

Abstract
Although the classical antibiotic spectinomycin is a potent bacterial protein synthesis inhibitor, poor antimycobacterial activity limits its clinical application for treating tuberculosis. Using structure-based design, a novel semisynthetic series of spectinomycin analogs was generated with selective ribosomal inhibition and excellent narrow-spectrum antitubercular activity. In multiple murine infection models, these spectinamides were well tolerated, significantly reduced lung mycobacterial burden and increased survival. In vitro studies demonstrated a lack of cross-resistance with existing tuberculosis therapeutics, activity against MDR/XDR-tuberculosis, and an excellent pharmacological profile. Key to their potent antitubercular properties was their structural modification to evade the Rv1258c efflux pump, which is upregulated in MDR strains and is implicated in macrophage induced drug tolerance. The antitubercular efficacy of spectinamides demonstrates that synthetic modifications to classical antibiotics can overcome the challenge of intrinsic efflux pump-mediated resistance and expands opportunities for target based tuberculosis drug discovery.
Introduction
Nearly seventy years since the discovery of streptomycin, the first antibiotic used to treat tuberculosis, this disease remains a formidable cause of human mortality with 1.4 million deaths/year.1 Multidrug-resistant (MDR) strains cause an estimated 300,000 deaths/year and extensively drug-resistant (XDR) tuberculosis has spread to 84 countries with some strains reportedly resistant to all available drugs.2 With the exception of rifamycins, antitubercular therapeutic regimes consist of unaltered natural products or completely synthetic molecules. This is in contrast to the therapeutic regimes for other bacterial infections, which are dominated by semisynthetic derivatives of natural products. The success of semisynthetic drugs is attributed to the high structural diversity of their antibiotic cores not found in purely synthetic collections and the synthetic modifications that maximize potency, safety and distribution in humans.3,4 With the expanding structural and molecular information available for drug targets, reevaluation of existing antibacterial classes underutilized for tuberculosis may provide opportunities for synthetic modifications that retain or improve target affinity whilst circumventing native resistance mechanisms, such as efflux.
One structurally distinct antibiotic that has not yielded any approved semi-synthetic analogs, and has limited activity against M. tuberculosis, is spectinomycin. It has historically been used as a second-line agent to treat gonorrheal infections. Although spectinomycin is chemically similar to aminoglycosides, it binds to a separate site within the 16S bacterial ribosomal subunit, designated helix 34, and blocks ribosome translocation.5,6 Unlike aminoglycosides, spectinomycin has a high safety margin, having only minor side effects (injection site soreness, chills, and nausea) with no nephrotoxicity or ototoxicity when administered for a short term at high therapeutic doses.7,8
In this study, we describe the design, synthesis, and evaluation of a class of novel semisynthetic spectinomycin analogs, the spectinamides, as antitubercular agents. These studies were inspired by: 1) the finding that researchers in the 1980's were able to generate spectinomycin analogs with increased activity against gram positive pathogens;9–12 2) the recently available crystal structures of spectinomycin bound to the ribosome;6 and 3) the design approach used for tigecycline,13 a semisynthetic broad-spectrum tetracycline whose increased activity is ascribed to efflux avoidance by the key synthetic addition of a glycyl side chain.14 From this knowledge, we hypothesized that a structure based design cycle could be used to generate spectinomycin analogs that have increased ribosomal target affinity and/or avoid active efflux, thus gaining potency against M. tuberculosis at therapeutically achievable concentrations. The spectinamides described herein are potent, bacterial ribosomal inhibitors that avoid efflux by M. tuberculosis to achieve excellent antitubercular efficacy in vivo.
Results
Structure-guided modification of spectinomycin successfully increases antitubercular activity
Spectinomycin has a uniquely fused tricyclic architecture in which the diaminocyclitol moiety actinamine (ring A) is fused to a single sugar component actinospectose (ring C) via a β-glycosidic and a hemiketal linkage to form ring B (Fig. 1a).15 To determine how spectinomycin could be structurally modified while retaining ribosomal affinity, we analyzed past literature and recent structural studies, and built a homology model of the M. tuberculosis 16S helix 34 spectinomycin-binding site from the E. coli 30S spectinomycin structure.6 These analyses revealed that any modification of the spectinomycin core was most likely not possible via the aminocyclitol A ring,16–18 the oxocyclic B ring, or the B-C ring fusion, which are responsible for most of the key hydrogen bonding interactions within helix 34 of the 16S ribosomal RNA (Fig. 1b). However, stereospecific modification of the 3'keto group to an R-amine was well tolerated in our model predictions. From this we hypothesized that derivatization of the amine would allow introduction of functional groups that could: (i) potentially make extra ribosomal contacts adjacent to helix 34 and with protein loop RpsE; (ii) modulate transporter affinity and overall physicochemical properties.
Figure 1.

(a-b) The structure and ribosomal binding site of spectinomycin. (a) Tricyclic structure of spectinomycin sketched manually with ring numbering indicated. (b) Spectinomycin bound to helix 34 with hydrogen bonding interactions indicated by yellow dashes derived from crystal structure PDB 2QOU. The nearby protein loop of RpsE is shown in magenta. From this analysis, the 3'keto functionality (indicated by arrow) on the C ring is clearly available for modification, as indicated by the arrow. (c–d) The stable conformational complex of the spectinamides in the ribosomal spectinomycin-binding site as determined by 5ns molecular dynamics simulation. (c) Compound 1544 modeled into the spectinamide helix 34 ribosomal binding site. (d) Display of the proposed binding interactions of the spectinamide side chain. E. coli numberings are shown in the figure. Corresponding numberings in M. tuberculosis for RpsE are V55(V24), V56(V25), R60(R29), R61(R30), F62(F31) and 16S are G1054(G1064), C1056(C1066), G1058(G1068), A1182(A1191), C1183(C1192), G1184(G1193). (e) In organello mitochondrial expression of S35 methionine labeled COX1 in the presence of inhibitors at indicated doses as determined by densitometry of autoradiograms. Representative results from three independent experiments are shown.
Consequently, an initial panel of 16 substituted spectinamides was synthesized and tested for antitubercular potency, antibacterial spectrum of activity, and protein synthesis inhibition (Supplementary Table 1). The synthesis was achieved in a convergent 4-step sequence from spectinomycin10 (Supplementary Scheme 1). From this compound set, 1329 was discovered as the initial lead (Table 1), which showed good MIC (minimal inhibitory concentration) activity specific to M. tuberculosis (1.6 μg/ml) and inhibition of mycobacterial ribosomal translation (1.2 μg/ml). Interestingly, several analogs in this compound set, such as 1351, inhibited mycobacterial ribosomes in in vitro translation assays at low concentrations (IC50 0.37 μg/ml) but had poor antitubercular activity (25 μg/ml). This suggested that like spectinomycin these compounds were not concentrating in the cell19, whereas 1329 accumulated intracellularly.
Table 1.
Structures and activities of representative spectinamides.
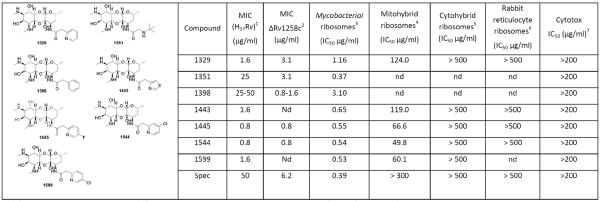
|
Abbreviations: MIC, minimum inhibitory concentration; Spec, spectinomycin; nd, not determined.
1,2MIC activity against M. tuberculosis H37Rv and Rv1258c Tap pump knockout strains (determined using method 1); 3translation inhibition against M. smegmatis (mycobacterial) ribosomes;4,5 bacterial ribosomes mutated to contain the mitochondrial (mitohybrid), and cytosolic (cytohybrid) helix 34 sequences;6 rabbit reticulocyte ribosomes representing native eukaryotic ribosomes;7 mammalian cytotoxicity determined against the Vero and J774 cell lines.
Lead spectinamides benefit from additional contacts to the ribosomal binding site
Spectinamide binding was further rationalized by in silico docking into our active site model using Glide,20 and performing 5 ns molecular dynamics simulations, accounting for conformational flexibility within helix 34 and the nearby protein loop of RpsE. Molecular dynamics simulations suggested that lead spectinamides formed a stable complex in the spectinomycin-binding site, with the side chain making contacts in a previously unexplored pocket located adjacent to helix 34 and RpsE (Fig. 1c). This structure-based approach also helped to rationalize the SAR observed during development of the series: first, the need for a 2-pyridyl group that makes a H-bond with the free ribosyl hydroxyls of C1192 or G1193 with an averaged calculated acceptor-donor distance of 3.04 Å; second, the need for an acetamide spacer which orientates the pyridyl ring appropriately to participate in these interactions.
Simulations indicated that introduction of halogen substituents into the meta and para positions of the pyridyl ring of 1329 would increase complex stability by locking the pyridyl nitrogen H-bonding interaction with G1193 and forming favorable interactions with backbone amide groups in the RpsE loop (Fig. 1d).21 This structure-guided modification of 1329 successfully produced optimized leads (1445, 1544, and 1599) with improved antitubercular potency (Table 1).
Lead spectinamides remain on target and are not cross resistant with frontline therapeutics
All lead spectinamides retained activity against a panel of M. tuberculosis strains including susceptible strains, strains mono-resistant to frontline therapeutics (Table 2), and MDR- and XDR-tuberculosis clinical isolates (Table 3, Supplementary Tables 2,3). Spontaneous mutants of M. tuberculosis arose at the following frequencies: 1329 (1.9–3.7 × 10−6), 1544 (1.3–3.1 × 10−6), and 1599 (1.6–7.4 × 10−7), which are comparable to isoniazid (0.14–3.2 × 10−7) and other frontline anti-tubercular drugs. Sequencing helix 34 in 6 spontaneous spectinamide-resistant mutants revealed changes at key nucleotides (C1066A, C1066G, C1192A, and A1191G), demonstrating that this modified series remained on molecular target.22 Importantly, spectinamide-resistant mutants displayed no cross-resistance with other protein synthesis inhibitors used to treat tuberculosis, including streptomycin, amikacin, kanamycin, capreomycin and linezolid. These results are consistent with the distinct ribosomal binding site for spectinomycin and its lack of inactivation by tubercular aminoglycoside-modifying enzymes such as acetyltransferase Eis.23
Table 2.
Activity of spectinamides against various mono-resistant typed M. tuberculosis strains
| Strain | Resistance | MIC (μg/ml) | ||||||
|---|---|---|---|---|---|---|---|---|
| 1329 | 1443 | 1445 | 1544 | 1599 | Spec | Strep | ||
| H37Rv | None | 1.6 | 0.8 | 1.6 | 1.6 | 1.6 | 100 | 1.6 |
| ATCC 358221 | Isoniazid (katG S315T) | 3.1 | 1.6 | 3.1 | 1.6 | 1.6 | 100 | 1.6 |
| ATCC 35820 | Streptomycin (rpsl K43R) | 3.1 | 1.6 | 6.3 | 1.6 | 1.6 | 200 | >200 |
| ATCC 358382 | Rifampin (rpoB S351L) | 1.6 | 6.3 | 0.8 | 1.6 | 1.6 | 100 | 1.6 |
| ATCC 358373 | Ethambutol | 3.1 | 1.6 | 6.3 | 1.6 | 1.6 | 100 | 1.6 |
| H37Rv-HL-144 | 1329 | >200 | >200 | >200 | >200 | >200 | >200 | 1.6 |
| H37Rv-HL-154 | 1329 | >200 | >200 | >200 | >200 | >200 | >200 | 1.6 |
Abbreviations: MIC, minimum inhibitory concentration; Spec, spectinomycin; Strep, streptomycin.
Isoniazid MIC = >12.5μg/ml;
Rifampin MIC = >25.0 μg/ml;
Ethambutol MIC = >25.0 μg/ml.
Representative spontaneous spectinamide-resistant mutants with base changes in 16S rRNA were not cross-resistant to linezolid, capreomycin, streptomycin, kanamycin, isoniazid, ethambutol, and rifampicin. MICs were determined using method 1.
Table 3.
Activity of spectinamides against multiple isolates of MDR- and XDR-tuberculosis1 (n= 24)
| Compound | MIC90 (μg/ml)2 | MIC50 (μg/ml)2 | MIC range (μg/ml)3 |
|---|---|---|---|
| 1329 | 3.8 | 2.3 | 1.6 – 7.5 |
| 1445 | 3.8 | 1.6 | 0.8 – 3.8 |
| 1544 | 1.8 | 1.6 | 0.8 – 1.8 |
| 1599 | 1.9 | 0.8 | 0.3 – 1.9 |
The panel consisted of 19 MDR and 5 XDR strains; MDR strains were resistant to isoniazid, rifampicin, and at least 2 other antitubercular antibiotics, whereas XDR strains were resistant to isoniazid, rifampicin, ofloxacin, and kanamycin. All strains were resistant to at least 4 different classes of antimycobacterial drugs; this included 8 streptomycin- and 7 kanamycin-resistant strains.
MIC90 and MIC50 refer to the MIC value at which 90% and 50% of the strains are susceptible.
The range refers to MIC values across the whole MDR/XDR panel of 24 strains. See Supplementary Table 3 for details. MICs were determined using methods 1 and 2.
Spectinamides are narrow spectrum inhibitors with activity against non-replicating M. tuberculosis bacilli
Significant antibacterial activity was only seen against our test set of mycobacteria from the tuberculosis complex and the closely related pathogens M. ulcerans and M. marinum (Supplementary Table 4) but not against an extensive panel of gram-positive and -negative bacteria (Tables S1). The only exception was activity observed against an E. coli strain deficient in TolC, a component of the major efflux pump AcrAB-TolC (Supplementary Table 1). Subsequent analysis showed lead spectinamides efficiently inhibit E. coli protein synthesis in coupled transcription/translation assays (Supplementary Fig. 1). As the spectinomycin-binding site is highly conserved across bacterial species, these results indicate active efflux or poor cellular uptake as major causes of compound inactivity in other species.
The potential for spectinamides to act against latent M. tuberculosis infections was assessed by comparing the potency of 1599 to frontline drugs isoniazid and rifampin under hypoxic conditions24 (Supplementary Table 5). As anticipated, a large proportion (>50%) of bacteria survived treatment with isoniazid, whereas less than 0.01% survived treatment with rifampin. Most encouragingly 1599 was also highly active as only 0.06% of bacteria survived treatment, indicating that spectinamides may be suitable to sterilize and act against persistent infections in vivo.
Spectinamides overcome efflux-mediated intrinsic resistance
In M. tuberculosis, efflux pumps are increasingly being found to play a role in the intrinsic resistance to many antimicrobial agents19. Recent studies demonstrated that deletion of multidrug transporter Rv1258c reduces intrinsic spectinomycin resistance.25 We suspected this transporter could influence the activity of spectinamides, since 1351, a glycyl substituted spectinamide is a potent ribosomal inhibitor but is only weakly antitubercular, whereas 1329, a 2-pyridyl spectinamide, is a less potent ribosomal inhibitor but has much stronger antitubercular activity (Supplementary Table 1). This suggested that chemical variations on the spectinamide substituent could overcome efflux mechanisms in M. tuberculosis.
To further explore this idea, spectinamides with good ribosomal inhibition and either excellent (1329, 1445, and 1544) or poor (1351 and 1398) antitubercular MICs were tested against an Rv1258c deficient strain of M. tuberculosis. A significantly lower MIC was observed in the Rv1258c knock-out mutant for 1351 and 1398 as well as spectinomycin, but 1329, 1445, and 1544 potencies were unaffected (Table 1). Complementation of the knock-out mutant with Rv1258c restored the MIC for 1351 and 1398 to wild type levels (Supplementary Table 6), thus confirming the role of Rv1258c in their efflux. Inactivation of all other pumps tested did not significantly affect the susceptibility to spectinomycin or the spectinamides tested (Supplementary Table 6).
Spectinamides do not inhibit mammalian mitochondrial or cytoplasmic translation
There was no detectable cytotoxicity for the lead compounds against two mammalian cell lines (Table 1). Concerned with the close homology of bacterial and human mitochondrial ribosomes, the well-documented mitochondrial toxicities and consequent side effects of many antibacterial protein synthesis inhibitors,26–29 we studied the effect of spectinamides on eukaryotic cytosolic and mitochondrial translation. In our in organello mitochondrial translation assay (Fig. 1e), linezolid had an IC50 value of 13.5 μM, which is similar to previously reported values.30 In contrast, spectinomycin and 1544 did not inhibit in organello mitochondrial translation, even up to 1 mM. To further validate target specificity and exclude off-target effects related to compound permeability, we compared the compounds' selective activity for bacterial and eukaryotic ribosomes in cell-free translation assays (Table 1).31,32 Compounds were first tested against rabbit reticulocyte ribosomes, which represent native eukaryotic ribosomes, and second, against constructed bacterial hybrid ribosomes with the bacterial helix 34 drug-binding pocket replaced by the corresponding human mitoribosomal and cytoribosomal homologs (Supplementary Fig. 2). Similar to spectinomycin, the spectinamides showed excellent target selectivity by demonstrating antiribosomal activity that spares both the eukaryotic cytosolic ribosome (selectivity index ratio of IC50 cytohybrid ribosome: IC50 bacterial ribosome > 500) and the eukaryotic mitochondrial ribosome (selectivity index ratio of IC50 mitohybrid ribosome: IC50 bacterial ribosome > 100) (Table 1). Leads 1544 and 1599 underwent an extensive in vitro profiling screen against 68 primary human molecular targets and the 5 major human cytochrome P450 enzymes to detect any relevant off-target pharmacologic effects (Supplementary Table 7). There was no significant response, including no inhibition of CYP450 enzymes and no interaction with the cardiac hERG potassium ion channel.
Lead spectinamides have favorable pharmacokinetic profiles
In vitro assays showed that spectinamides had low plasma protein-binding properties and were stable against hepatic microsomal metabolism (Table 4). Pharmacokinetic studies in rats revealed extensive renal elimination in the unchanged form for all compounds, with a disposition pattern similar to that of other aminoglycoside antibiotics (Table 4). A pharmacokinetic study performed in mice for 1599 indicated similar dose normalized systemic exposure between intravenous administration in rats and subcutaneous administration in mice, thereby bridging the results of the pharmacokinetic studies in rats to the subsequent in vivo efficacy studies in mice (Supplementary Table 8). The postantibiotic effect of spectinamides ranged from moderate (15 – 27 hours) for 1329, 1443, and 1445 to long (75 – 132 hours) for the chlorinated compounds 1544 and 1599, with the postantibiotic effect of 1599 being similar to that of streptomycin (132 hours). In pharmacodynamic time-kill experiments, compound 1445 demonstrated time-above–MIC dependent killing in contrast to the Cmax-dependent killing of aminoglycosides (Supplementary Fig. 3).33
Table 4.
Pharmacokinetic and pharmacodynamic properties of lead spectinamides.
| Property | Measurement | 1329 | 1443 | 1445 | 1544 | 1599 | Spectinomycin (unless noted) |
|---|---|---|---|---|---|---|---|
| Protein serum binding (Rat) | % bound | 28.6 (8.9) | 13.1 (0.3) | 27.7 (4.3) | 15.0 (9.9) | 12.5 (1.7) | 12.7 (7.5)2 |
| Microsomal metabolic stability (Rat) | % remaining after 90 min incubation | 83.7 (16.4) | 109 (2.7) | 102 (1.2) | 67.3 (16.3) | 88.0 (4.2) | 80.4 (19.9) |
| IV Pharmacokinetics (Rat) | t½ (h)1 | 0.52 (6.8) | 0.44 (0.8) | 0.45 (7.7) | 1.06 (9.2) | 0.58 (20.8) | 0.75 (49.3)2 |
| Vd (L/kg) | 1.15 (11.9) | 0.46 (10.3) | 0.57 (7.7) | 0.87 (22.9) | 0.82 (34.5) | 0.76 (45.2)2 | |
| CL (L/h/kg) | 0.72 (10.8) | 0.89 (6.8) | 0.56 (8.9) | 0.94 (6.3) | 1.22 (14.3) | 0.60 (11.5)2 | |
| % excreted unchanged in urine | 45.6 (9.7) | 86.9 (12.5) | 108 (8.3) | 51.8 (9.9) | 88.5 (11.5) | 55.3 (27.0)2 | |
| Postantibiotic effect | at 10 × MIC (h) | 18.8 (7.9) | 26.9 (48.9) | 15.7 (22.5) | 75.4 (38.4) | 133 (19.6) | Strep 132 |
Values represent means (% coefficient of variation).
Abbreviations: t½: half life; Vd: volume of distribution; CL: clearance; Strep: streptomycin. All lead spectinamides had excellent aqueous solubility >1mM.
t½ is based on decline of plasma concentration in the therapeutically relevant concentration range.
Indicates values published in reference number45 and included for comparison.
Activity is sustained in vivo against both acute and chronic tuberculosis infection models
Initial evaluation of the compounds in vivo was performed in an acute M. tuberculosis infection model to provide an assessment of drug efficacy primarily against rapidly replicating bacteria (Fig. 2a; Supplementary Table 9).34 The four lead compounds (1329, 1445, 1544, and 1599) demonstrated significant in vivo efficacy in reducing bacterial burden in lungs when compared to saline controls (P < 0.001), showing more than one log10CFU reduction versus the control after nine consecutive days of treatment (Supplementary Table 9). There were no statistical differences between the activity of the spectinamide derivatives and streptomycin when administered at similar doses (P > 0.05). Five independent acute infection mouse studies were performed for 1544 and 1599, which all showed similar results. To assess activity against a chronic tuberculosis infection, 1544 and 1599 were evaluated in Balb/c mice after 28 days of treatment (Fig. 2b).35 Subcutaneous administration of 1544 or 1599 significantly decreased lung bacterial burden (P<0.001) (Supplementary Table 10) with no apparent toxicity. The activity of both compounds was similar to that of streptomycin (P > 0.05) and isoniazid (P > 0.05).
Figure 2.

In vivo efficacy trial data showing bacterial burden (log10 CFU) in the lungs of M. tuberculosis infected mice (a) M. tuberculosis–acutely infected gamma-interferon receptor knockout [IFNγ KO mice; n=5] treated with lead spectinamides and comparator drugs, all subcutaneously dosed at 200 mg/kg BID for 9 days (SEM). (b) M. tuberculosis– chronically infected immunocompetent mice (n=6) treated with lead spectinamides and streptomycin, all subcutaneously dosed at 200 mg/kg QD for 28 days (SEM). INH control was dosed at 25 mg/kg QD by oral gavage. (c) Dose ranging study on M. tuberculosis infected mice with a high bacterial load (n=6) and treated with increasing doses [mg/kg] of spectinamide 1599 or streptomycin for 28 days (SEM). (d) Intrapulmonary delivery of 1599 and streptomycin three times a week (TIW) [mg/kg] compared to rifampin dosed orally daily in M. tuberculosis– chronically infected immunocompetent mice (n=5). In panels a, b and d asterisks (*) indicate P <0.001 by pairwise multiple comparison procedures (Tukey Test).
A dose escalation efficacy trial was then performed with 1599 in Balb/c mice infected with a high and ultimately fatal tuberculosis burden (Fig. 2c). All untreated mice and mice receiving 1599 at 50 mg/kg/d became moribund by day 18 and were humanely euthanized. The bacterial burden in the lungs of surviving mice showed a statistically significant reduction after drug treatment when compared to the start of treatment controls (P < 0.001) (Fig. 2c and Supplementary Table 11). Efficacy of 1599 in this model was not statistically different from streptomycin at similar doses (P > 0.05). No apparent drug toxicity was noted over the 28 day treatment period. Recently, lead 1599 has been included in one additional lethal dose and two additional chronic trials. In each case, results were similar to those presented in Fig. 2b–c.
The efficacy of 1599 was also determined via intrapulmonary delivery in which drug was administered intratracheally to chronically infected mice using a liquid microsprayer (Fig. 2d). In this trial, 1599 demonstrated excellent efficacy in a dose dependent manner. Treatment resulted in a 2.2 log CFU reduction in total lung tuberculosis burden versus the control group after 4 weeks of treatment at the highest dose (200 mg/kg, 3 days per week). The reduction in bacterial load by 1599 was comparable to that of rifampin (P > 0.05), and statistically superior to that of streptomycin delivered a similar dose, route and schedule as 1599 (P = 0.004 comparing 1599 and streptomycin treatment groups) (Supplementary Table 12). 1599 treated mice had continued reduction in bacterial load in the lungs between weeks 2 and 4 of treatment (P < 0.05), while no additional killing was observed for streptomycin after 2 weeks of treatment. In this trial 1599 was well tolerated by intrapulmonary delivery and no apparent toxicity (no severe weight loss or death) was noted.
Discussion
The devastating socioeconomic and public health impact of tuberculosis, the emergence of MDR- and XDR-strains, and the known toxicity of many existing antitubercular drugs underscores the need for novel drug candidates with excellent antitubercular activity and a good safety profile. Herein we have reported the discovery of a novel antitubercular spectinamide series generated by the synthetic modification of spectinomycin. We demonstrated that: (1) the potency of this series is a product of high affinity for the mycobacterial ribosome and avoidance of efflux; (2) efflux mediated drug resistance in M. tuberculosis can be overcome through chemical modification of the substrate antibiotic; and (3) spectinamides are excellent preclinical drug candidates for tuberculosis with potent in vivo efficacy as well as a safe in vitro pharmacological profile.
Structural studies revealed a tight SAR for this series, with absolute need for an R-amide at the 3' position of the spectinamine C ring coupled to a 2-pyridyl or 2-thiazole ring through an acetamide linker to maximize ribosomal affinity and most importantly avoid pump-mediated efflux. Compounds were further optimized by introducing halogen substituents to the 4 and 5 positions of the pyridyl ring, which according to computational modeling resulted in additional stabilizing interactions with backbone residues of the nearby RpsE protein loop.36 The requirement for a 2-pyridyl or bioisostere 2-thiazole group appears twofold: (1) the ring nitrogen is well positioned to form a hydrogen bond with the free ribosyl hydroxyl group on C1192/G1193 in helix 34; and (2) this nitrogen appears key to avoiding efflux, since the phenyl analog 1398 has poor MIC values despite good in vitro protein synthesis inhibition. One explanation for this requirement is that this heteroaryl nitrogen atom may allow the formation of a favored intramolecular hydrogen bond in solution between it and the amide NH,37 thus masking some of the molecule's polarity and helping the spectinamides avoid Rv1258c-mediated efflux.
The Rv1258c efflux pump (also known as Tap efflux pump) is induced in macrophage-resident M. tuberculosis and may promote intracellular survival by allowing the organism to escape killing by the host immune response.38 It is also responsible for the efflux of spectinomycin, resulting in high MICs that prevent its clinical utility.19,39 We established that Rv1258c pump-mediated efflux can be avoided by structural modification of spectinomycin, thereby significantly reducing the MIC. This challenges the historical notion that M. tuberculosis is intrinsically resistant to many standard antibiotics due to its greasy coat,40 which excludes polar antibiotic substances. It is becoming clear that intrinsic efflux transporters, such as Rv1258c, are important in reducing intracellular drug concentrations and may play an important role in persistence in M. tuberculosis.38,39 Thus, compounds designed to avoid pump-mediated efflux may have superior activity against hard-to-kill, drug-tolerant bacteria, which may lead to better in vivo efficacy.
Microbial and pharmacological testing of the spectinamides has demonstrated they possess many properties attractive for preclinical candidacy. First, spectinamides possesses a narrow spectrum of activity which is most likely associated with pump-mediated efflux, or possibly limited uptake, in other species. Therefore, long-term treatment of tuberculosis with spectinamides is unlikely to select for resistance in non-targeted pathogens. Second, the site of action of spectinamides within the ribosome is advantageous as it does not overlap with other protein synthesis inhibitors and drug binding is specific to the bacterial ribosome. This is evident in the lack of cross resistance with other clinically relevant protein synthesis inhibitors and in the pharmacology of the series. Studies demonstrated no appreciable cytotoxicity and no in vitro activity against eukaryotic cytosolic or mitochondrial ribosomal translation. This reduces the potential for side effects such as ototoxicity27,41 and myeloid suppression29,30 commonly associated with other protein synthesis inhibitors. Finally, potential concerns of drug–drug interactions during future use in combination pharmacotherapy regimens are reduced based on in vitro metabolic profiling and pharmacokinetic studies. Extensive in vitro testing against mammalian enzymes and receptors showed that our spectinamide leads have little potential for off-target effects leading to adverse reactions, including those associated with drug metabolism and cardiac toxicity.
Pharmacokinetic studies showed good distribution throughout the body, low plasma protein binding, and elimination mediated largely by unchanged excretion into the urine. This is in contrast to most of the other new tuberculosis drug candidates derived from phenotypic screening, which tend to be highly lipophilic and highly protein bound. Thus, the spectinamides afford the possibility of reaching subpopulations unattainable by lipophilic antituberculosis agents.42 These properties combined with the high safety index of the parent drug spectinomycin7,8 suggest the spectinamides will have a safe pharmacological profile favorable for preclinical drug development.
Most exciting, our lead compounds demonstrated potent antitubercular activity in vivo, in multiple tuberculosis infection mouse models. Excellent antitubercular activity was observed in both acute and chronic tuberculosis models. In addition, twice daily treatment with 1599 was equivalent to streptomycin in a high bacterial burden model, rescuing mice from an otherwise lethal tuberculosis infection. Finally, intrapulmonary administration of 1599 provided excellent protection, reducing the bacterial load throughout the 4 week treatment period suggesting sterilizing activity whereas streptomycin apparently did not show further killing beyond the first 2 weeks of treatment. The excellent efficacy shown by 1599 following intrapulmonary delivery may reflect good distribution of free drug within the lung coupled with its ability to avoid Rv1258c drug tolerance which has been shown to be induced in alveolar macrophages.38
Our study supports the further development of spectinamides as novel antitubercular agents, owing to their tight SAR, a narrow antimicrobial spectrum, low synthesis costs, and excellent in vivo efficacy and in vitro safety profiles. The potential safety and efficacy of this series represent a significant advancement over most second-line agents used to treat MDR tuberculosis. Widespread use of second-line drugs such as aminoglycosides has been limited due to their renal and auditory toxicities, as well as narrow therapeutic window. Resistance to most second-line agents has also emerged, warranting the development of other therapeutic options. Further, spectinamides overcome clearance by efflux pump Rv1258c, which has been implicated in phenotypic drug tolerance and often up-regulated in MDR tuberculosis strains.38,43,44 These benefits and the demonstrated potency via intrapulmonary delivery for this class of compounds overshadow the general lack of oral bioavailability of aminocyclitols (including the spectinamides), which is a common limitation of many second-line agents forming the core of MDR and XDR therapy. This study represents the first steps toward developing an antitubercular drug class using a semisynthetic approach to overcome intrinsic pump-mediated resistance mechanisms. Studies toward the clinical development of spectinamides as anti-tubercular agents are ongoing.
Online Methods
Molecular modeling
A homology model of the M. tuberculosis 16S helix 34 spectinomycin-binding site was built utilizing the crystal structure of E. coli in complex with spectinomycin (PDB 2QOU).6 To reduce the computational cost a ≈ 15Å truncated sphere centered on the spectinomycin-binding site was used throughout the simulation. Residues including nucleotide A1081G of helix 34 and residues T24V and I30R of the RpsE protein loop were mutated to the M. tuberculosis sequence, whilst contacts within a 5Å radius of spectinomycin are fully conserved. Terminal residues were harmonically restrained to their X-ray positions. The spectinamide series was sketched manually and each geometry was optimized in Jaguar, version 7.8 (Schrodinger, LLC, Portland, OR) at the B3LYP/6–31G** level of approximation. The compounds were then docked into the binding active site using Glide,20 and top poses were retained. Molecular dynamics calculations were carried out with the AMBER11 program,46 using FF03 force field for the RNA and protein and General Amber Force Field for the ligand. The whole complex was solvated in an octahedron box of TIP3P water molecules and neutralized by adding sodium ions while keeping crystal ions. Periodic boundary conditions, particle-mesh Ewald treatment of the long-term electrostatics and SHAKE-enabled 2 fs time steps were employed. A two-stage energy minimization was performed followed by a gradual heating of the complex system from 0K to 300K over 60 ps and a 50 ps equilibration. An additional 0.5 ns simulation at 300K was performed to further optimize the system. All production runs were performed with the NPT ensemble for 5 ns.
General methods to synthesize spectinamides
The spectinamides were synthesized from spectinomycin (Waterstone Technology, IN) according to the procedure of Woitun,10 except that the final deprotection step was modified using either catalytic hydrogenation and palladium on carbon (10% Pd/C) in 1.25M HCl in methanol for 2 h or acid hydrolysis (compounds with hydrogenation-sensitive side chains) by reaction with 48% HBr solution at room temperature for 2 h (see Supplementary Information for full analytical data).
Minimum inhibitory concentration (MIC) and cytotoxicity testing
MICs for aerobic bacteria were performed according to CLSI methods, as described previously.47,48 Detailed methodology for M. tuberculosis susceptibility testing can be found in the Supplementary Methods. Cytotoxicity was determined as described,49 using Vero cells (ATCC CCL-81) and J774 cell lines and detection of viability with CellTiter96 ® Assay (Promega).
Mycobacterium tuberculosis susceptibility testing
Method 1
Compounds were dissolved in 100% DMSO at a concentration of 10 mg/ml. Two-fold serial dilutions of compounds were prepared in 100 μl in of Middlebrook 7H9 broth supplemented with 10% albumin-dextrose complex and 0.05% (v/v) Tween80. The optical density at 600nm (OD600) of a mid-log phase M. tuberculosis culture was determined and inoculum prepared by adjusting OD600 to 0.01 in supplemented Middlebrook 7H9 broth. To each well of the 96-well, round bottom assay plate 100 μl of inoculum was added to achieve a final drug concentration range of 200 – 0.2 μg/mL, with no drug in column 12. Plates were placed in sealed bags and incubated at 37°C for 7 days in ambient air, at which point the MIC was recorded as the lowest concentration of drug that prevented visible growth. In all assays, reference antibiotics (typically streptomycin and spectinomycin) were included for quality control.
Method 2
MICs were determined as described in method 1, except that the units of drug concentrations ranges were in molar concentrations. MIC values were then converted to units of μg/mL for comparison with method 1 results.
Method 3
MICs were determined using an MGIT 960 equipped with TBeXiST software (Becton-Dickinson) as described.50
Rapid anaerobic development model of dormancy.24
10 ml aliquots of M. tuberculosis H37Rv (diluted 1:100 from a mid-log phase culture) were delivered to glass cultures tubes containing stir bars. Tubes were capped with rubber septa and stirred rapidly (~800 rpm) in a 37°C incubator for 7 days, at which point the methylene blue indicator, (1.5 μg/ml), was reduced to yield a colorless culture. Using a sterile syringe, 100 μl of 1 mg/ml test compound or carrier DMSO were injected through septa. Cultures were incubated for an additional 7 days prior to enumeration of viable bacilli by titration of colony forming units. Results from test cultures were normalized to the carrier treated group. Results presented represent the average of 2–3 independent experiments.
Construction of mutant ribosomes
M. smegmatis mc2155 ΔrrnBΔrrnAattB::rrnB was used for all genetic constructions.31 Hybrid rRNA ribosomes (Supplementary Fig. 2) were constructed using PCR mutagenesis to generate mutant rDNA fragments, cloned into the rRNA operon of integration-proficient plasmid pMV361 hyg-rrswt to result in plasmid pMV361 hyg-rrshybrid.32 Plasmid pMV361 hyg-rrshybrid was transformed into M. smegmatis ΔrrnBΔrrnAattB::rrnB. Successful gene replacement of the wt rRNA operon by the hybrid rRNA operon via plasmid exchange was determined by sequence analysis. Ribosomes were purified from bacterial cell pellets as previously described.51
Cell-free luciferase translation assays
Purified 70S hybrid bacterial ribosomes and rabbit reticulocyte lysate (RRL, Promega) were used in translation reactions. Firefly luciferase mRNA was produced in vitro by using T7 RNA polymerase. Translation reactions were carried out as previously described.51 The IC50 values represent the drug concentration that inhibits luciferase activity by 50%.
Mitochondrial in organello translation
Mitochondria were isolated from HEK293 cells52 and in organello translation was done with slight modifications as described.30,53,54 In brief, the mitochondria-enriched pellet was resuspended in 1 ml of mitochondria reaction buffer containing cycloheximide (100 mg/L) to inhibit cytosolic translation and S35-methionine [15 μl; 370 MBq (10mCi)/ml, specific activity 37TBq(1000Ci)/mmol, Hartmann Analytic KSM-01] to label proteins synthesized by the mitochondrial ribosome. The suspension was split into equal aliquots of 54 μL, and drugs were added to a final reaction volume of 60 μl. Reaction mixtures were incubated for 2 h at 30°C with shaking, centrifuged for 5 min at 15000g, and the pellets washed and resuspended in H2O. S35-methionine labelled proteins were resolved by 18% SDS polyacrylamide gel electrophoresis. COX1 was identified by autoradiography on the basis of its molecular weight of 39 kDa and by the absence of the corresponding band when we used COX1 nonsense mutant cells55 in in organello translation. Bands were quantified using Aida Image Analyzer (Fuji). The IC50 values represent the drug concentration that inhibits COX1 synthesis by 50%. Every experiment was repeated three times.
Post antibiotic effect (PAE)
PAE studies were performed in M. bovis BCG, as described previously.48 The difference between the time required to reach 50% saturation OD600 (1.1 +/− 0.2) for the untreated and treated cultures was calculated as the PAE. Each treatment was tested in at least two biologically independent experiments.
Protein binding
Rat plasma protein binding was determined at 500 ng/ml and 5 μg/ml by equilibrium dialysis at 37°C using the RED® device (Thermo Scientific, Rockford, IL), as described previously.56 Pre-and post-dialysis sample concentrations were determined by LC-MS/MS analysis. Average results of triplicate reactions from a single experiment are presented.
In vitro microsomal metabolic stability
In vitro microsomal metabolic stability was assessed in pooled rat liver microsomal preparations (Cellzdirect, Austin, TX), according to the manufacturers protocol. Disappearance of the parent compound was monitored by sampling at 0, 15, 30, 45, 60, 75, and 90 min during the incubation period and subsequent LC-MS/MS analysis.56 Average results of triplicate reactions from a single experiment are presented.
Pharmacokinetic studies
The Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center approved protocol (number 1463), was followed for all pharmacokinetic studies. Compounds were administered to groups of catheterized (jugular and femoral veins) male Sprague-Dawley rats (n= 4–5) intravenously at a dose of 10 mg/kg. Thirteen serial blood samples (~250 μL) were collected at predefined time points for 48 h post-dose. Plasma was separated immediately by centrifugation (9,300g for 10 min at 4°C). Cumulative urine samples were collected for 48 h. For pharmacokinetic bridging studies in mice, C57BL/6 mice (n=24) were dosed subcutaneously at 200 mg/kg. Blood samples were collected by cardiac puncture pre-dose and at 7 pre-defined time points post-dose. Plasma was separated immediately by centrifugation as above and stored at −80°C until analysis. Compound concentrations in plasma and urine were quantified by LC-MS/MS analysis. Pharmacokinetic profiles of compounds were analyzed by standard non-compartmental procedures,56 using Phoenix WinNonlin 6.2 (Pharsight Corporation, Mountain View, CA).
Quantification of compound concentrations by liquid chromatography - tandem mass spectrometry (LC-MS/MS)
50 μl aliquots of plasma and urine samples were prepared by protein precipitation with 200 μl methanol (spiked with internal standard 3'-Dihydro-3'-deoxy-3'(R)- isopropylacetylamino spectinomycin) followed by centrifugation at 10,000g for 10 min at 4°C. Chromatographic separation of the supernatant was carried out on a Luna 3μM HILIC, 100 × 4.6 mm column(Phenomenex, Torrance, CA) using a gradient mobile phase of methanol and 10 mM ammonium formate pH 2.75 at a flow rate of 0.4 ml/min. Detection was performed with an API 3000 triple-quadruple mass spectrometer (ABI-Sciex, Foster City, CA) with electrospray ionization in multiple reaction monitoring mode, using the compound specific mass transfers of m/z 453.3→247.3 for 1329, m/z 459.2→207.2 for 1443, m/z 471.3/207.1 for 1445, m/z 487.2/128.3 for 1544, m/z 487.2/207.1 for 1599, and m/z 418.3/207.1 for 1369. A calibration curve ranging from 1.95–5,000 μg/L was constructed for each test compound and validated with spiked samples of rat or mouse plasma or urine. The peak area ratios of analyte to internal standard were linear over the concentration range tested for all compounds, with correlation coefficients (weighted least-square linear regression analyses) >0.997. Accuracy (deviation of analysed quality control samples from nominal values) was within ±3% over the entire range of the calibration curve, while precision (coefficient of variation of repeated measurements of quality control samples) was <2% for all compounds.
Pharmacodynamic time-kill studies
Using a previously reported in vitro PK/PD model system57 time-kill experiments were performed on M. bovis BCG Pasteur cultures in the presence of 1445. Total daily dose simulations (0.4, 2, 10, 50 mg/kg/day) were administered once daily (QD), twice daily (BID), and thrice daily (TID). The number of viable bacteria in each sample was quantified by luminescence (BacTiter-Glo Promega, Madison, WI) and by standard CFU enumeration.
In vivo efficacy of spectinamides in four mouse tuberculosis aerosol infection models
All animal efficacy studies were performed according to guidelines of the Colorado State University Institutional Animal Care and Use Committee, and approved under Protocol Number: 13-4263A. For all mouse infection models, mice (5–6/group) were challenged with an aerosol of M. tuberculosis Erdman [TMC 107, ATCC 35801] via a GlasCol aerosol chamber in a certified ABSL-3 laboratory according to guidelines of the Institutional Animal Care and Use Committee. Drugs were delivered by intrapulmonary administration in 50 μl or by subcutaneous injection in 200 μl volume of either saline for streptomycin or in high salt buffer (156 mM sodium phosphate 137 mM NaCl), which was required to increase the pH of spectinamide acid salts to a final pH range 6.5–7.5. For endpoint analysis, mice were euthanized and lungs collected. The left lung lobe was homogenized for enumeration of CFUs by plating dilutions of the organ homogenates on non-selective 7H11 agar plates.58 The CFUs were converted to logarithms, which were then evaluated by a one-way analysis of variance, followed by a multiple comparison analysis of variance between all mouse groups, to include control and treatment groups, by a one-way Tukey test (SigmaPlot, Systat Inc. San Jose, CA). For the high burden infection trial, multiple comparison analysis of variance was conducted by Tukey test between treatment groups only as no control mice survived without treatment intervention. Differences were considered significant at the 95% level of confidence
Model 1
Efficacy in an acute infection model was tested using a low-dose aerosol (LDA; 100 CFU per mouse) in female IFN gamma knock-out (GKO) mice (Jackson Laboratories, Bar Harbor, ME) as described.58 Starting 13 days after LDA, mice received drug (200 mg/kg subcutaneously) twice daily 9 h apart for 9 consecutive days. Lungs were harvested at day 22 after LDA. The model yielded similar results in 5 independent experiments and results from a representative experiment presented.
Model 2
Evaluation of compounds in a chronic tuberculosis model was performed in wild type female Balb/c mice (Charles River Labs, Wilmington, MA) infected with a LDA. 35,59–61 After 21 days, mice were treated daily (5 days a week) with 200 mg/kg drug subcutaneously. After 28 days of treatment followed by two days of drug clearance, lungs were harvested.
Model 3
Efficacy in a lethal infection model was tested by establishing a high bacterial burden of 108 Log CFU in lungs prior to treatment. Female Balb/c mice were challenged with a high-dose aerosol inoculum (3.5 Log10CFU per mouse) which results in death without treatment intervention around 18–20 days after aerosol challenge. At 13 days post exposure, mice were dosed either daily or twice daily (5 days a week) for up to 28 days with 1599 at 12.5, 25, 50, 100, 200 mg/kg; streptomycin was used as the control, treated 200 mg/kg in the same manner. Mice that became moribund were (humanely) euthanized. For all remaining mice, lungs were harvested after 28 days of treatment followed by two days of drug clearance.
Model 4
To determine efficacy via intrapulmonary nebulized drug delivery, BALB/c female mice were infected with a LDA as in the chronic tuberculosis model. At 24 days after aerosol exposure, mice received drug via oral gavage (rifampin 10 mg/kg daily 5 days a week) or intrapulmonary aerosol delivery (1599 at 45 or 200 mg/kg; streptomycin 200 mg/kg; saline control) three times a week as previously described.62 Mice were anaesthetized using isoflurane and aerosols delivered employing a Microsprayer (model IA-C; PennCentury, Philadelphia, PA) attached to an FMJ-250 high pressure-syringe device (PennCentury) in a volume of 50 μl per dose as described.63–65 After 2 or 4 weeks of therapy, lungs were harvested without a drug free interval.
Supplementary Material
Acknowledgements
This study was supported by the National Institutes of Health grant AI090810, the NIAID IDIQ Contract Task Order HHSN272201000009I/01, the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children's Research Hospital (SJCRH), and in part by the Intramural Research Program of the NIAID, NIH and the Spanish Government (grant BIO-2009-09405). We thank Dr. Lei Yang and Dr. Jerrod Scarborough from SJCRH for their help with the analysis of the final compounds, Mr. Marcus Maddox from SJCRH for technical assistance in determining MIC values, Dr. Josiah Ryman from UTHSC for technical assistance in the performance of pharmacokinetic studies in mice, Dr. Michelle Butler from Microbiotix for coordination of the MDR tuberculosis testing, and Dr. Elaine Tuomanen from SJCRH for critical evaluation of this manuscript.
Footnotes
Author contributions: R.E.L. designed the compound series. J.G.H., D.F.B, R.B.L. and H.I.B. performed MIC testing and microbiology studies. J.L., J.Q., R., S.L.W, D.S. performed the medicinal chemistry. T.M., R.A., E.C.B. designed and performed MIC testing and ribosome inhibition studies. M.S.S., M.R.M., M.G.J, A.J.L. designed and performed the in vivo efficacy trials. P.K.V., C.R., D.M., A.T., and B.M. designed and performed the pharmacokinetic analysis. Z.Z. and S.D. performed the molecular modelling experiments. C.V., D.F.B. and J.A. designed and performed the efflux mutant testing. All authors discussed and analysed the data. R.E.L., E.C.B., A.J.L., R.B.L., D.F.B., J.A. and B.M. wrote the manuscript.
References
- 1.Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. N Engl J Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 2.WHO . Global Tuberculosis Report 2012. WHO; Geneva, Switzerland: 2012. [Google Scholar]
- 3.Silver LL. Challenges of antibacterial discovery. Clin Microbiol Rev. 2011;24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. Journal of natural products. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter AP, et al. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 6.Borovinskaya MA, Shoji S, Holton JM, Fredrick K, Cate JH. A steric block in translation caused by the antibiotic spectinomycin. ACS Chem Biol. 2007;2:545–552. doi: 10.1021/cb700100n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Novak E, Schlagel CA, LeZotte LA, Pfeifer RT. The tolerance of high dose intravenous spectinomycin therapy in man. J Clin Pharmacol. 1974;14:442–447. doi: 10.1002/j.1552-4604.1974.tb02326.x. [DOI] [PubMed] [Google Scholar]
- 8.Akiyoshi M, Yano S, Ikeda T. Ototoxicity of spectinomycin. Jpn J Antibiot. 1976;29:771–782. author's transl. [PubMed] [Google Scholar]
- 9.Thomas RC, Fritzen EL. Spectinomycin modification. III. Spectinomycin analogs with C-3'-branched chain sugars. J Antibiot (Tokyo) 1985;38:208–219. doi: 10.7164/antibiotics.38.208. [DOI] [PubMed] [Google Scholar]
- 10.Woitun E, Maier R, Wetzel B, Reuter W, Lechner U. Modification of spectinomycin. 2. Derivatives of 4-dihydro-4-deoxy-4(R)-aminospectinomycin. J Antibiot (Tokyo) 1981;34:22–27. doi: 10.7164/antibiotics.34.22. [DOI] [PubMed] [Google Scholar]
- 11.Maier R, Woitun E, Reuter A, Reuter W, Wetzel B. Modification of spectinomycin. 1. Synthesis of 4-aminospectinomycins. J Antibiot (Tokyo) 1981;34:16–21. doi: 10.7164/antibiotics.34.16. [DOI] [PubMed] [Google Scholar]
- 12.Zurenko GE, Yagi BH, Vavra JJ, Wentworth BB. In vitro antibacterial activity of trospectomycin (U-63366F), a novel spectinomycin analog. Antimicrob Agents Chemother. 1988;32:216–223. doi: 10.1128/aac.32.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Noskin GA. Tigecycline: a new glycylcycline for treatment of serious infections. Clin Infect Dis. 2005;41(Suppl 5):S303–314. doi: 10.1086/431672. [DOI] [PubMed] [Google Scholar]
- 14.Projan SJ. Preclinical pharmacology of GAR-936, a novel glycylcycline antibacterial agent. Pharmacotherapy. 2000;20:219S–223S. doi: 10.1592/phco.20.14.219s.35046. discussion 224S–228S. [DOI] [PubMed] [Google Scholar]
- 15.Wiley PF, Argoudelis AD, Hoesema H. The Chemistry of Actinospectacin. IV. The Determination of the Structure of Actinospectacin. Journal of the American Chemical Society. 1963;85:2652–2659. [Google Scholar]
- 16.Foley L, Lin JT, Weigele M. Spectinomycin chemistry. II.) 9-Deoxy-4(R)-dihydrospectinomycin and 9-deoxyspectinomycin. J Antibiot (Tokyo) 1978;31:979–984. doi: 10.7164/antibiotics.31.979. [DOI] [PubMed] [Google Scholar]
- 17.Foley L, Lin JT, Weigele M. Preparation of 7-deoxyspectinomycin and 7-deoxy-8-epi-4(R)-dihydrospectinomycin. J Antibiot (Tokyo) 1979;32:418–419. doi: 10.7164/antibiotics.32.418. [DOI] [PubMed] [Google Scholar]
- 18.Rosenbrook W, Jr., Carney RE. Spectinomycin modification. I 7-EPI-9-deoxy-4(R)-dihydrospectinomycin. J Antibiot (Tokyo) 1975;28:953–959. doi: 10.7164/antibiotics.28.953. [DOI] [PubMed] [Google Scholar]
- 19.Balganesh M, et al. Efflux pumps of Mycobacterium tuberculosis play a significant role in antituberculosis activity of potential drug candidates. Antimicrob Agents Chemother. 2012;56:2643–2651. doi: 10.1128/AAC.06003-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 21.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. Principles and applications of halogen bonding in medicinal chemistry and chemical biology. J Med Chem. 2013;56:1363–1388. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 22.Galimand M, Gerbaud G, Courvalin P. Spectinomycin resistance in Neisseria spp. due to mutations in 16S rRNA. Antimicrob Agents Chemother. 2000;44:1365–1366. doi: 10.1128/aac.44.5.1365-1366.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zaunbrecher MA, Sikes RD, Jr., Metchock B, Shinnick TM, Posey JE. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009;106:20004–20009. doi: 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honaker RW, Dhiman RK, Narayanasamy P, Crick DC, Voskuil MI. DosS responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. Journal of Bacteriology. 2010;192:6447–6455. doi: 10.1128/JB.00978-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramon-Garcia S, et al. Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis BCG. Antimicrobial Agents and Chemotherapy. 2012;56:2074–2083. doi: 10.1128/AAC.05946-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bottger EC, Springer B, Prammananan T, Kidan Y, Sander P. Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO reports. 2001;2:318–323. doi: 10.1093/embo-reports/kve062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hobbie SN, et al. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A. 2008;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hobbie SN, et al. Mitochondrial deafness alleles confer misreading of the genetic code. Proc Natl Acad Sci U S A. 2008;105:3244–3249. doi: 10.1073/pnas.0707265105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barnhill AE, Brewer MT, Carlson SA. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial components. Antimicrob Agents Chemother. 2012;56:4046–4051. doi: 10.1128/AAC.00678-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother. 2006;50:2042–2049. doi: 10.1128/AAC.01411-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hobbie SN, et al. Engineering the rRNA decoding site of eukaryotic cytosolic ribosomes in bacteria. Nucleic Acids Res. 2007;35:6086–6093. doi: 10.1093/nar/gkm658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shcherbakov D, et al. Directed mutagenesis of Mycobacterium smegmatis 16S rRNA to reconstruct the in-vivo evolution of aminoglycoside resistance in Mycobacterium tuberculosis. Mol Microbiol. 2010 doi: 10.1111/j.1365-2958.2010.07218.x. [DOI] [PubMed] [Google Scholar]
- 33.Drusano GL. Antimicrobial pharmacodynamics: critical interactions of `bug and drug'. Nature reviews. Microbiology. 2004;2:289–300. doi: 10.1038/nrmicro862. [DOI] [PubMed] [Google Scholar]
- 34.Lenaerts AJ, Gruppo V, Brooks JV, Orme IM. Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrobial Agents and Chemotherapy. 2003;47:783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenaerts AJ, et al. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother. 2005;49:2294–2301. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bissantz C, Kuhn B, Stahl M. A medicinal chemist's guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhn B, Mohr P, Stahl M. Intramolecular hydrogen bonding in medicinal chemistry. J Med Chem. 2010;53:2601–2611. doi: 10.1021/jm100087s. [DOI] [PubMed] [Google Scholar]
- 38.Adams KN, et al. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell. 2011;145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramon-Garcia S, et al. Functional and genetic characterization of the tap efflux pump in Mycobacterium bovis BCG. Antimicrob Agents Chemother. 2012;56:2074–2083. doi: 10.1128/AAC.05946-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee RE, Brennan PJ, Besra GS. Mycobacterium tuberculosis cell envelope. Curr Top Microbiol Immunol. 1996;215:1–27. doi: 10.1007/978-3-642-80166-2_1. [DOI] [PubMed] [Google Scholar]
- 41.Akbergenov R, et al. Molecular basis for the selectivity of antituberculosis compounds capreomycin and viomycin. Antimicrob Agents Chemother. 2011;55:4712–4717. doi: 10.1128/AAC.00628-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dartois V, Barry CE., 3rd A medicinal chemists' guide to the unique difficulties of lead optimization for tuberculosis. Bioorg Med Chem Lett. 2013;23:4741–4750. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louw GE, et al. Rifampicin reduces susceptibility to ofloxacin in rifampicin-resistant Mycobacterium tuberculosis through efflux. Am J Respir Crit Care Med. 2011;184:269–276. doi: 10.1164/rccm.201011-1924OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siddiqi N, et al. Mycobacterium tuberculosis isolate with a distinct genomic identity overexpresses a tap-like efflux pump. Infection. 2004;32:109–111. doi: 10.1007/s15010-004-3097-x. [DOI] [PubMed] [Google Scholar]
- 45.Madhura DB, Lee R, Meibohm B. Pharmacokinetic profile of spectinomycin in rats. Pharmazie. 2013;68:675–676. [PMC free article] [PubMed] [Google Scholar]
- 46.Case DA, et al. AMBER 11. University of California; San Francisco: 2010. [Google Scholar]
- 47.CLSI . Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard M7-A7. Clinical Laboratory Standards Institute; Wayne, PA: 2006. [Google Scholar]
- 48.Hurdle JG, et al. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother. 2008;62:1037–1045. doi: 10.1093/jac/dkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mallari JP, et al. Development of potent purine-derived nitrile inhibitors of the trypanosomal protease TbcatB. Journal of Medicinal Chemistry. 2008;51:545–552. doi: 10.1021/jm070760l. [DOI] [PubMed] [Google Scholar]
- 50.Springer B, Lucke K, Calligaris-Maibach R, Ritter C, Bottger EC. Quantitative drug susceptibility testing of Mycobacterium tuberculosis by use of MGIT 960 and EpiCenter instrumentation. J Clin Microbiol. 2009;47:1773–1780. doi: 10.1128/JCM.02501-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bruell CM, et al. Conservation of bacterial protein synthesis machinery: initiation and elongation in Mycobacterium smegmatis. Biochemistry. 2008;47:8828–8839. doi: 10.1021/bi800527k. [DOI] [PubMed] [Google Scholar]
- 52.Pallotti F, Lenaz G. Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods in cell biology. 2001;65:1–35. doi: 10.1016/s0091-679x(01)65002-7. [DOI] [PubMed] [Google Scholar]
- 53.McKee EE, Grier BL, Thompson GS, McCourt JD. Isolation and incubation conditions to study heart mitochondrial protein synthesis. The American journal of physiology. 1990;258:E492–502. doi: 10.1152/ajpendo.1990.258.3.E492. [DOI] [PubMed] [Google Scholar]
- 54.Fernandez-Silva P, Acin-Perez R, Fernandez-Vizarra E, Perez-Martos A, Enriquez JA. In vivo and in organello analyses of mitochondrial translation. Methods in cell biology. 2007;80:571–588. doi: 10.1016/S0091-679X(06)80028-2. [DOI] [PubMed] [Google Scholar]
- 55.Hornig-Do HT, et al. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. The EMBO journal. 2012;31:1293–1307. doi: 10.1038/emboj.2011.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Budha NR, et al. Pharmacokinetically-guided lead optimization of nitrofuranylamide anti-tuberculosis agents. AAPS J. 2008;10:157–165. doi: 10.1208/s12248-008-9017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Budha NR, Lee RB, Hurdle JG, Lee RE, Meibohm B. A simple in vitro PK/PD model system to determine time-kill curves of drugs against Mycobacteria. Tuberculosis. 2009;89:378–385. doi: 10.1016/j.tube.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lenaerts AJM, Gruppo V, Brooks JV, Orme IM. Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrobial Agents and Chemotherapy. 2003;47:783–785. doi: 10.1128/AAC.47.2.783-785.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lenaerts AJ, Degroote MA, Orme IM. Preclinical testing of new drugs for tuberculosis: current challenges. Trends in Microbiology. 2008;16:48–54. doi: 10.1016/j.tim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 60.De Groote MA, et al. Comparative studies evaluating mouse models used for efficacy testing of experimental drugs against Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2011;55:1237–1247. doi: 10.1128/AAC.00595-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Groote MA, et al. Importance of confirming data on the in vivo efficacy of novel antibacterial drug regimens against various strains of Mycobacterium tuberculosis. Antimicrobial Agents and Chemotherapy. 2012;56:731–738. doi: 10.1128/AAC.05701-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalez-Juarrero M, Woolhiser LK, Brooks E, DeGroote MA, Lenaerts AJ. Mouse model for efficacy testing of antituberculosis agents via intrapulmonary delivery. Antimicrob Agents Chemother. 2012;56:3957–3959. doi: 10.1128/AAC.00464-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ordway D, et al. XCL1 (lymphotactin) chemokine produced by activated CD8 T cells during the chronic stage of infection with Mycobacterium tuberculosis negatively affects production of IFN-gamma by CD4 T cells and participates in granuloma stability. J Leukoc Biol. 2007;82:1221–1229. doi: 10.1189/jlb.0607426. [DOI] [PubMed] [Google Scholar]
- 64.Rosas-Taraco AG, et al. Intrapulmonary delivery of XCL1-targeting small interfering RNA in mice chronically infected with Mycobacterium tuberculosis. Am J Respir Cell Mol Biol. 2009;41:136–145. doi: 10.1165/rcmb.2008-0363OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosas-Taraco AG, et al. Local pulmonary immunotherapy with siRNA targeting TGFbeta1 enhances antimicrobial capacity in Mycobacterium tuberculosis infected mice. Tuberculosis (Edinb) 2011;91:98–106. doi: 10.1016/j.tube.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.