combined inhibition of nitric oxide and vasodilating prostaglandins. J Physiol 589: 3671–3683, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dufour SP, Patel RP, Brandon A, Teng X, Pearson J, Barker H, Ali L, Yuen AH, Smolenski RT, Gonzalez-Alonso J. Erythrocyte-dependent regulation of human skeletal muscle blood flow: role of varied oxyhemoglobin and exercise on nitrite, S-nitrosohemoglobin, and ATP. Am J Physiol Heart Circ Physiol 299: H1936–H1946, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edmunds NJ, Marshall JM. The roles of nitric oxide in dilating proximal and terminal arterioles of skeletal muscle during systemic hypoxia. J Vasc Res 40: 68–76, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology 24: 107–116, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferguson SK, Hirai DM, Copp SW, Holdsworth CT, Allen JD, Jones AM, Musch TI, Poole DC. Impact of dietary nitrate supplementation via beetroot juice on exercising muscle vascular control in rats. J Physiol 591: 547–557, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Golub AS, Pittman RN. Bang-bang model for regulation of local blood flow. Microcirculation 20: 455–483, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91: 1046–1055, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Alonso J, Richardson RS, Saltin B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol 530: 331–341, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanada A, Sander M, Gonzalez-Alonso J. Human skeletal muscle sympathetic nerve activity, heart rate and limb haemodynamics with reduced blood oxygenation and exercise. J Physiol 551: 635–647, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart EC, Charkoudian N, Wallin BG, Curry TB, Eisenach J, Joyner MJ. Sex and ageing differences in resting arterial pressure regulation: the role of the beta-adrenergic receptors. J Physiol 589: 5285–5297, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinonen IH, Kemppainen J, Kaskinoro K, Peltonen JE, Borra R, Lindroos M, Oikonen V, Nuutila P, Knuuti J, Boushel R, Kalliokoski KK. Regulation of human skeletal muscle perfusion and its heterogeneity during exercise in moderate hypoxia. Am J Physiol Regul Integr Comp Physiol 299: R72–R79, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol 583: 855–860, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim-Shapiro DB, Schechter AN, Gladwin MT. Unraveling the reactions of nitric oxide, nitrite, and hemoglobin in physiology and therapeutics. Arterioscler Thromb Vasc Biol 26: 697–705, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111: 220–230, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kneale BJ, Chowienczyk PJ, Brett SE, Coltart DJ, Ritter JM. Gender differences in sensitivity to adrenergic agonists of forearm resistance vasculature. J Am Coll Cardiol 36: 1233–1238, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Lundberg JO, Carlstrom M, Larsen FJ, Weitzberg E. Roles of dietary inorganic nitrate in cardiovascular health and disease. Cardiovasc Res 89: 525–532, 2011 [DOI] [PubMed] [Google Scholar]
- 33.Marshall JM. Adenosine and muscle vasodilatation in acute systemic hypoxia. Acta Physiol Scand 168: 561–573, 2000 [DOI] [PubMed] [Google Scholar]
- 34.Marshall JM. Roles of adenosine and nitric oxide in skeletal muscle in acute and chronic hypoxia. Adv Exp Med Biol 502: 349–363, 2001 [DOI] [PubMed] [Google Scholar]
- 35.McCullough WT, Collins DM, Ellsworth ML. Arteriolar responses to extracellular ATP in striated muscle. Am J Physiol Heart Circ Physiol 272: H1886–H1891, 1997 [DOI] [PubMed] [Google Scholar]
- 36.Mortensen SP, Gonzalez-Alonso J, Bune LT, Saltin B, Pilegaard H, Hellsten Y. ATP-induced vasodilation and purinergic receptors in the human leg: roles of nitric oxide, prostaglandins, and adenosine. Am J Physiol Regul Integr Comp Physiol 296: R1140–R1148, 2009 [DOI] [PubMed] [Google Scholar]
- 37.Nicholson WT, Vaa B, Hesse C, Eisenach JH, Joyner MJ. Aging is associated with reduced prostacyclin-mediated dilation in the human forearm. Hypertension 53: 973–978, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roach RC, Koskolou MD, Calbet JA, Saltin B. Arterial O2 content and tension in regulation of cardiac output and leg blood flow during exercise in humans. Am J Physiol Heart Circ Physiol 276: H438–H445, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Rowell LB, Saltin B, Kiens B, Christensen NJ. Is peak quadriceps blood flow in humans even higher during exercise with hypoxemia? Am J Physiol Heart Circ Physiol 251: H1038–H1044, 1986 [DOI] [PubMed] [Google Scholar]
- 40.Seddon MD, Chowienczyk PJ, Brett SE, Casadei B, Shah AM. Neuronal nitric oxide synthase regulates basal microvascular tone in humans in vivo. Circulation 117: 1991–1996, 2008 [DOI] [PubMed] [Google Scholar]
- 41.Shepherd JT. Circulation to skeletal muscle. In: Handbook of Physiology. The Cardiovascular System. Peripheral Circulation and Organ Blood Flow. Bethesda, MD: Am. Physiol. Soc, 1983, sect. 2, vol. III, pt. 1, chapt. 11, p. 319–370 [Google Scholar]
- 42.Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 276: 2034–2037, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Weisbrod CJ, Eastwood PR, O'Driscoll G, Walsh JH, Best M, Halliwill JR, Green DJ. Vasomotor responses to hypoxia in type 2 diabetes. Diabetes 53: 2073–2078, 2004 [DOI] [PubMed] [Google Scholar]
- 44.Welch HG, Bonde-Petersen F, Graham T, Klausen K, Secher N. Effects of hyperoxia on leg blood flow and metabolism during exercise. J Appl Physiol 42: 385–390, 1977 [DOI] [PubMed] [Google Scholar]
- 45.Wilkins BW, Pike TL, Martin EA, Curry TB, Ceridon ML, Joyner MJ. Exercise intensity-dependent contribution of beta-adrenergic receptor-mediated vasodilatation in hypoxic humans. J Physiol 586: 1195–1205, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkins BW, Schrage WG, Liu Z, Hancock KC, Joyner MJ. Systemic hypoxia and vasoconstrictor responsiveness in exercising human muscle. J Appl Physiol 101: 1343–1350, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]