

Abstract
The mammalian target of rapamycin (mTOR) signaling pathway is a master regulator of cell growth and metabolism. Deregulation of the mTOR pathway has been implicated in a number of human diseases such as cancer, diabetes, obesity, neurological diseases and genetic disorders. Rapamycin, a specific inhibitor of mTOR, has been shown to be useful in the treatment of certain diseases. Here we discuss its mechanism of action and highlight recent findings regarding the effects and limitations of rapamycin monotherapy and the potential utility of combination therapy with rapamycin.
An introduction to rapamycin: history and mechanism of action
Rapamycin was initially discovered as an antifungal metabolite produced by Streptomyces hygroscopicus from a soil sample of Easter Island (also known as Rapa Nui). Subsequently, rapamycin was found to possess immunosuppressive and anti-proliferative properties in mammalian cells, spurring an interest in identifying the mode of action of rapamycin. Rapamycin was shown to be a potent inhibitor of S6K1 activation, a serine/threonine kinase activated by a variety of agonists (Chung et al., 1992; Kuo et al., 1992; Price et al., 1992) and an important mediator of PI3 kinase signaling (Chung et al., 1994). Concurrently, the target of rapamycin (TOR) was identified in yeast and animal cells (Laplante and Sabatini, 2012; Loewith and Hall, 2011). Rapamycin forms a gain-of-function complex with the 12-kDa FK506-binding protein (FKBP12), and this complex binds and specifically acts as an allosteric inhibitor of mammalian TOR (mTOR, also known as mechanistic TOR) complex 1 (mTORC1).
Biochemical and genetic analysis of mTOR has demonstrated that it is present in two functionally distinct complexes. The core components of mTORC1 consist of mTOR, mammalian lethal with sec-13 protein 8 (mLST8) and regulatory associated protein of TOR (raptor). Additional components include DEP-domain containing mTOR interacting protein (DEPTOR) and Proline–rich Akt substrate 40kDa (PRAS40). The mTOR complex 2 (mTORC2) core is composed of mTOR, rapamycin insensitive companion of mTOR (rictor), stress-activated protein kinase-interacting protein 1 (mSIN1) and mLST8. Protein observed with rictor 1/2 (protor 1/2) and DEPTOR are additional regulatory components (Cornu et al., 2013; Laplante and Sabatini, 2012). S6 kinase 1 (S6K1) and eukaryotic inhibition factor eIF4E binding protein 1 (4E-BP1) are two well characterized substrates of mTORC1 (Ma and Blenis, 2009). Only mTORC1 is acutely sensitive to inhibition by rapamycin. However, long-term exposure to rapamycin inhibits mTORC2 in some cell types by sequestering newly synthesized mTOR molecules (Laplante and Sabatini, 2012).
Over the past two decades, significant progress has been made in understanding the complexity of mTORC1 regulation and its roles in disease. mTORC1 is a signal integrator responding to multiple signals from growth factors, nutrients, energy and oxygen status to control processes that are required for cell growth and proliferation, including mRNA biogenesis, protein, lipid and nucleotide synthesis, energy metabolism and autophagy (Figure 1). Improper regulation of the mTORC1 pathway is frequently found in cancers as well as in several genetic disorders. Recent evidence indicates that mTORC1 is also an important modulator for aging and age-related diseases (Johnson et al., 2013a). In contrast to mTORC1, much less is known about mTORC2. mTORC2 participates in cell survival via activation of Akt and SGK1. mTORC2 also regulates organization of the actin cytoskeleton through activation of PKCα, paxillin and small GTPases, Rho and Rac (Laplante and Sabatini, 2012)
Figure 1.
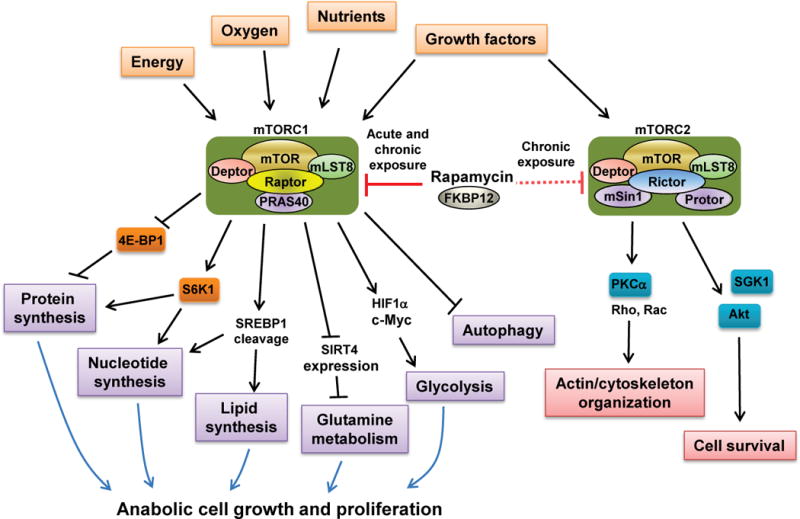
The two mTOR complexes and the regulation of key cellular processes. mTOR exists in two functionally distinct complexes termed mTORC1 and mTORC2. mTORC1 integrates multiple signals from growth factors, oxygen, energy levels and nutrients such as amino acids to promote cell growth and proliferation by activation of anabolic processes such as protein, lipid and nucleotide synthesis, stimulation of energy metabolism such as glycolysis and glutaminolysis, and inhibition of catabolic process such as autophagy. Unlike mTORC1, mTORC2 only responds to growth factors and regulates actin/cytoskeleton organization and cell survival through the pathways as shown above. Rapamycin acutely inhibits mTORC1 whereas chronic exposure to rapamycin can also inhibit mTORC2.
Effects of rapamycin in cancer
Increased activation of mTORC1 is observed in numerous human cancers due to gain-of-function mutations in oncogenes (i.e., PI3K, AKT, or Ras) and/or loss-of-function mutations in tumor suppressors (i.e., PTEN, LKB1 or TSC1/2), upstream regulators of mTORC1. These mutations provide cancer cells with a selective growth advantage in comparison to normal cells (Menon and Manning, 2008). In order to meet the high demands of proliferation, cancer cells often have fundamental alterations in nutrient uptake and energy metabolism, processes that are directly controlled by the mTORC1 pathway. Accordingly, in addition to driving protein synthesis, oncogenic activation of mTORC1 promotes a gene expression program that is involved in cancer cell metabolic reprogramming. Activation of mTORC1 promotes glycolysis via upregulation of Hypoxia-inducible factor alpha (HIF1α) and c-Myc; stimulates lipid biosynthesis and the pentose phosphate pathway through sterol regulatory element binding protein 1 (SREBP-1) (Yecies and Manning, 2011); and positively controls glutamine metabolism by SIRT4 repression (Csibi et al., 2013). Thus, drugs that selectively target mTORC1, like rapamycin, are expected to impair cancer metabolism and are considered promising anti-cancer therapies.
The poor solubility and pharmacokinetics of rapamycin triggered the development of several rapamycin analogs (rapalogs). Two water-soluble derivatives of rapamycin, temsirolimus and everolimus, were approved by the Food and Drug Administration (FDA) in 2007 and 2009, respectively, for the treatment of advanced renal cancer carcinoma (RCC). In 2011, the FDA approved the use of everolimus for patients with progressive neuroendocrine tumors of pancreatic origin (PNET). Additionally, temsirolimus was evaluated in several clinical trials for the treatment of advanced neuroendocrine carcinoma (NEC), advanced or recurrent endometrial cancer, and relapsed or refractory mantle cell lymphoma (MCL, approved in the European Union in 2009). Moreover, a few trials of everolimus were conducted in patients with advanced gastric cancer, advanced non-small cell lung cancer (NSCLC), and advanced hepatocellular carcinoma. Ridaforolimus, a rapamycin analog, was also examined in clinical trials for advanced bone and soft-tissue sarcomas as well as a variety of advanced solid tumors (Wander et al., 2011). Overall, however, rapalogs have only achieved modest effects in major solid tumors in the clinic. The reasons for the limited clinical success of rapalogs have not been established, but are likely related to the large number of mTORC1-regulated negative feedback loops that suppress upstream signaling systems such as activation of receptor tyrosine kinases, PI3K-Akt signaling and Ras-ERK pathway and which can be re-activated with rapamycin (discussed more extensively below). In order to overcome these limitations, alternative strategies have been explored in the past few years. For instance, a number of ATP-competitive mTOR inhibitors have been developed, blocking both mTORC1 and mTORC2 activity. Due to high sequence homology shared between mTOR and PI3K, some compounds that were originally identified as PI3K inhibitors were later shown to inhibit mTOR. Unlike rapamycin, which is a specific allosteric inhibitor of mTORC1, these ATP-competitive inhibitors target the catalytic site and prevent the feedback-mediated PI3K/Akt activation (described below), and therefore can potentially offer broader, more potent and sustained mTOR inhibition (Benjamin et al., 2011) (Figure 2).
Figure 2.
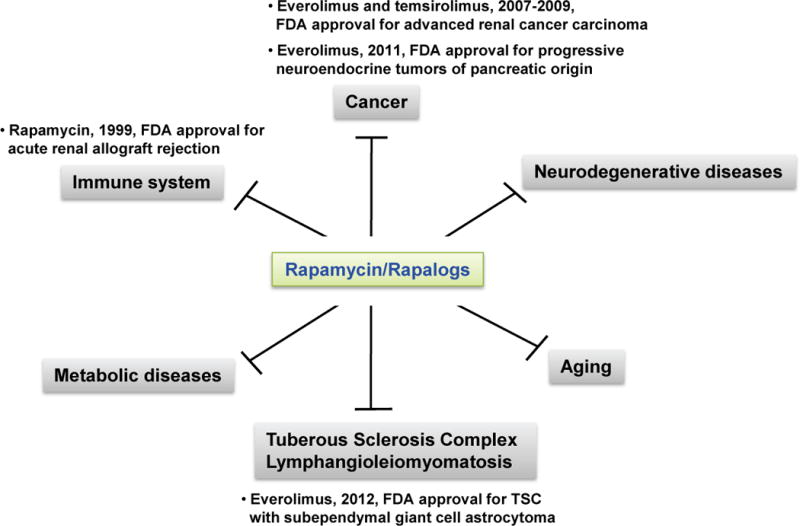
Effects of rapamycin in various diseases. A growing body of evidence has demonstrated that although limited, rapamycin or rapalogs are beneficial for treating various diseases including cancer, diabetes, tuberous sclerosis complex, lymphangioleiomyomatosis, neurodegenerative diseases and aging. FDA-approved cases are described.
Effects of rapamycin in Lymphangioleiomyomatosis (LAM), Tuberous Sclerosis Complex (TSC), and neurological diseases
LAM is a rare lung disease characterized by abnormal proliferation of smooth muscle-like cells. Infiltration of the lung with these cells by a mysterious mechanism referred to as benign metastasis, can result in an anti-inflammatory response, cyst formation and ultimately respiratory failure (Yu and Henske, 2010). LAM arises almost exclusively in women during their childbearing age, and occurs either as an isolated disorder (sporadic LAM) or in association with tuberous sclerosis complex (TSC). TSC is a genetic disorder characterized by the development of benign hamartomatous tumors involving multiple organ systems. Both LAM and TSC are caused by loss-of-function mutations in the TSC genes (TSC1 or TSC2), whose protein products function as a complex to inhibit the activity of mTORC1. Thus, hyperactive mTORC1 signaling is directly linked to the unregulated cell growth that drives the clinical manifestations. Hence, the use of mTORC1 inhibitors could potentially benefit LAM and TSC patients. In the past few years, several clinical trials were conducted to examine the efficacy and safety of rapamycin-based therapy. For example, in 2011, McCormack et al reported a double-blind, randomized, placebo controlled trial in which 89 patients with LAM who had moderate lung impairment were assigned to either sirolimus (also known as rapamycin) or placebo for 1 year, followed by 1-year observation period. Their study demonstrated that treatment with sirolimus for 1-year stabilized lung function and improved quality of life in patients with LAM. However, once patients stopped taking sirolimus, their lung function deteriorated again. The authors concluded that additional trials are required to investigate the duration of treatment and long-term safety of sirolimus (McCormack et al., 2011). Approximately 80% of all TSC patients and 50% of women with LAM also develop angiomyolipomas (AML), which are benign tumors of the kidney. Bissler et al conducted two separate trials to evaluate the use of sirolimus or everolimus for AML in TSC or LAM. Their results showed that sirolimus or everolimus decreased AML volume (Bissler et al., 2013; Bissler et al., 2008). However, the tumors returned to their original states when sirolimus was discontinued, underscoring the cytostatic and not cytotoxic effects of rapamycin and rapalogs. Since rapamycin-based therapy suppresses immune function and may cause serious side effects such as thrombocytopenia and hyperlipidemia, impaired wound healing, nephrotoxicity, and altered insulin sensitivity, the safety of long-term use still remains uncertain.
A growing body of evidence suggests that the mTORC1 pathway may also be involved in a number of neurological disorders. For instance, patients with TSC are prone to a wide range of neurological diseases, such as epilepsy, neurocognitive dysfunction and autism. Therefore, inhibition of mTORC1 is a potential therapeutic option. In a mouse model of TSC, rapamycin treatment was shown to suppress epilepsy, and shortly after, the study by Muncy et al demonstrated that rapamycin reduced seizure frequency in a 10-year-old girl with TSC (Muncy et al., 2009). A recent study by Sato et al reported that rapamycin reverses impaired social interactions associated with autism spectrum disorder in a mouse model of TSC. Their data suggested that the abnormal behaviors displayed by TSC-deficient mice were correlated with altered gene and protein expressions involved in the mTOR signaling (Sato et al., 2012). In 2012, the FDA approved the use of everolimus for the treatment of patients with TSC who have subependymal giant cell astrocytoma (SEGA), a neurological manifestation of TSC. Currently, there is an ongoing phase 2 trial assessing the possible neurocognitive benefits from treatment with everolimus (NCT01289912). Future therapies involving the use of ATP-competitive mTOR inhibitors or in combinations with other drugs, such as estrogen antagonists for the treatment of LAM are being explored.
Finally, a number of studies have indicated that pharmacological inhibition of mTORC1 can provide neuroprotection in several in vivo models of neurodegenerative diseases, including Alzheimer’s, Parkinson’s, Huntington’s and spinocerebellar ataxia type 3 (Bove et al., 2011). Mounting evidence suggested that accumulation of misfolded and aggregated proteins is a common feature of these diseases, possibly caused by mTORC1-driven protein synthesis and defective autophagic degradation. Thus, suppression of protein synthesis and induction of autophagy by rapamycin may prevent or diminish protein aggregation. Moreover, a recent study has shown that rapamycin improves survival and attenuates disease progression in a mouse model (Ndufs4−/−) of Leigh syndrome, which is a neurological disorder characterized by a genetic defect that disrupts mitochondrial function (Johnson et al., 2013b). For more detailed descriptions of the molecular mechanisms underlying how rapamycin exerts its neuroprotective effect, we refer the readers to a comprehensive review (Bove et al., 2011) (Figure 2).
Effects of rapamycin on longevity and aging
mTOR signaling is an important player in longevity regulation. Genetic or pharmacological inhibition of mTOR signaling has been found to extend lifespan of invertebrates including yeast, nematodes and fruit flies (Lamming et al., 2013). In 2009, Harrison et al showed that rapamycin extends both median and maximal lifespan of male and female genetically heterogeneous mice when beginning treatment at 9 or 20-months of age (1.5–2 years duration) (Harrison et al., 2009), representing the first demonstration in mammals. Subsequent work by other groups confirmed the positive effect of rapamycin on lifespan in mice with different genetic backgrounds and other model organisms (Lamming et al., 2013). Two classes of explanations may account for these observations: 1). rapamycin increases lifespan by slowing aging; or 2). rapamycin inhibits detrimental metabolic diseases or lethal neoplastic diseases, independent of aging effects. To test the hypothesis that rapamycin might retard aging in mice, Wilkinson et al used a genetically heterogeneous mouse model and analyzed a number of age-related pathologies as well as age-dependent spontaneous activity of mice upon rapamycin treatment beginning at 9-months of age (1 year duration). Their results suggested that age-dependent changes occur more slowly in rapamycin-treated mice, including alterations in heart, liver, endometrium, adrenal gland, and tendon elasticity. Rapamycin was also shown to diminish age-related decline in spontaneous activity of mice (Wilkinson et al., 2012).
As rapamycin is known to have modest anti-proliferative properties in many forms of cancer, lifespan extension by rapamycin could also be caused by suppression of specific life-limiting pathologies (e.g., cancer). In a recent article by Neff et al 2013, several concerns were raised with regard to the previous reports on the effect of rapamycin in slowing aging. For example, cancer is the main cause of death in mice including the mice strain used in the study by Wilkinson et al. In addition, aging-independent effects by rapamycin were not examined previously. In the study by Neff et al 2013, the authors dosed male C57BL/6j mice with rapamycin or vehicle control for 1 year at 3 different treatment onsets: young adulthood (i.e., at 4 months), midlife (i.e., at 13 months), and late in life (i.e., at 20–22 months). After completing the treatment, they performed a large assessment of diverse structural and functional aging phenotypes in a variety of cell types, tissues and organ systems. Intriguingly, while rapamycin did extend lifespan in mice, age-related traits were largely unaffected. Although rapamycin was able to rescue a subset of aging-dependent phenotypes, such as spatial learning and memory impairments, as well as declined exploratory activity, similar positive effects on many of these attributes were also observed in young mice, indicating an age-independent effect. They reasoned that the discrepancy in findings could be due to different mouse models (genetic backgrounds) used in the studies or technical variations in the analysis (Neff et al., 2013), which, however were challenged by Blagosklonny (Blagosklonny, 2013). Clearly, future studies with other mouse strains and gender are warranted (Figure 2).
Effects of rapamycin on metabolism
In response to nutrients and growth factors, mTOR signaling serves as a key regulator of cell metabolism to coordinate the balance between anabolic and catabolic processes. When fasting, muscle and liver produce glucose via glycogenolysis (glycogen breakdown) and gluconeogenesis (glucose synthesis), and adipose tissue generates fatty acids through lipolysis, whereas, upon feeding, glycogenesis (glycogen synthesis) is favored in muscle and liver, and lipid uptake is favored in adipose tissue. Dysregulation of mTOR signaling has been linked to the development of a few metabolic diseases, such as diabetes and obesity (Laplante and Sabatini, 2012). Inhibition of the mTOR pathway by rapamycin has been demonstrated to have both beneficial and detrimental effects on metabolism. For example, acute rapamycin treatment improves insulin sensitivity in vitro and in vivo by disrupting a S6K-mediated feedback loop (described below) (Krebs et al., 2007; Tremblay and Marette, 2001). Moreover, rapamycin treatment inhibits human adipocyte differentiation in vitro (Bell et al., 2000) and protects against high fat diet-induced obesity in C57BL/6J mice (16 weeks) (Chang et al., 2009a). As discussed above, rapamycin appears capable of extending the lifespan of mice and preventing the onset of many age-related diseases, however, deleterious metabolic effects associated with rapamycin were also reported in other studies. For instance, Fradenkel et al found that rapamycin treatment (2 weeks) worsened hyperglycemia in a nutrition-dependent type 2 diabetes mouse model (Fraenkel et al., 2008). Similarly, the study by Chang et al demonstrated that rapamycin administration (6 weeks) exacerbates glucose intolerance in diet-induced obesity KK/HIJ mice (Chang et al., 2009b). Moreover, the study by Houde et al reported that rapamycin treatment (2 weeks) promoted insulin resistance and hyperlipidemia in rats (Houde et al., 2010). It is currently unclear how rapamycin can have both positive and negative effects. In 2012, Lamming et al showed that rapamycin exerts different effects via separate mechanisms. They found that while reduced mTORC1 increases longevity and maintains normal glucose homeostasis, disruption of mTORC2 contributes to insulin resistance in vivo (Lamming et al., 2012). Additionally, Fang and colleagues suggested that these seemingly controversial findings might be explained by the duration of rapamycin treatment (Fang et al., 2013). Their study compared various metabolic effects in male mice after 2, 6, or 20 weeks of rapamycin treatment. After 2 weeks of rapamycin treatment, the mice displayed smaller pancreas and enlarged liver. However, with prolonged treatment, these features returned to normal levels while adiposity, body weight (BW), and food consumption were significantly reduced. More strikingly, insulin sensitivity was altered with respect to different lengths of rapamycin treatment. Under normal physiological conditions, insulin suppresses hepatic gluconeogenesis while increasing lipogenesis, but in individuals with impaired insulin sensitivity, excess insulin is secreted to compensate for the insulin resistance. In agreement with previous findings by Houde et al (Houde et al., 2010), insulin levels were increased after 2 weeks of rapamycin treatment, and the treated mice became glucose intolerant and insulin resistant. Interestingly, after 6 weeks of treatment the mice exhibited a transition to improved insulin sensitivity and by 20 weeks of rapamycin treatment, insulin levels were decreased while insulin sensitivity increased. Additional metabolic effects including lipid profile, oxygen consumption and ketogenesis were also altered in a manner relative to treatment durations (Fang et al., 2013). Together, these remarkable findings may provide insight into how long-term treatment of rapamycin extends longevity in mice. However, how these studies will translate to human aging is unclear (Figure 2).
Limitations of monotherapy
The beneficial effects of rapamycin have been largely shown in pre-clinical animal models. However, the clinical success of rapamycin has been associated with only a few benign and malignant cancers. Several factors may contribute to the modest outcome in the clinic. 1). Rapamycin and rapalogs are generally cytostatic instead of cytotoxic. Clinical trials have demonstrated that while rapamycin treatment can induce tumor regression, tumors regrow upon the cessation of treatment (Bissler et al., 2008; Marsh et al., 2008) 2). Rapamycin fails to completely inhibit a subset of mTORC1-mediated processes, such as protein synthesis and autophagy. Choo et al reported that rapamycin differentially regulates S6Ks versus 4E-BP1. In several cell types, short-term treatment of rapamycin de-phosphorylates 4E-BP1, however, phosphorylation of 4E-BP1 remerges after 12+ hours treatment, resulting in a recovery in cap-dependent translation despite continued inhibition of S6K (Choo et al., 2008). In addition, although it was known that rapamycin effectively activates autophagy in yeast, the induction of autophagy is only mild in mammalian cells (Thoreen and Sabatini, 2009). These partial inhibitory effects may offer a potential explanation for the poor efficacy of rapamycin in clinical trials. However, it is puzzling why mTORC1 substrates have differential sensitivities to rapamycin treatment in cells. In a recent study, Kang and colleagues found that mTORC1 phosphorylation sites are one determinant for the differential responses to rapamycin. It was shown that the ability of mTORC1 to phosphorylate a specific site in vitro has a strong correlation with the respective rapamycin-resistant site in cells. For example, mTORC1 potently phosphorylated the synthetic peptides encompassing 4E-BP1 (T37) and (T46), which are rapamycin resistant. In contrast, mTORC1 weakly phosphorylated S6K1 (T389), which is rapamycin sensitive. Furthermore, their findings indicated that the sequence composition of mTORC1 phosphorylation sites is one essential factor in determining substrate quality within cells. Lastly, Kang et al were able to control mTORC1 activity toward a substrate phosphorylation site in vitro, and subsequently alter substrate sensitivity to rapamycin, nutrient and growth factor levels in cells as well (Kang et al., 2013). 3). Another reason for the limited clinical success of rapalogs is the existence of several feedback loops involved in cell survival responses. For instance, under normal circumstances, mTORC1-mediated activation of S6K1 promotes degradation of insulin receptor substrate (IRS) and subsequent downregulation of PI3K signaling. Inhibition of mTORC1 by rapamycin relieves this negative feedback loop, thereby increasing PI3K signaling. Likewise, mTORC1-mediated signaling via S6K1 also inhibits mTORC2 by phosphorylation of Rictor. Rapamycin blocks this feedback loop, resulting in mTORC2-mediated AKT activation. Additionally, mTORC1 negatively regulates growth factor receptor-bound protein 10 (Grb10), an inhibitor of PI3K signaling. Thus, rapamycin released mTORC1-mediated inhibition of Grb10, leading to PI3K pathway activation in certain cell types (Laplante and Sabatini, 2012) (Figure 3). 4). Recent studies also demonstrate that inhibition of PI3K/AKT/mTOR pathway can lead to reactivation of many receptor tyrosine kinases and their downstream signaling systems (Chandarlapaty et al., 2011) 5). Compensatory pathways exist that are often activated in cancer cells or up-regulated in response to chronic rapamycin treatment (discussed above). For example, other AGC kinase family members can compensate for S6K1 inhibition such as RSK and Akt (Mendoza et al., 2011). 6). Finally, eIF4E expression can be up-regulated with chronic inhibition of mTOR (Alain et al., 2012). One of the major unanswered questions in the field is why these various compensatory mechanisms and negative feedback loops are differentially regulated in different organisms or in different cancer cell types. Fully addressing this question will lead to an improved understanding as to why rapalogs work better in some cases than in others, and that should lead to improved therapeutic responses with mTOR inhibitors in monotherapy or combination therapy.
Figure 3.
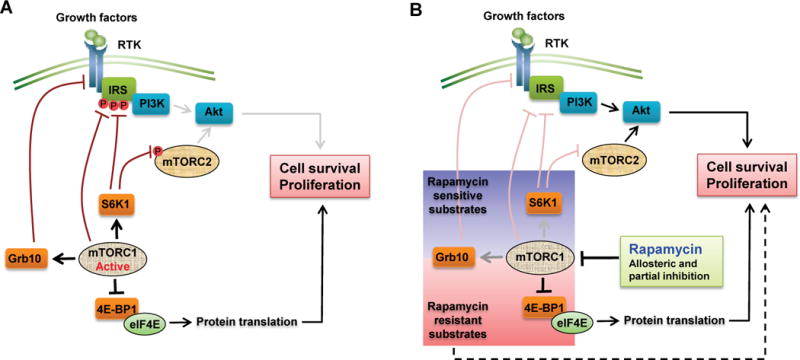
(A) Many negative feedback loops in mTOR signaling. When mTORC1 is active, Akt activation is attenuated by inhibition of the RTK-PI3K signaling pathway through multiple mechanisms including inhibitory phosphorylation events on IRS by mTORC1 or S6K1 and Rictor by S6K1, and stimulation of inhibitory adaptor protein Grb10 by mTORC1. (B) Rapamycin relieves negative feedback loops and promotes activation of PI3K signaling. The inhibition of mTORC1 by rapamycin reduces the mTORC1-driven negative feedback effects, and thus activates the PI3K-Akt pathway and promotes cell survival. Additionally, partial inhibition of mTORC1 substrates by rapamycin such as 4E-BP1 may also contribute to cell survival.
Rapamycin in combination therapy
The modest effect of rapamycin-based therapy has prompted investigators to consider other therapeutic options. Combination therapy with rapamycin or rapalogs has become a promising approach as various strategies have the potential of improving drug efficacy by inhibiting multiple targets including those activated by removal of feedback loops or those involved in parallel pathways. Additionally, combination approaches may delay the emergence of drug resistance.
In 2012, everolimus was approved by the FDA in combination with exemestane (aromatase inhibitor) for the treatment of postmenopausal women with advanced hormone receptor-positive HER2-negative breast cancer who experience recurrence or progression after treatment with letrozole or anastrozole (aromatase inhibitors) alone. In addition, multiple clinical trials are currently evaluating the efficacy of rapalogs with several chemotherapeutic agents, including paclitaxel, doxorubicin and capecitabine. Furthermore, pre-clinical and clinical data suggested that rapalogs work effectively with targeted therapies to enhance anti-tumor activity, such as receptor tyrosine kinase (RTK) targeted therapy, hormone-based therapy, angiogenesis inhibitors and IGF-IR inhibitor. For example, the combination of rapamycin and erolitinib, (epidermal growth factor receptor (EGFR) inhibitor), resulted in a synergistic effect in vitro and significantly reduced tumor growth in vivo compared to single agent treatment. This may be linked to the activation of RTKs by inhibition of the PI3K/Akt/mTOR pathway (Chandarlapaty et al., 2011). In addition, a phase I trial examined the combination of everolimus with pasireotide, a somatostatin analog, in patients with advanced neuroendocrine tumors. The preliminary data showed that the combination induced antitumor activity and further studies are underway (Zaytseva et al., 2012). Furthermore, everolimus was shown to potently inhibit growth in many tumor types with activated mTOR in combination with lower doses of the catalytic mTOR inhibitor BEZ235, used to reduce survival via feedback loops activating Akt (Nyfeler et al., 2012). Many more clinical trials are currently investigating different combination approaches. Exciting outcomes from these studies are anticipated in the near future.
Conclusion and future perspective
Since the serendipitous discovery of rapamycin, considerable achievements have been made in understanding the mechanism of action and unraveling the intricate signaling network of mTOR. Uncontrolled mTORC1-mediated signaling is often observed in human diseases. Therefore, it was thought that pharmacological inhibition of mTOR by rapamycin would have a substantial and wide range of clinical effects. Although rapamycin-based therapy has shown benefits for patients with RCC, TSC and LAM-related tumors, the use of rapamycin monotherapy in a broad spectrum of metabolic diseases, especially in treating cancers, is limited due to its modest efficacy. This may be explained by the inability of rapamycin to completely block mTORC1-mediated signaling events, the presence of several feedback loops and the up-regulation of compensatory pathways that promote cell survival and growth. Thus, there is a critical need to further define these signaling processes and to develop new strategies that can overcome these drawbacks. The recent emergence of combination therapy with rapamycin may further increase efficacy and bypass feedback activation of survival pathways. Efforts that focus on exploring novel drug combinations with optimal doses will have great potential to yield an improvement of efficacy and safety profiles. Additionally, significant promise remains for the discovery of new pathway inhibitors as well as the possibility that existing bioactives may directly or indirectly reduce mTORC1 and/or mTORC2 activity in monotherapy or in combination.
Acknowledgments
We apologize to those whose work was not discussed and cited in the review due to limitations in space and scope. We thank the Blenis Lab members Drs. Michal Nagiec, Alfredo Csibi, Gina Lee, Bin Xue and Sang-Oh Yoon for helpful discussions and comments on this manuscript. JL is a Tuberous Sclerosis Alliance Postdoctoral Fellow. JB is an Established Investigator of the LAM Foundation. NIH Grants GM51405 and CA46595 provide research support for the Blenis laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alain T, Morita M, Fonseca BD, Yanagiya A, Siddiqui N, Bhat M, Zammit D, Marcus V, Metrakos P, Voyer LA, et al. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer research. 2012;72:6468–6476. doi: 10.1158/0008-5472.CAN-12-2395. [DOI] [PubMed] [Google Scholar]
- Bell A, Grunder L, Sorisky A. Rapamycin inhibits human adipocyte differentiation in primary culture. Obesity research. 2000;8:249–254. doi: 10.1038/oby.2000.29. [DOI] [PubMed] [Google Scholar]
- Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nature reviews Drug discovery. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV. Rapamycin extends life- and health span because it slows aging. Aging. 2013;5:592–598. doi: 10.18632/aging.100591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell. 2011;19:58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GR, Chiu YS, Wu YY, Chen WY, Liao JW, Chao TH, Mao FC. Rapamycin protects against high fat diet-induced obesity in C57BL/6J mice. Journal of pharmacological sciences. 2009a;109:496–503. doi: 10.1254/jphs.08215fp. [DOI] [PubMed] [Google Scholar]
- Chang GR, Wu YY, Chiu YS, Chen WY, Liao JW, Hsu HM, Chao TH, Hung SW, Mao FC. Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol. 2009b;105:188–198. doi: 10.1111/j.1742-7843.2009.00427.x. [DOI] [PubMed] [Google Scholar]
- Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Grammer TC, Lemon KP, Kazlauskas A, Blenis J. PDGF- and insulin-dependent pp70S6k activation mediated by phosphatidylinositol-3-OH kinase. Nature. 1994;370:71–75. doi: 10.1038/370071a0. [DOI] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69:1227–1236. doi: 10.1016/0092-8674(92)90643-q. [DOI] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Current opinion in genetics & development. 2013;23:53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, Jeong SM, Dempsey JM, Parkhitko A, Morrison T, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell. 2013;153:840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Westbrook R, Hill C, Boparai RK, Arum O, Spong A, Wang F, Javors MA, Chen J, Sun LY, et al. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metab. 2013;17:456–462. doi: 10.1016/j.cmet.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57:945–957. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013a;493:338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A, et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science. 2013b doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013;341:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, et al. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56:1600–1607. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- Kuo CJ, Chung J, Fiorentino DF, Flanagan WM, Blenis J, Crabtree GR. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature. 1992;358:70–73. doi: 10.1038/358070a0. [DOI] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. The Journal of clinical investigation. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011;189:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Trahair TN, Martin JL, Chee WY, Walker J, Kirk EP, Baxter RC, Marshall GM. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5:357–361. doi: 10.1038/ncponc1112. [DOI] [PubMed] [Google Scholar]
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in biochemical sciences. 2011;36:320–328. doi: 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Manning BD. Common corruption of the mTOR signaling network in human tumors. Oncogene. 2008;27(Suppl 2):S43–51. doi: 10.1038/onc.2009.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, et al. Rapamycin extends murine lifespan but has limited effects on aging. The Journal of clinical investigation. 2013;123:3272–3291. doi: 10.1172/JCI67674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyfeler B, Chen Y, Li X, Pinzon-Ortiz M, Wang Z, Reddy A, Pradhan E, Das R, Lehar J, Schlegel R, et al. RAD001 enhances the potency of BEZ235 to inhibit mTOR signaling and tumor growth. PloS one. 2012;7:e48548. doi: 10.1371/journal.pone.0048548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DJ, Grove JR, Calvo V, Avruch J, Bierer BE. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science. 1992;257:973–977. doi: 10.1126/science.1380182. [DOI] [PubMed] [Google Scholar]
- Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, Mizuguchi M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292. doi: 10.1038/ncomms2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5:725–726. doi: 10.4161/auto.5.5.8504. [DOI] [PubMed] [Google Scholar]
- Tremblay F, Marette A. Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. The Journal of biological chemistry. 2001;276:38052–38060. doi: 10.1074/jbc.M106703200. [DOI] [PubMed] [Google Scholar]
- Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of clinical investigation. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson JE, Burmeister L, Brooks SV, Chan CC, Friedline S, Harrison DE, Hejtmancik JF, Nadon N, Strong R, Wood LK, et al. Rapamycin slows aging in mice. Aging Cell. 2012;11:675–682. doi: 10.1111/j.1474-9726.2012.00832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer research. 2011;71:2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Henske EP. mTOR activation, lymphangiogenesis, and estrogen-mediated cell survival: the “perfect storm” of pro-metastatic factors in LAM pathogenesis. Lymphatic research and biology. 2010;8:43–49. doi: 10.1089/lrb.2009.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaytseva YY, Valentino JD, Gulhati P, Evers BM. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319:1–7. doi: 10.1016/j.canlet.2012.01.005. [DOI] [PubMed] [Google Scholar]