

SUMMARY
The serine esterase monoacylglycerol lipase (MGL) is primarily responsible for deactivating the signaling lipid 2-arachidonoylglycerol (2-AG), an endocannabinoid with full agonist activity at both principal cannabinoid receptors. Although MGL is recognized as a potential therapeutic target, the paucity of structural information on this enzyme has hindered development of MGL-selective inhibitors. Previously, we overexpressed and purified human MGL as the hexa-histidine-tagged recombinant protein (hMGL) and showed that it catalyzed the hydrolysis of both 2-AG and novel fluorogenic reporters. We now characterize by mass spectroscopy the hMGL active site using two chemically distinct inhibitors as direct probes: 5-((biphenyl-4-yl)methyl)-N,N-dimethyl-2H-tetrazole-2-carboxamide (AM6701) and N-arachidonylmaleimide (NAM). Suitable conditions were established for hMGL inhibition by AM6701, and the inhibitor-treated enzyme was subjected to trypsin digestion. The tryptic digest of AM6701-inhibited hMGL was analyzed by MALDI-TOF and tandem MS, which showed that AM6701 had carbamylated the serine in a GXSXG motif of the putative MGL catalytic triad. These results provide the first direct confirmation of the essential role of this serine residue for catalysis and establish the mechanism of AM6701 as a high-affinity, covalent hMGL inhibitor. When applied to NAM-treated hMGL, our direct, ligand-assisted approach revealed that partial alkylation of cysteine residues 215 and/or 249 was sufficient to achieve ~ 80% hMGL inhibition. Further alkylation at cysteine 39 did not increase the extent of enzyme inhibition. Although Cys215 and/or Cys249 mutations to alanine(s) did not affect hMGL’s ability to hydrolyze reporter substrate, as compared to nonmutated hMGL the C215A mutant was more sensitive to NAM, whereas the C249A mutation reduced the enzyme’s sensitivity to NAM. These data conclusively demonstrate a sulfhydryl-based mechanism underlying MGL inhibition by this fatty alkyl-maleimide substrate analog in which Cys249 is of paramount importance. Identification of amino acids critical to catalysis by and pharmacological modulation of hMGL provides information useful in the design of selective MGL inhibitors as potential drugs.
INTRODUCTION
Cannabinoid receptors, their endogenous ligands (“endocannabinoids”), and the enzymes responsible for endocannabinoid synthesis and deactivation comprise a ubiquitous mammalian signaling system that regulates diverse physiological functions. The principal endocannabinoids, N-arachidonoylethanolamine (anandamide, AEA) (Devane et al., 1992) and 2-arachidonoylglycerol (2-AG) (Mechoulam et al., 1995; Sugiura, et al., 1995), are produced on-demand as cannabinoid-receptor activating ligands having distinctive pharmacological profiles. Since unregulated endocannabinoid signaling can have adverse consequences, endocannabinoid concentrations are tightly and redundantly controlled at points of synthesis, transport, and biotransformation of the associated signaling lipids (Di Marzo et al., 2007). Enzymatic endocanabinoid deactivation is key to attenuating endocannabinoid signaling. Fatty acid amide hydrolase (FAAH) is an integral-membrane amidase primarily responsible for AEA hydrolysis that may exist in multiple isoforms (Cravatt et al., 1996; Wei et al., 2006). Although FAAH also metabolizes 2-AG in vitro (Goparaju et al., 1998), soluble monoacylglycerol lipase (MGL) serves as the major enzyme for 2-AG deactivation in cells (Dinh et al., 2004), perhaps along with subsidiary MGL-like esterases (Muccioli et al., 2007; Blankman et al., 2007).
Agents that modulate endocannabinoid-system transmission are actively being sought as therapeutics to treat important behavioral, metabolic, and neurological diseases (Vemuri et al., 2007; Pacher et al., 2006). For therapeutic upregulation of endocananbinoid signaling, pharmacological inhibition of endocannabinoid deactivating enzymes may offer more selectivity and less risk of unwanted psychotropic side-effects as compared to cannabinoid-receptor agonists (Malan et al., 2003). Specifically, targeted inhibition of 2-AG deactivation is considered an attractive therapeutic approach against pain, inflammation, and neurodegenerative and immune disorders (Pacher et al., 2006; Saario and Laitinen, 2007). Although many available FAAH inhibitors also act on MGL (Deutsch et al., 1997; Ghafouri, et al., 2004), high-affinity ligands that potently and selectively inhibit MGL are currently lacking. The molecular mechanisms of known MGL inhibitors are speculative, having been inferred mainly by analogy from MGL modeling studies with or without virtual screening (Karlsson et al., 1997; Saario et al., 2006). A putative MGL catalytic triad {Ser122…Asp239…His269} has been predicted from homology with other serine hydrolases and limited site-directed mutagenesis experiments (Karlsson et al., 1997). However, direct experimental demonstration of the involvement of these (or other) amino acid residues in pharmacological MGL inhibition is lacking.
First described as a putative competitive inhibitor of AEA transport (IC50 = 270 pM in a cellular assay) with analgesic activity in rodents (Moore et al., 2005), LY2183240 was subsequently shown to target mouse-brain FAAH (IC50 ≈ 13 nM) and recombinant MGL (IC50 ≈ 5.3 nM) in a time-dependent manner (Alexander and Cravatt, 2006). FAAH inhibition by LY2183240 was postulated to involve carbamylation of active-site Ser241 (Alexander and Cravatt, 2006). However, the molecular details of MGL inhibition by LY2183240 remain unknown. In addition, Ortar et al. (Ortar et al., 2007) have recently demonstrated that LY2183240 consists of two isomers, as we have confirmed in the course of LY2183240 synthesis and purification for the present work (data not shown).
Substituted maleimide derivatives including NAM inhibit incompletely MGL-like enzymatic activity in rat cerebellar membranes (Saario et al., 2005) and rat adipocyte subcellular fractions (Sakurada and Noma, 1981). As with inhibition of MGL by LY2183240, no experimental evidence is available regarding the mechanism of MGL inhibition by NAM. The conjugated double bond of N-substituted maleimides can undergo a facile Michael addition to nucleophilic groups of amino acids and, particularly, the thiol group of cysteine (Smyth et al., 1960). Consequently, NAM’s inhibition of MGL in rat cerebellar membranes was hypothesized from molecular modeling studies to reflect an interaction between NAM and either Cys242 or Cys208, these two of the enzyme’s six cysteine residues having been modeled proximally to the putative substrate-binding site (Saario et al., 2005). There are four cysteine residues in human MGL, and their thiol groups are not involved in disulfide bond formation (Zvonok et al., 2008).
In the present study, we show that the 2,5-LY2183240 isomer, 5-((biphenyl-4-yl)methyl)-N,N-dimethyl-2H-tetrazole-2-carboxamide (AM6701), is a more potent inhibitor of recombinant hexa-histidine-tagged human MGL (hMGL) than the 1,5-isomer, 5-((biphenyl-4-yl)methyl)-N,N-dimethyl-1H-tetrazole-1-carboxamide (AM6702). We have therefore characterized AM6701-inhibited hMGL through a comprehensive mass-spectrometric (MS) analysis in order to identify directly the amino acid residue(s) involved. We have also successfully applied this ligand-assisted approach to obtain the first direct structural information on hMGL inhibition by NAM, which we have augmented with site-directed mutagenesis data. The results are of value in identifying specific amino acid residues critical to MGL catalysis and pharmacological modulation, informing thereby the rational design of selective MGL inhibitors with potential therapeutic utility.
RESULTS AND DISCUSSION
hMGL Inhibition by AM6701 and NAM
SDS-PAGE followed by either Coomassie staining or western blot anti-5-His antibody detection demonstrated that the hMGL overexpressed in E. coli and isolated by immobilized metal-affinity chromatography is a single monomeric protein with an estimated molecular mass, ≈ 35 kDa, that approximates the calculated mass of 34,123 kDa (Figures 1A and 1B). The catalytic properties of purified hMGL are similar to those of the homologous, soluble crude rMGL using either 2-AG or a novel fluorogenic reporter, arachidonoyl,7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE), as substrate (Zvonok et al., 2008). With a sensitive fluorescence-based assay, the inhibitor profiles of both LY2183240 isomers and NAM were first characterized using E. coli lysate (containing hMGL) and soluble crude rMGL as enzyme sources. After 3-hr incubation, the LY2183240 isomers AM6701 and AM6702 inhibited hMGL in the nM range with concentration-dependence, whereas NAM was markedly less potent (Figure 1C). Only the LY2183240 2,5-regioisomer (AM6701) inhibited both hMGL and soluble crude rMGL with low-nM IC50’s, making AM6701 the more potent LY2183240 isomer as well as the most potent MGL inhibitor profiled (Figure 1D). The high potency of AM6701 as MGL inhibitor relative to both AM6702 and NAM is supported by published studies utilizing 2-AG as substrate, albeit with varying sources of crude MGL(-like) activity (Oktar et al., 2007; Saario et al., 2005) (Figure 1D).
Figure 1.
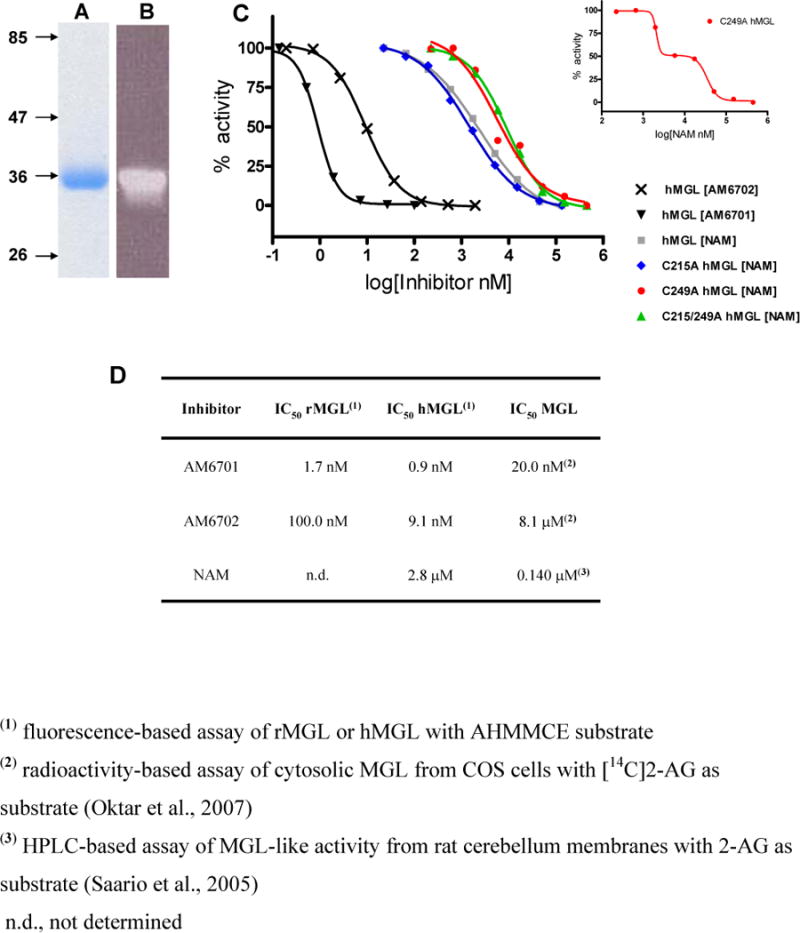
Coomassie stained 10% PAGE SDS gel (A) and western blot (B) analysis of IMAC-purified, recombinant hexa-histidine-tagged human MGL (hMGL). (C) Best-fit, one-site plots of the concentration-dependent inhibition by AM6701, AM6702, and NAM of hMGL and designated hMGL cysteine mutants to hydrolyze AHMMCE. The inset highlights the biphasic nature of the hMGL C249A mutant by NAM as a two-site plot. (D) IC50 values for inhibition of soluble crude rMGL, hMGL, and MGL prepared from COS cells and rat cerebellum by AM6701, AM6702, and NAM.
We next defined conditions that would elicit maximal hMGL inhibition by either AM6701 or NAM so as to generate, efficiently, enzyme preparations useful for identifying the amino acid residue(s) critical to each inhibitor’s action by MALDI-TOF MS. At a fixed substrate concentration of 50 μM AHMMCE, inhibition of purified hMGL was dependent upon the inhibitor:hMGL molar ratio (Table 1). Even at an equimolar ratio with hMGL, AM6701 markedly (by ~ 72%) inhibited the enzyme. Maximal (~ 90%) inhibition of hMGL by AM6701 was attained at an 8.3-fold molar excess of AM6701 over hMGL. In contrast, an equimolar ratio of NAM to hMGL resulted in only ~ 50% inhibition. A 3.3:1 NAM:hMGL molar ratio potentiated enzyme inhibition to ~ 80%, whereas up to a 33-fold molar excess of NAM elicited little further hMGL inhibition. Identical results were obtained at a fixed substrate concentration of 100 μM AHMMCE (data not shown). Consequently, we elected to incubate hMGL for 1 hr with an 8.3-fold molar excess of AM6701 to characterize by MALDI-TOF MS all inhibitor-related enzyme modification(s). Because of the relatively more pronounced concentration-dependence by which NAM inhibited hMGL hydrolysis of fluorogenic reporter AHMMCE (Table 1), the mechanism of hMGL inhibition by NAM was studied at the various NAM:hMGL molar ratios specified in Tables 1 and 2.
Table 1.
Concentration-dependence of hMGL Inhibition by AM6701 and NAM
| Inhibitor | Inhibitor:hMGL molar ratio |
hMGL Inhibition (%) 10 min |
|---|---|---|
| AM6701 | 1:1 | 72 |
| 3.3:1 | 85 | |
| 8.3:1 | 91 | |
| NAM | 1:1 | 51 |
| 3.3:1 | 80 | |
| 8.3:1 | 82 | |
| 33:1 | 84 |
Reporter substrate (AHMMCE) concentration was 50 μM.
Table 2.
Relative Content of NAM-modified, Cysteine-containing Peptides Identified by MS in Tryptic Digest of NAM-treated hMGL
| hMGL cysteine residues | m/z | Relative Alkylation NAM:hMGL ratio |
|
|---|---|---|---|
| 8.3:1 | 33:1 | ||
| Cys39 | 3144.62; 3300.72 | 0% | 100%(a) |
| Cys208 | 2007.05 | 0% | 0%(a) |
| Cys215 | 2158.28 | 26%(b) | 22%(b) |
| Cys249 | 2445.38; 3534.97 | 74%(b) | 78%(b) |
percent alkylation of Cys39 relative to Cys208
percent alkylation of Cys215 relative to Cys249
MS Analysis of hMGL Inhibition by AM6701
Desalted samples of untreated hMGL and enzyme that had been inhibited > 90% by a 1hr preincubation with an 8.3-fold molar excess of AM6701 were subjected overnight to in-solution trypsin digestion prior to MALDI-TOF MS analysis. A comparison of the spectra of the tryptic digest from naïve and AM6701-inhibited hMGL revealed in the latter only one peptide with a 71-Da mass increase, which was provisionally attributed to the addition of a single dimethylcarbamyl group to the enzyme (Figure 2A). Importantly, the carbamylation product was sensitive to the dithiothreitol-iodoacetamide treatment typically employed to disrupt disulfide bonds and alkylate free sulfhydryls prior to trypsin digestion. To avoid complete removal of the carbamyl group from AM6701-inhibited hMGL, a limited, 30-min dithiothreitol-iodoacetamide treatment (Figure 2A) was conducted prior to MALDI-TOF MS. Under these mild conditions, ~ 30% of the peptide carbamylation observed in non-reduced, non-alkylated, AM6701-inhibited hMGL was retained (Figure 2B).
Figure 2.
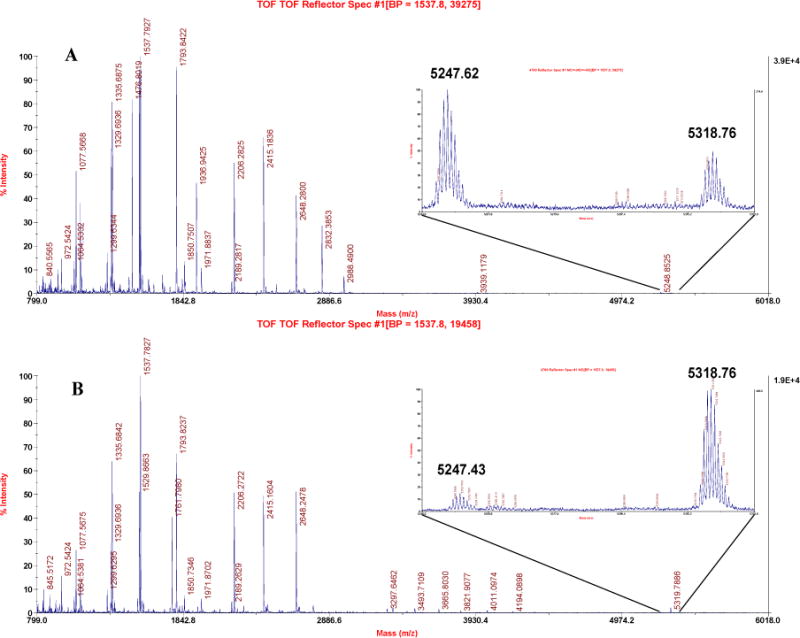
The tryptic digest of the AM6701-inhibited hMGL was peptide fingerprinted using MALDI-TOF MS. (A) The enzyme was subjected to mild reduction-alkylation before trypsin digestion. (B) The enzyme was digested with trypsin without reduction-alkylation. High-resolution MS spectra of the precursor ions are shown for the unmodified (m/z 5247.62) and carbamylated (m/z 5318.76) peptide DYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAK (position 117–172).
Figure 3 depicts the MS/MS spectra of the 5247.87 m/z and 5318.79 m/z peptide ions derived from trypsin digests of naïve (Figure 3A) and AM6701-inhibited (Figure 3B) hMGL, respectively. Although the precursor ions are intensive (Figure 2A and 2B), MS/MS spectral optimization is complicated by the high molecular weights involved, as reflected in the signal-to-noise ratio of the MS/MS spectra in Figures 3A and 3B. Nonetheless, the same fragmentation ions corresponding to the y-ion for the sequence IAGGMSHG (Figure 3A) and the sequence IAGGM (Figure 3B) are identified. Notably, the masses for the last three residues (SHG) in the digest of AM6701-inhibited hMGL (Figure 3B) are 71 m/z greater than in the naïve enzyme (Figure 3A). The high-resolution spectrum of the unmodified precursor ion at m/z 5247.87 (Figure 3A) corresponds to the tryptic peptide DYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAK (position 117–172), and the second ion at m/z 5318.79 is the carbamylated (+71 Da) product (Figure 3B). Since there are three serine residues in the tryptic fragment, tandem MS was performed on both the unmodified and modified ions to determine the exact serine residue modified by AM6701. Tandem MS unambiguously identified Ser129 as the hMGL residue carbamylated by AM6701. Mechanistically, the carbamylation product may arise from an attack by the Ser129 hydroxyl group on the AM6701 carbonyl moiety, the resultant carbamylated serine accounting for a 71-Da increase in the mass of the peptide (Figure 3C). Figure 3D, left panel, depicts schematically the spatial disposition of the amino acid residues Ser129…Asp246…His276 in the hMGL catalytic triad (Ser122…Asp239…His269 in human MGL) in an energetically favorable orientation with AM6701 before its reaction with enzyme. The carbamylated Ser129 residue in AM6701-inhibited hMGL is depicted in Figure 3D, right panel. These data clearly demonstrate the essential role of Ser129 in the conserved G127XSXG131 motif (i.e., G120XSXG124 in human MGL) of the catalytic triad for hMGL esterase activity.
Figure 3.
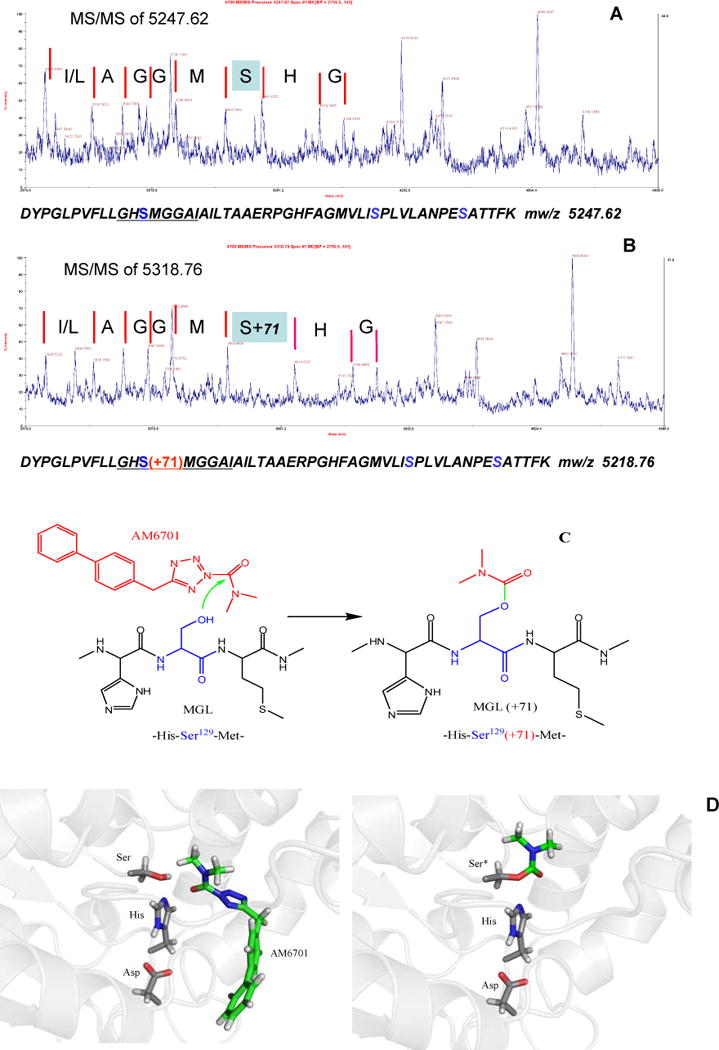
Tandem MALDI-TOF MS/MS analysis of unmodified (A) and modified (B) peptides identified Ser129 as the hMGL residue carbamylated by AM6701. The fragmentation ions corresponding to the y-ion sequence IAGGMSHG and IAGGMS(+71)HG are underscored. (C) Illustration of the carbamylation reaction between the Ser129 of hMGL and AM6701. (D) Schematic presentation of the hMGL catalytic triad within which AM6701 is oriented before (left panel) and after (right panel) hMGL inhibition by Ser129 carbamylation (Ser*). The carbons of AM6701 and the MGL peptide bonds are depicted in green and gray, respectively. Blue, nitrogen; white, hydrogen; red, oxygen
MS and Mutational Analyses of hMGL Inhibition by NAM
The graded concentration-dependence by which NAM inhibited hMGL activity (Figure 1 and Table 1) suggests the presence of high-affinity inhibitor binding site(s) that are in close proximity to cysteine residues capable of interacting covalently with NAM and that may affect catalysis differentially. This suggestion is directly supported by MALDI-TOF MS analysis of cysteine-containing peptides in tryptic digests of NAM-inhibited hMGL (Figure 4A and 4B). At a NAM:hMGL molar ratio of 1 to 8.3, both unmodified and NAM-modified peptides at Cys215 or Cys249 were identified (Table 2), the latter evidencing a 369.27-Da mass increase. Even at the maximal, 33-fold NAM molar excess employed, comparable incomplete (84%) hMGL inhibition was observed, and Cys215 or Cys249 was still partially alkylated (Figure 4B). Based on the intensity of peptide signals in the MS spectra, the relative cysteine alkylation at positions 215 and 249 was similar at NAM:hMGL molar ratios of 8.3:1 (Cys215, 26% and Cys249, 74%) and 33:1 (Cys215, 22% and Cys249, 78%) (Table 2). Thus, a ~ 4-fold increase in NAM concentration affected neither the relative alkylation of Cys215 and Cys249 nor the degree of hMGL inhibition by NAM (Table 1), suggesting that NAM may modify either Cys249 or Cys215, but not both in the same hMGL molecule. At the 33:1 NAM:hMGL molar ratio, Cys39 also became alkylated, whereas Cys208 remained unmodified at all NAM:hMGL molar ratios examined (Table 2). MS/MS analysis of the 1789.04, 2158.28, 2445.37, 3144.55, 3300.75, and 3534.93 m/z peptide ions identified Cys215, Cys249, and Cys39 as the sites of hMGL alkylation by NAM (data not shown). Figure 4C depicts the alkylation reaction between a cysteine residue of hMGL and NAM.
Figure 4.
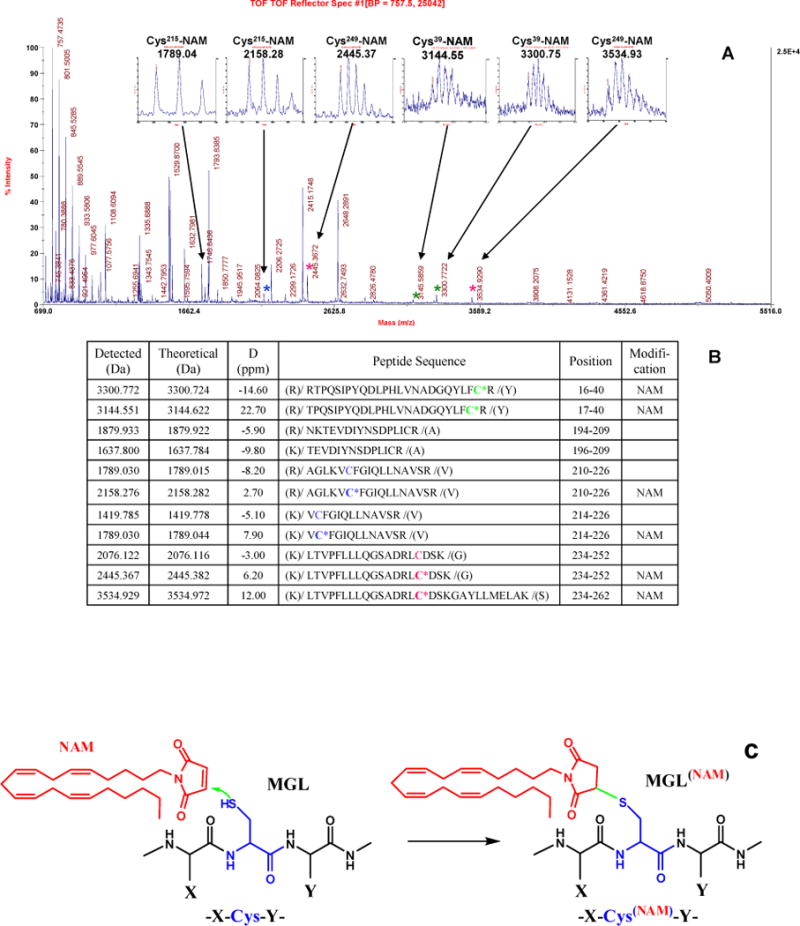
(A) The tryptic digest of hMGL inhibited with NAM (at a 33:1 NAM:hMGL molar ratio) was peptide fingerprinted using MALDI-TOF MS. High-resolution MS spectra of NAM-modified peptide ions are shown in the insets. (B) As identified by MS, the cysteine containing peptides without or with NAM attached are presented in the table. The cysteines at positions 39, 215, and 249 modified with NAM are shown in bold (C*) and highlighted in green, blue, and red, respectively. (C) Schematic illustration of the interaction between MGL alkylation by NAM.
Additional evidence regarding the mechanism of MGL inhibition by NAM was obtained through characterization of cysteine-to-alanine C215A and C249A single and double C215/249A hMGL mutants. Our data indicate that the catalytic activity of the single C215A and C249A mutants and the double C215/249A hMGL mutant did not differ significantly from that of nonmutated hMGL, as indexed by their ability to hydrolyze AHMMCE (data not shown). However, we did observe differences among hMGL and the mutant enzymes in their susceptibility to NAM inhibition (Figure 1C). As evaluated by one-site regression analysis (Figure 1C), elimination of Cys249 in either the C249A (IC50 = 6.4 μM) or the C215/249A (IC50 = 8.5 μM) hMGL mutant reduced the enzyme’s sensitivity to NAM as compared to nonmutated hMGL (IC50 = 2.8 μM). Due to its biphasic appearance, the NAM concentration-response of the C249A mutant was further analyzed with a two-site equation. The portion of this concentration-response curve (Figure 1C, inset) below the inflection region at ~ 50% enzyme activity may reflect the partial activity of the C249A mutant alkylated by NAM at Cys215. In marked contrast, the C215A mutant itself was two-fold more sensitive to NAM (IC50 = 1.4 μM) than hMGL (IC50 = 2.8 μM). These mutational analyses overall suggest the greater importance of Cys249 vs. Cys215 in hMGL inhibition by NAM.
Our MS and mutation data demonstrate conclusively that hMGL inhibition by NAM depends upon the differential Michael addition of an arachidonylmaleimide group to select cysteine residues in the enzyme. Partial alkylation of hMGL at Cys215 and/or Cys249 with NAM is sufficient to inhibit hMGL by ~ 80%, Cys249 alkylation being favored, whereas Cys39 and Cys208 may not be critical. Yet large molar NAM excesses do inhibit hMGL completely (Figure 1C). We hypothesize that, at an 8.3-to-33-fold molar excess of NAM, either Cys249 or Cys215, but not both, becomes alkylated in any given hMGL molecule, Cys249 preferentially (Table 2). As a consequence of this initial alkylation event, accessibility of the other, unmodified cysteine residue to another NAM molecule is restricted. Since the C215A hMGL mutant was more sensitive to NAM than the nonmutated hMGL, whereas the C249A mutant was less sensitive to NAM, Cys249 alkylation may be the more decisive determinant of hMGL inhibition by NAM (relative to Cys215 alkylation). The partial inhibition of MGL in rat cerebellar membranes (Saario et al., 2005) and rat adipocyte fractions (Sakurada and Noma, 1981) by assorted maleimides suggests that our MS and mutational characterization of hMGL-NAM interactions might offer some mechanistic context for MGL inhibition by other N-substituted maleimides across species.
SIGNIFICANCE
Detailed mechanistic understanding of the enzymes that deactivate endocannabinoids is a prerequisite for the effective pharmacotherapeutic modulation of the endocannabinoid signaling system. Although extant structural information on MGL relies heavily upon provisional modeling studies, the present work has utilized a ligand-assisted, MS-based approach augmented with mutational analysis to characterize at the molecular level the mechanisms by which AM6701 and NAM inhibit this critical endocannabinoid-system enzyme responsible for deactivating 2-AG, the most abundant brain endocannabinoid and a full agonist at both cannabinoid receptors. Potent inhibition of hMGL by the carbamyl tetrazole, AM6701, was shown to involve a covalent interaction resulting in selective enzyme carbamylation at Ser129 (i.e., Ser122 in human MGL) in the GXSXG motif of the MGL catalytic triad. This finding constitutes the first direct confirmation of the essential role of Ser129 for MGL hydrolytic activity. Appreciable (~ 80%) hMGL inhibition by NAM was shown to involve covalent Michael addition of an N-arachidonylmaleimide group to Cys215 and/or Cys249 in hMGL (i.e., Cys208 and Cys242 in human MGL), somewhat preferentially to the latter cysteine residue. Cys39 and Cys208 in hMGL (i.e., Cys32 and Cys201 in human MGL) appear not to play an essential role in partial hMGL inhibition by NAM. Direct demonstration of the involvement of Ser129 in hMGL inhibition by AM6701 and Cys249 and Cys215 in hMGL inhibition by NAM is useful in designing potent, highly selective MGL-targeted inhibitors as drug candidates to treat disorders (e.g., anxiety, pain, inflammation, various neurodegenerative diseases) for which potentiating endocannabinoid-system activity might be therapeutic.
EXPERIMENTAL PROCEDURES
Materials
Standard laboratory chemicals, culture media, isopropyl-β-D-thio-galactopyranoside, lysozyme, and DNase I were purchased from Sigma Chemical Co (St. Louis, MO, USA) and Fisher Chemical (Pittsburgh, PA, USA), unless otherwise specified. SDS–PAGE supplies and Bio Spin™ P-6 columns were from Bio-Rad (Hercules, CA, USA). MS-grade trypsin (Trypsin Gold) was from Promega (Madison, WI, USA). AM6701, AM6702, and AHMMCE were synthesized at the Center for Drug Discovery, Northeastern University, by standard routes that will be detailed elsewhere. Soluble crude rMGL was prepared essentially as described (Goparaju et al., 1998).
Mutant hMGL Preparation and Profiling
The cysteine to alanine single C215A and C249A hMGL mutants and the double C215/249A hMGL mutant were generated using the Stratagene QuickChange site-directed mutagenesis kit (La Jolla, CA, USA). The DNA primary structure of all mutants was confirmed by sequencing. The hMGL mutant enzymes were expressed in E. coli and assayed for hydrolytic activity with AHMMCE in the absence or presence of NAM at known concentrations, as described below.
Preparative hMGL Purification by Immobilized Metal Affinity Chromatography
A single colony of E. coli BL21 (DE3) cells containing the plasmid pET45His6hMGL was inoculated into 12 ml of Luria broth supplemented with ampicillin (100 μg/ml) and grown with shaking at 250 rpm overnight at 37 °C. The next morning, 10 ml of this culture was inoculated into 1 L of Luria both-ampicillin medium, which was incubated with shaking at 37 °C until reaching an OD600 of 0.6–0.8. Protein expression was induced by adding 1 mM (final conc.) isopropyl-β-D-thio-galactopyranoside. After 5 hr induction, E. coli cells were harvested by centrifugation (5,000 g, 10 min, 4 °C), washed with PBS buffer, and stored at −80°C. Five g (wet-weight) of cells was resuspended in 50 ml lysis buffer (50 mM Tris, pH 8.0, containing 100 mM NaCl and 0.5% Triton X-100) supplemented with lysozyme (0.2 mg/ml) and DNase I (25 μg/ml) and disrupted on ice by sonication. The sonicate was centrifuged (10,000 g, 30 min, 4°C), and the supernatant was incubated with 1.0 ml (bed volume) pre-equilibrated BD Talon™ metal affinity resin (Takara, Otsu, Shiga, Japan) at room temperature in a rotator. After 1 hr, the suspension was transferred to a gravity-flow column and allowed to settle. The resin was washed with 15 ml lysis buffer, then with 15 ml lysis buffer containing 0.1% Triton X-100 and 10 mM imidazole to elute unbound material. The recombinant hexa-histidine-tagged human MGL (hMGL) was finally eluted with lysis buffer containing 0.1% Triton X-100 and 200 mM imidazole, and the eluate was collected in 500-μl fractions that were analyzed by SDS-PAGE.
SDS-PAGE and Western Blot Analysis
Samples were denatured at 70°C for 5 min in Laemmli buffer containing 5% β-mercaptoethanol and resolved on 10% PAGE SDS gels. Gels were either stained with Commassie blue or transferred to PVDF membranes for immunodetection with anti-5His horseradish peroxidase conjugate following manufacturer’s directions (QIAexpress; Qiagen, Valencia, CA, USA). Protein bands on the blots were visualized using the ECL Western Blotting Analysis System (GE Healthcare, Piscataway, NJ, USA). A FluorChem Imaging System (Alpha Innotech, San Leandro, CA, USA) was used to photograph developed gels and blots.
Assay of Soluble Crude rMGL and hMGL Hydrolytic Activity
The assay to quantify soluble crude rMGL and hMGL enzymatic activity was based on the hydrolysis of the novel fluorogenic reporter substrate AHMMCE and was performed in a 96-well plate (Costar 3650) format using a Synergy HT Plate Reader (BioTek Instruments, Winooski, Vermont, USA). To determine overall concentration-response profiles (Figure 1C, 1D), 8 μl E. coli lysate containing hMGL (175 ng total protein) with or without designated test compound at specified concentrations in 50 mM Tris-HCl (pH 7.4) containing 8% DMSO was pre-incubated for 15 min at room temperature. AHMMCE was next added to a final concentration 20 μM in a total volume 200 μl. The incubation was continued for 3 hr, during which fluorescence readings at 360 nm/460 nm (λexcitation/λemission) were taken every 15 min. Relative fluorescence units were converted to the amount of 7-hydroxy-4-methylcoumarin (HMC) formed from AHMMCE enzymatic hydrolysis based on a standard fluorescence curve of known amounts of HMC. The HMC formed after 2 h (i.e., within the linear assay response) was plotted against test-compound concentration, and a nonlinear regression equation was used to determine IC50 values (Figure 1D) (Prism software, Version 4; GraphPad, San Diego, CA, USA).
Assay of hMGL Inhibition Using AHMMCE
The concentration-dependence of hMGL inhibition (Table 1) was evaluated after incubation of purified hMGL with test compound for 1 hr at room temperature. The assay was performed in a 96-well format (Costar #3795) in 10 mM Tris-HCl (pH 8.0) containing 5% DMSO (final volume 20 μl) with hMGL alone (1 ng) and hMGL (3 ng) that had been incubated with test compound at a known molar ratio to hMGL enzyme, as specified in Table 1. AHMMCE (50 or 100 μM) was then added as fluorogenic reporter substrate. The plate was exposed to ultraviolet light (360 nm) for 3 sec, and images were recorded at the start of the assay and every 10 min thereafter for 1 hr using a FluorChem Imaging System. The brightness of each well was quantified with FluorChem Imaging System Software and normalized to enzyme concentration, and the percent enzyme inhibition was determined. The intensity of light emission was proportional to the amount of functional enzyme and correlated negatively with the test-compound’s effectiveness as hMGL inhibitor.
Interaction of Inhibitors with hMGL and Preparation of Peptide Hydrolysates for MALDI-TOF MS
Purified hMGL (3.1 μg, 3 μM) in 10 mM Tris-HCl, pH 8.0, (30 μl) was incubated for 1 hr at room temperature without or with test compound present at a known molar ratio to hMGL. After evaluation of hMGL inhibition by AM6701 or NAM (above), the incubation was terminated by rapid desalting with a Bio-Spin™ 6 Column in 50 mM ammonium bicarbonate buffer containing 0.02% CYMAL, pH 8.0. The desalted hMGL was digested with trypsin (200 ng) overnight either directly or after reduction-alkylation under mild conditions at room temperature with dithiothreitol (17 mM for 30 min) and iodoacetamide (55 mM for 1 hr in the dark) (Zvonok et al., 2007).
MALDI-TOF MS Analysis
All MS data were acquired on a 4800 Maldi TOF/TOF™ mass spectrometer (Applied Biosystems, Framingham, MA, USA) fitted with a 200Hz solid-state ultraviolet laser (wavelength 355 nm) from samples spotted on Opti-TOF® 384-well plate inserts. Each sample of protein digest was crystallized by spotting 0.5 μl tryptic digest and 0.5 μl α-cyano-4-hydroxycinnamic acid matrix (5 mg/mL in aqueous 60% acetonitrile/0.1% trifluoroacetic acid). After crystallization, the dried spot was washed with 5 μl aqueous 0.1% trifluoroacetic acid for rapid desalting. In those cases where trace residual salt and detergent suppressed signal, the digest was diluted 10-fold with matrix solution, and 1 μl of the diluted digest was spotted for analysis. Spectra were acquired by accumulating data from several positions within each sample well to determine the ions present. All MS spectra were externally calibrated using a mixture of peptide standards [des-Arg1-bradykinin at MH+ 904.4681; angiotensin I at MH+ 1296.6853; Glu-fibrino peptide at MH+ 1570.6774; ACTH (clip 1–17) at MH+ 2093.0867; ACTH (clip 18–39) at MH+ 2465.1989; and ACTH (clip 7–38) at MH+ 3657.9294]. MS/MS spectra were acquired on selected ions of interest. The instrument was calibrated in the MS/MS mode using five daughter ions (at m/z 175.119, 684.346, 813.389, 1056.475 and 1441.634) generated from the fragmentation of Glu-fibrino peptide (MH+ 1570.6774.) MS/MS spectra were acquired under the following conditions: precursor isolation resolution of 200; collision energy of 2kV; cell pressure of 2 × 10−5 torr; air as collision gas. Accumulation was performed until spectra were of optimal quality.
Data analyses were performed by comparing the monoisotopic peaks with the theoretical molecular weights corresponding to the expected peptide digestion products. The maximum error allowed was set to 100 ppm. Theoretical molecular weights of expected peptides after digestion were calculated using MS-digest (UCSF MS facility, San Francisco, CA, USA), FindPept and FindMod tools (ExPASy server, Swiss Institute of Bioinformatics, Geneva, Switzerland).
Acknowledgments
This work has been supported by grants from the National Institute on Drug Abuse, National Institutes of Health: DA09158, DA00493, DA03801, DA07215, and DA07312 (AM).
Abbreviations Used
- AEA
(anandamide), N-arachidonoylethanolamine
- 2-AG
2-arachidonoylglycerol
- AHMMCE
arachidonoyl,7-hydroxy-6-methoxy-4-methylcoumarin ester
- AM6701
5-((biphenyl-4-yl)methyl)-N,N-dimethyl-2H-tetrazole-2-carboxamide
- AM6702
5-((biphenyl-4-yl)methyl)-N,N-dimethyl-1H-tetrazole-1-carboxamide
- FAAH
fatty acid amide hydrolase
- hMGL
recombinant hexa-histidine-tagged human MGL
- HMC
7-hydroxy-4-methylcoumarin
- MALDI-TOF MS
matrix-assisted laser desorption/ionization time-of-flight mass spectrometry
- MGL
monoacylglycerol lipase
- m/z
mass to charge ratio
- MS
mass spectrometry
- NAM
N-arachidonylmaleimide
- rMGL
rat-brain monoacylglycerol lipase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexander JP, Cravatt BF. The putative endocannabinoid transport blocker LY2183240 is a potent inhibitor of FAAH and several other brain serine hydrolases. J Am Chem Soc. 2006;128:9699–9704. doi: 10.1021/ja062999h. [DOI] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol. 2007;18:129–140. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- Deutsch DG, Lin S, Hill WA, Morse KL, Salehani D, Arreaza G, Omeir RL, Makriyannis A. Fatty acid sulfonyl fluorides inhibit anandamide metabolism and bind to the cannabinoid receptor. Biochem Biophys Res Commun. 1997;231:217–221. doi: 10.1006/bbrc.1997.6072. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Kathuria S, Piomelli D. RNA interference suggests a primary role for monoacylglycerol lipase in the degradation of the endocannabinoid 2-arachidonoylglycerol. Mol Pharmacol. 2004;66:1260–1264. doi: 10.1124/mol.104.002071. [DOI] [PubMed] [Google Scholar]
- Ghafouri N, Tiger G, Razdan RK, Mahadevan A, Pertwee RG, Martin BR, Fowler CJ. Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol. 2004;143:774–784. doi: 10.1038/sj.bjp.0705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju SK, Ueda N, Yamaguchi H, Yamamoto S. Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett. 1998;422:69–73. doi: 10.1016/s0014-5793(97)01603-7. [DOI] [PubMed] [Google Scholar]
- Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. cDNA Cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. J Biol Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- Malan TP, Jr, Ibrahim MM, Lai J, Vanderah TW, Makriyannis A, Porreca F. CB2 cannabinoid receptor agonists: pain relief without psychoactive effects? Curr Opin Pharmacol. 2003;3:62–67. doi: 10.1016/s1471-4892(02)00004-8. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- Moore SA, Nomikos GG, Dickason-Chesterfield AK, Schober DA, Schaus JM, Ying BP, Xu YC, Phebus L, Simmons RMA, Li D, Iyengar S, Felder CC. Identification of a high-affinity binding site involved in the transport of endocannabinoids. Proc Natl Acad Sci USA. 2005;102:17852–17857. doi: 10.1073/pnas.0507470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli GG, Xu C, Odah E, Cudaback E, Cisneros JA, Lambert DM, López Rodríguez ML, Bajjalieh S, Stella N. Identification of a novel endocannabinoid-hydrolyzing enzyme expressed by microglial cells. J Neurosci. 2007;27:2883–2889. doi: 10.1523/JNEUROSCI.4830-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortar G, Cascio MG, Moriello AS, Camalli M, Morera E, Nalli M, Di Marzo V. Carbamoyl tetrazoles as inhibitors of endocannabinoid inactivation: A critical revisitation. Eur J Med Chem. 2007;43:62–72. doi: 10.1016/j.ejmech.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Jarvinen T, Niemi R. Characterization of the sulfhydryl-sensitive site in the enzyme responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Chem Biol. 2005;12:649–656. doi: 10.1016/j.chembiol.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Saario SM, Salo OM, Nevalainen T, Poso A, Laitinen JT, Jarvinen T, Niemi R. Fatty acid amide hydrolase inhibitors from virtual screening of the endocannabinoid system. J Med Chem. 2006;49:4650–4656. doi: 10.1021/jm060394q. [DOI] [PubMed] [Google Scholar]
- Saario SM, Laitinen JT. Therapeutic potential of endocannabinoid-hydrolysing enzyme inhibitors. Basic Clin Pharmacol Toxicol. 2007;101:287–293. doi: 10.1111/j.1742-7843.2007.00130.x. [DOI] [PubMed] [Google Scholar]
- Sakurada T, Noma A. Subcellular localization and some properties of monoacylglycerol lipase in rat adipocytes. J Biochem (Tokyo) 1981;90:1413–1419. doi: 10.1093/oxfordjournals.jbchem.a133607. [DOI] [PubMed] [Google Scholar]
- Smyth DG, Nagamatsu A, Fruton JS. Some reactions of N-ethylmaleimide. J Am Chem Soc. 1960;82:4600–4604. [Google Scholar]
- Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Res Commun. 1995;215:89–97. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- Vemuri VK, Janero DR, Makriyannis A. Pharmacotherapeutic targeting of the endocannabinoid signaling system: Drugs for obesity and the metabolic syndrome. Physiol Behav. 2007 doi: 10.1016/j.physbeh.2007.11.012. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem. 2006;281:36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- Zvonok N, Yaddanapudi S, Williams J, Dai S, Dong K, Rejtar T, Karger BL, Makriyannis A. Comprehensive proteomic mass spectrometric characterization of human cannabinoid CB2 receptor. J Proteome Res. 2007;6:2068–2079. doi: 10.1021/pr060671h. [DOI] [PubMed] [Google Scholar]
- Zvonok N, Yaddanapudi S, Williams J, Dai S, Dong K, Rejtar T, Karger BL, Makriyannis A. Full mass spectrometric characterization of human monoacylglycerol lipase generated by large-scale expression and single-step purification. J Proteome Res. 2008;7:2158–2164. doi: 10.1021/pr700839z. [DOI] [PMC free article] [PubMed] [Google Scholar]