

Abstract
CXCR4 is expressed by basal keratinocytes (KCs), but little is known about its function in inflamed skin. We crossed K14-Cre and CXCR4flox/flox (f/f) transgenic mice, resulting in mice with specific loss of the CXCR4 gene in K14-expressing cells (K14-CXCR4KO), including basal KCs. K14-CXCR4KO pups had no obvious skin defects. We compared K14-CXCR4KO and CXCR4f/f control mice in an IL-23-mediated psoriasisform dermatitis model and measured skin edema, histologic, and immunohistological changes. IL-23-treated K14-CXCR4KO mice showed a 1.3-fold increase in mean ear swelling, 2-fold increase in epidermal thickness, and greater parakeratosis. IL-23-treated WT mice showed weak CXCR4 expression in areas of severe epidermal hyperplasia, but strong CXCR4 expression in non-hyperplastic regions, suggesting CXCR4 may regulate keratinocyte proliferation. To test this hypothesis, we overexpressed CXCR4 in HaCaT keratinocyte cells and treated them with IL-22 and/or CXCL12. CXCL12 blocked IL-22-mediated HaCaT cell proliferation in vitro and synergized with IL-22 in upregulating SOCS3, a key regulator of STAT3. SOCS3 was required for CXCR4-mediated growth inhibition. In human psoriatic skin, both CXCR4 and SOCS3 were upregulated in the junctional region at the border of psoriatic plaques. Thus, CXCR4 plays an unexpected role in inhibiting KC proliferation and mitigating the effects of proliferative Th17 cytokines.
Introduction
Chemokine receptors comprise four homologous families of seven-transmembrane-spanning, G protein-coupled receptors that activate key intracellular signaling pathways controlling cell shape, migration (chemotaxis), and proliferation (Nestle et al, 2009). In inflammatory skin disease, chemokines and chemokine receptors also play important roles in immune cell migration into skin (Lonsdorf et al, 2009). Keratinocytes (KCs) are considered a rich source of chemokines such as CXCL8 and CCL20, which are abundant in psoriatic plaques (Schon and Boehncke, 2005), but the role of chemokine receptors that are expressed by KCs has not been fully explored. CXCR4, a chemokine receptor that plays multiple roles in cancer metastasis (Balkwill, 2004), vasculogenesis (Urbich and Dimmeler, 2004, Yamaguchi et al, 2003), stem cell recruitment (Petit et al, 2002), and HIV infection (Bleul et al, 1996), has been detected in proliferating KCs after burn injury (Avniel et al, 2006). Interestingly, inhibition of CXCR4 appeared to increase the rate of re-epithelialization following burn injury (Avniel et al, 2006), suggesting a regulatory role for CXCR4 in skin repair or re-epithelialization.
In the psoriasis field, clinicians have long been aware of the Koebner phenomenon (Weiss et al, 2002), and biochemical evidence has also suggested a link between wound healing and psoriatic plaques (Romanowska et al, 2010, Mansbridge and Knapp, 1987). The above-mentioned link between CXCR4 and wound healing prompted us to explore the role of CXCR4 in the context of a well-known model of psoriasiform dermatitis (Chan et al, 2006, Hedrick et al, 2009, Mabuchi et al, 2011) that is induced by repeated injections of the proinflammatory cytokine IL-23, a cytokine that is critical for the maintenance of Th17 cells (Fitch et al, 2007).
To determine the role of CXCR4 signaling in epidermis in the setting of psoriasiform dermatitis, we specifically deleted CXCR4 in murine keratin 14 (K14)-positive basal KCs by crossing mice carrying floxed CXCR4 alleles with those expressing Cre recombinase under the transcriptional control of the K14 promoter. We next induced skin inflammation via repeated injections of IL-23, resulting in psoriasiform dermatitis (Hedrick et al, 2009, Wilson et al, 2007). Surprisingly, targeted deletion of CXCR4 in basal KCs resulted in a markedly exaggerated response to IL-23 in the skin of K14-CXCR4KO mice. Furthermore, our in vitro studies indicated that CXCR4 signaling abrogates the proliferative response of KCs to IL-22 (Zheng et al, 2007) through a mechanism that requires SOCS3. In aggregate, our data strongly suggest that CXCR4 may have an unsuspected role in regulating epidermal proliferation in some inflammatory skin conditions.
Results
Targeted deletion of CXCR4 in basal KCs results in exaggerated psoriasiform changes after IL-23 treatment
We first confirmed that K14-CXCR4KO mice were homozygous for the floxed CXCR4 gene and Cre gene using PCR (Supplementary Figure S1). Because we were principally interested in the expression of CXCR4 in epidermal keratinocytes after IL-23 treatment, we separated the epidermis from dermis by standard techniques. Whereas epidermal CXCR4 mRNA was upregulated in CXCR4f/f control mice after IL-23 treatment, K14-CXCR4KO mouse skin showed minimal expression of epidermal CXCR4 (Figure 1A). Combining the results of three experiments, the P value (after combining 3 experiments) did not reach significance (P=0.062). It is known that Langerhans cells (LC), which do not express keratin 14, express high levels of CXCR4 upon activation, thus accounting for measurable (and variable) levels of CXCR4 in the epidermis of K14-CXCR4KO mice. Minimal levels of CXCR4 protein, possibly from LC, were detected in the basal keratinocytes of K14-CXCR4 KO mice vs. CXCR4f/f mice by antibody staining (Supplementary Figure S2). CXCR4 was not detected in PBS-injected CXCR4f/f (or WT) mice epidermis (data not shown). Thus, epidermal CXCR4 expression in CXCR4f/f control mice is upregulated following repeated injection of IL-23, but only minimally observed in CXCR4KO mouse skin (possibly due to incomplete deletion of the CXCR4 gene by the Cre recombinase or unaffected expression of CXCR4 by LC).
Figure 1. CXCR4 expression in CXCR4f/f control mice (CXCR4f/f-Cre− mice) and K14-CXCR4KO mice following IL-23 injection.
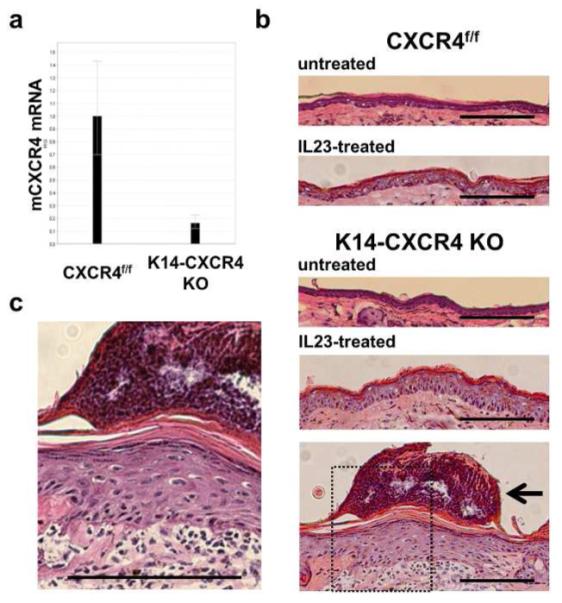
CXCR4 KO mice and CXCR4f/f control mice ears were injected with IL-23 every other day for 5 days and collected at day 5. Abrogation of CXCR4 expression was shown using real-time PCR (2-3 ears). Representative results from 1 of 3 experiments with similar results are shown (A). H&E staining was performed for ear sections from K14-CXCR4KO mice and control mice treated with or without IL-23. Neutrophils in the stratum corneum are indicated by a black arrow in (B) and shown at higher magnification in (C). Scale bars, 100 μm. Representative results from 1 of 3 experiments with similar results are shown (B, C).
Histologically, IL-23-treated ears of K14-CXCR4KO mice exhibited exaggerated psoriasiform changes compared to control mice (Figure 1C, D). Specifically, they displayed a thicker epidermis (Figure 1B), increased parakeratosis with greater accumulation of neutrophils in the cornified layer, and a dense inflammatory infiltrate in the dermis (Figure 1C). The neutrophilic aggregates were subjectively observed more frequently in the K14-CXCR4KO mice. While both control mice and K14-CXCR4KO mice showed epidermal thickening after IL-23 treatment, K14-CXCR4KO mice ears showed a 2-fold increase compared to CXCR4f/f control mice (Figure 2A). We also counted infiltrating cells in the dermis (Figure 1C) and found that the number of infiltrating cells increased 1.5 fold in K14-CXCR4KO mice compared with CXCR4f/f control mice (Figure 2B). Ear swelling, a measurement of dermal edema and inflammation, during IL-23 treatment was significantly increased (following ANOVA analysis) in K14-CXCR4KO mice on days 4 and 5 compared to CXCR4f/f control mice, which showed an increase above day 0 only on day 5 (Figure 2C). Thus, targeted deletion of CXCR4 in basal KCs results in exaggerated psoriasiform inflammatory changes following treatment with IL-23.
Figure 2. Epidermal thickening and psoriasiform changes in CXCR4f/f control and K14-CXCR4KO mice following IL-23 injection.
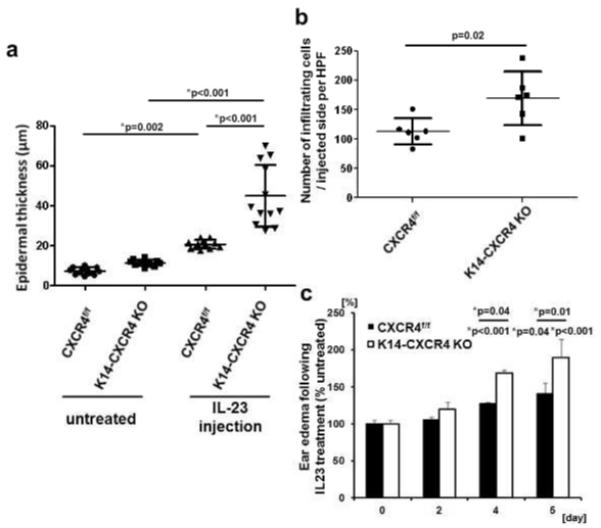
Ear sections from K14-CXCR4KO mice (n=3) and CXCR4f/f control mice (n=3) treated with IL-23 every other day for 5 days were H&E stained. The subjectively thickest epidermal regions were selected, and epidermal thicknesses were measured at four points in a blinded fashion for each mouse sample (A). Dermal infiltrating cells were counted on randomly selected sections of IL23 treated mice (n=3; B). Ear swelling was measured before injection (day 0, 2, 4) and harvesting ears (day 5; n=3). Representative data from one of two independent experiments with similar results are shown (A, B, C). *Indicates significance using Scheffé’s post hoc comparison test.
CXCR4 expression inversely correlates with epidermal hyperplasia in WT mice following IL-23 treatment
We observed that injection of IL-23 in WT C57BL/6 mice did not result in completely uniform epidermal hyperplasia throughout the entire epidermis above the injection site (Figure 3A). We hypothesized that the expression level of CXCR4 on basal KCs might regulate epidermal hyperplasia and thus examined CXCR4 expression by immunofluorescence microscopy in basal KCs of WT mice treated with IL-23. Indeed, CXCR4 expression was enhanced in non-hyperplastic regions, whereas hyperplastic regions showed little or no expression of CXCR4 (Figure 3B).
Figure 3. Epidermal expression of CXCR4 and SOCS3 in WT mice following IL-23 treatment.
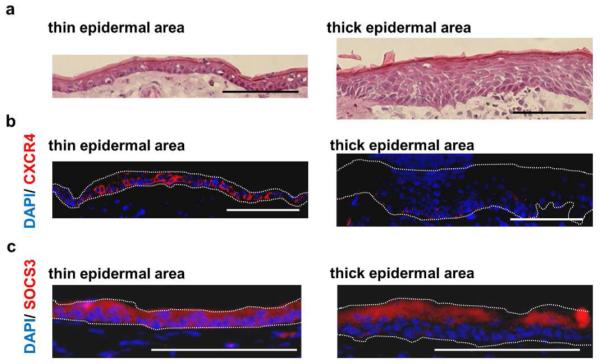
Ear sections from WT mice (n=3) treated with IL-23 were H&E stained and digitally photographed. Representative H&E stained sections from thick and thin epidermal regions of WT mice treated with IL-23 are shown (A). Skin from WT mice treated with IL-23 were stained with anti-CXCR4 (B) and anti-SOCS3 antibodies (C), and representative thick and thin epidermal regions were selected for illustration of differential antibody staining. Scale bars, 100 μm. The superior dotted lines in (B, C) represent the approximate level of the cornified layer while the inferior (or single) dotted lines in (B, C) represent the approximate level of the epidermal basement membrane.
In the IL-23 injection model, IL-22 is produced by immune cells such as conventional and γδ T cells (Mabuchi et al, 2011) and induces KC proliferation via signal transducer and activator of transcription 3 (STAT3) activation (Boniface et al, 2005). As a negative feedback control system for STAT3 signaling, suppressor of cytokine signaling 3 (SOCS3) is known to block the STAT3 phosphorylation by Janus kinase (JAK; (Brantley and Benveniste, 2008). Thus, we asked if SOCS3 expression differed between hyperplastic and non-hyperplastic regions. Similar to the expression pattern observed with CXCR4, SOCS3 expression in the basal epidermal layer was markedly reduced in hyperplastic regions compared to non-hyperplastic regions (Figure 3C). In summary, CXCR4 (and SOCS3) expression on basal KCs correlates with regions of reduced epidermal hyperplasia following IL-23 injection, suggesting that CXCR4 may negatively regulate KC proliferation.
IL-22 and CXCL12 synergistically upregulate SOCS3 in CXCR4-overexpressing HaCaT cells
IL-22 is a key downstream effector molecule that is produced by both αβ and γδ T cells (Mabuchi et al, 2012). It mediates epidermal hyperplasia and inflammation in response to IL-23. Keratinocytes are not believed to respond to IL-23 directly, but they do possess the IL-22 receptor, which acts through STAT3 to induce a variety of key changes found in psoriasis (Zheng et al, 2007). HaCaT cells, a keratinocyte-derived cell line, express the IL-22 receptor (Kreis et al, 2007) and are known to proliferate in response to IL-22 in vitro (Nakagawa et al, 2011). To determine if CXCR4 signaling regulated IL-22-mediated KC proliferation via SOCS3, we established CXCR4-overexpressing HaCaT (Boukamp et al, 1988) immortalized human KC cells (CXCR4-HaCaT) and cultured them in the presence of IL-22 with or without CXCL12. As expected, IL-22 increased CXCR4-HaCaT proliferation whereas CXCL12 alone had little effect on HaCaT cell proliferation (Figure 4A). Strikingly, co-addition of CXCL12 blocked the ability of IL-22 to stimulate HaCaT cell proliferation (Figure 4A), suggesting that CXCR4 activation functions as an inhibitor when IL-22 signaling is engaged.
Figure 4. Effects of CXCL12 on HaCaT cell proliferation, SOCS3 expression, and phosphorylation of STAT3.
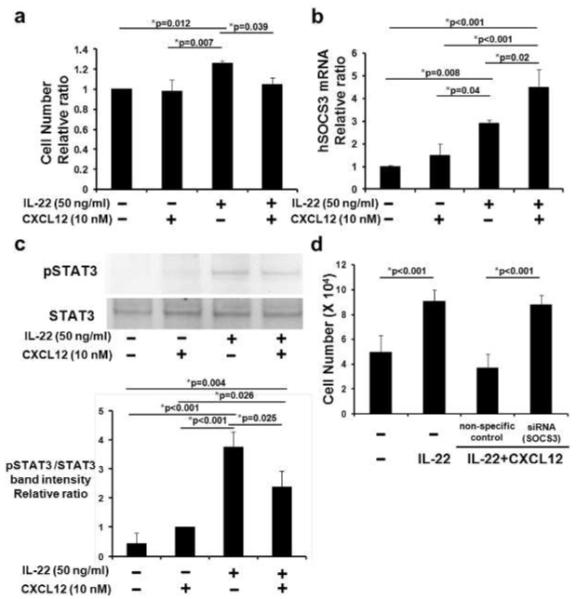
CXCR4-HaCaT cells were cultured for 1 day, then with human CXCL12 and/or human IL-22. After 2 more days (n=5 wells/condition), cells were counted. Summary of 3 independent experiments shown (A). CXCR4-HaCaT cells were stimulated with CXCL12 and/or IL-22 for 1 hr. SOCS3 mRNA expression was measured using real-time RT-PCR (n=3 wells/condition; 1 of 3 experiments shown, B). Phospho-STAT3 (pSTAT3) and total STAT3 were examined by Western blot (representative experiment shown, C). Analysis by densitometry (normalized using vehicle-treated cells) of 3 independent experiments is shown in the lower panel (C). CXCR4-HaCaT cells were transfected with SOCS3 siRNA and non-specific control siRNA, cultured with CXCL12 and IL-22 for 2 days, and counted (n=5; D). *Indicates significance using Scheffé’s post hoc comparison test.
The IL-22 receptor was only modestly upregulated in the presence of IL-22 and CXCL12 compared to IL-22 or CXCL12 alone (data not shown), indicating that CXCR4 did not directly regulate expression of this receptor. We next asked if inhibitors of JAK/STAT3 pathway were upregulated by co-culture of CXCL12 and IL-22. IL-22 induces phosphorylation of STAT3, thus promoting expression of JAK/STAT3 inhibitors such as SOCS3 and protein inhibitor of activated STAT3 (PIAS3) as potential negative feedback control mechanisms (Brantley and Benveniste, 2008). PIAS3 mRNA expression, however, was not affected by CXCL12, IL-22, or the combination of the two (data not shown). As expected based on previous reports, SOCS3 mRNA was upregulated in CXCR4-HaCaT cells cultured with IL-22 for 1 hr (Figure 4B). CXCL12 had little impact on SOCS3 expression by itself, but addition of CXCL12 in the presence of IL-22 synergistically enhanced SOCS3 mRNA level (Figure 4B). To confirm this result at the protein level, we saw enhanced SOCS3 upregulation in CXCR4-HaCaT cultured with IL-22 and CXCL12 using quantitative IF microscopy (Supplementary Figure S3).
We next asked if the observed SOCS3 upregulation inhibited the JAK/STAT3 pathway with the knowledge SOCS3 attenuates STAT3 phosphorylation at Tyr705 (Linke et al, 2010), a key activation site following exposure to IL-22 (Sestito et al, 2011). As shown in Figure 4C (full length blot is shown in Supplementary Figure S4), IL-22, but not CXCL12, induced marked phosphorylation of STAT3. The addition of CXCL12 with IL-22, however, reduced levels of phosphorylated STAT3 by 33.4±2.5% (mean±SD from 3 independent experiments) without affecting total STAT3. Thus, CXCR4-HaCAt cells cultured with IL-22 and CXCL12 show reduction in activated pSTAT3 at a key phosphorylation site.
We next asked if SOCS3 was required for CXCR4-mediated inhibition of CXCR4-HaCaT cell proliferation. CXCR4-HaCaT cells were transiently transfected with siRNA that specifically targeted SOCS3 and then cultured with IL-22 and CXCL12. We confirmed that SOCS3 siRNA inhibited SOCS3 expression in CXCR4-HaCaT cells cultured with IL-22 and CXCL12 (Supplementary Figure S5). Of note, CXCR4-HaCaT cell proliferation was restored when cells were treated with SOCS3-specific siRNA in the presence of IL-22 and CXCL12, but not with non-specific control siRNA (Figure 4D). Thus, CXCL12 in combination with IL-22 synergistically enhances SOCS3 expression in CXCR4-HaCaT cells, reduces phosphorylation of STAT3 at a critical tyrosine site, and blocks HaCaT cell proliferation via a SOCS3-dependent mechanism.
CXCR4 and SOCS3 are upregulated in the junctional zone between lesional and peri-lesional regions in human psoriatic skin
Lastly, we asked if CXCR4 was upregulated in human psoriatic skin lesions. The biopsied samples that we examined were taken from the border of psoriatic plaques and contained lesional hyperplastic areas as well as the transitional area (junctional zone) between clinically psoriatic and non-psoriatic skin (Figure 5A). The junctional zone showed moderate epidermal hyperplasia compared to peri-lesional regions (Figure 5A). Strong CXCR4 expression was observed in basal KCs in the junctional zone (Figure 5B; upper-left panel) between lesional and peri-lesional regions. CXCR4, however, was only focally present in lesional regions (Figure 5B; middle panel) and was nearly completely absent in peri-lesional regions (data not shown). We calculated the ratio of CXCR4-positive area to either the junctional or lesional zone areas of psoriatic epidermis and found a significant difference (Figure 5B; right panel). Control human skin from non-psoriatic, healthy patients showed little or no CXCR4 staining (Figure 5B; lower-middle panel). We next stained psoriatic skin sections with anti-SOCS3 Ab and noted increased SOCS3 expression in the junctional zone (Figure 5C; left panel). In samples taken from the same patient, lesional psoriasis regions that showed weak CXCR4 staining at the basal layer of the epidermis (Figure 5B; middle panel) also showed weak SOCS3 staining (Figure 5C; middle panel). The ratio of SOCS3-positive area to the entire junctional or lesional zones was also significantly higher in junctional zones (Figure 5C; right panel). Weak SOCS3 expression in normal human skin was shown to be almost the same as that in psoriasis lesional skin (Horiuchi et al, 2006). Similar results were noted in two additional psoriatic patients (data not shown). Thus, CXCR4 and SOCS3 were both upregulated at the junctional zone of psoriatic lesions, suggesting that expression of these two proteins may be a biomarker for the transition between hyperplastic psoriatic plaques and clinically non-affected skin.
Figure 5. CXCR4 and SOCS3 expression in human psoriatic skin.

Three human psoriasis skin samples containing the junctional zone between lesional and perilesional regions were biopsied (see Supplementary Table S1.). An example of H&E stainings showing these regions is shown (Patient 1; A). CXCR4 (B) and SOCS3 (C) stainings of the junctional and lesional area were performed and representative images of similar findings are depicted (Patient 2; See Supplementary Figure S6.). A healthy human skin sample was stained using anti-CXCR4 Ab (B; lower-left panel). CXCR4- and SOCS3-positive areas of 3 psoriasis patients were quantified using imageJ software and used for calculation of ratio (%) of marker-positive area to epidermal junctional or lesional zone areas (Right panels of B and C). Scale bars, 100 μm.
Discussion
CXCR4 has been shown in several cancer systems to positively regulate proliferation, survival, and metastasis (Dell’Agnola and Biragyn, 2007, Murakami et al, 2002, Murakami et al, 2003). Thus, we were surprised to see strikingly enhanced epidermal hyperplasia and parakeratosis in K14-CXCR4KO mice following IL-23 treatment. Previous reports showed that CXCR4 signaling can upregulate SOCS expression and inactive growth hormone function (Garzon et al, 2004). In our in vitro model, combined treatment with CXCL12 and IL-22 synergistically induced SOCS3 expression and reduced phosphorylation of STAT3. In contrast to the work of Garzone et al. (Garzon et al, 2004), CXCL12 alone induced little, or no, SOCS3 expression, which may be a reflection of the different cell type used in our system.
In human psoriasis skin samples, we observed strong CXCR4 expression in the junctional zone between lesional and peri-lesional regions. Increased SOCS3 expression was also detected in this area, further strengthening a possible relationship between CXCR4 and SOCS3 in controlling epidermal proliferation. We hypothesize that the coordinated upregulation of CXCR4 and SOCS3 at the border between affected and unaffected skin may provide a molecular explanation for the abrupt transition from affected to unaffected skin that is so highly characteristic of psoriasis lesions.
In this current work, we do not directly address which CXCR4 mechanisms regulate appears to be upregulated in the junctional zones of lesional skin. CXCR4, however, is known to be positively regulated by hypoxia-inducible factor ((Staller et al, 2003), which is strongly upregulated in psoriatic skin (Rosenberger et al, 2007). Our results suggest that CXCR4 (and SOCS3) upregulation may be a counter mechanism to balance the highly proliferative cytokine signals that are present in psoriatic lesions and to limit the circumferential expansion of psoriatic plaques. Given the limited amount of human tissue available to us that contained lesional, junction, and non-lesional areas, our results will need to be confirmed with larger numbers of psoriatic tissue samples. In aggregate, our data suggest that drugs that up-regulate CXCR4 function or expression might be helpful in the setting of psoriasis or other hyperproliferative diseases.
As illustrated in Figure 6A, upregulation of SOCS3 is a well-known negative feedback mechanism that balances the effect of STAT3 activation. In psoriatic lesions, however, SOCS3 is apparently not expressed at sufficient levels to block keratinocyte proliferation in the presence of STAT3-dependent inflammatory cytokines such as IL-22. CXCR4-mediated signaling, however, synergistically upregulates SOCS3, leading to reduced phosphorylation of STAT3, and, subsequently, to the reduced ability of HaCaT cells to proliferate in vitro in response to IL-22 (Figure 6B). Our in vitro data is complemented by our in vivo data showing profound loss of CXCR4 and SOCS3 expression in thick epidermal regions compared to the thin epidermal regions of IL-23-treated WT mice. CXCR4 may be essential for downregulating KC proliferation, and thus mitigating the effects of proliferative cytokines such as IL-23 and IL-22 under inflammatory conditions. It remains to be determined whether or not CXCR4 affects the degree of inflammation in other inflammatory skin diseases that do not show enhanced epidermal hyperplasia.
Figure 6. Schematic model of the role of CXCR4 and CXCL12 in IL-22-mediated keratinocyte proliferation.
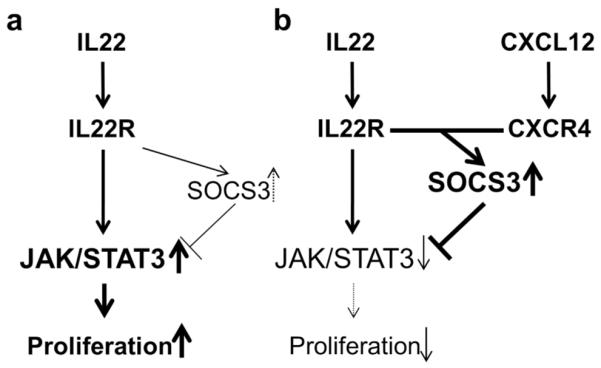
A schematic model for IL-22, CXCL12, and SOCS3 regulation of KC proliferation is shown. JAK/STAT3 pathway is activated by IL-22 and results in modest upregulation of SOCS3 that is insufficient to downregulate keratinocyte proliferation (A). SOCS3 expression, however, is synergistically upregulated in the presence of IL-22, CXCR4, and CXCL12 (B), resulting in the abrogation of STAT3 signaling and marked inhibition of keratinocyte proliferation.
Materials and Methods
Mice
C57BL/6 WT between 8 and 12 week old were purchased from Charles River Laboratories. Tissue-specific knockout mice were generated using a Cre-loxP approach (Zimmerman et al, 2011, Nagy, 2000). Heterozygous C57BL/6J mice carrying a floxed CXCR4 allele (CXCR4f/wt) were originally obtained from Dr. Daniel Littman (Howard Hughes Medical Institute, New York University, New York, NY). To selectively inactivate CXCR4 in basal KCs, CXCR4f/f mice were crossed with transgenic mice expressing the Cre recombinase under the transcriptional control of the K14 promoter (STOCK Tg (KRT14-cre) 1Amc/J; Jackson Laboratories). Animal protocols were approved by the Institutional Animal Care and Use Committee at the Medical College of Wisconsin.
Human psoriasis skin samples
Clinical protocols in compliance with Helsinki guidelines that approved collection of human psoriasis skin sample were obtained from the Institutional Ethical Committee of the Faculty of Medicine, University of Tokyo. All subjects signed written informed consent. Skin biopsies from the border of psoriatic lesions were taken from 3 patients (Supplementary Table S1) with typical psoriasis lesion by excisional biopsy. Skin samples were embedded in OCT compound, frozen, and then sectioned.
Genotyping of tissue specific knockout mice
Mouse DNA was extracted from tail tissue using DNeasy Blood & Tissue Kit (Qiagen). PCR using Taq DNA polymerase (Qiagen) was performed. The forward and reverse primers employed for PCR were shown in Supplementary Table S2.
Retroviral transduction of HaCaT human KC cell line
HaCaT cells were transduced with hCXCR4 cDNA using the pLNCX2 retroviral vector (Clontech, Mountain View, CA) as previously described (Fang et al, 2008). CXCR4-overexpressing HaCaT (CXCR4-HaCaT) were grown in MEM (Invitrogen) with 10% heat-inactivated FBS (Biowest).
Cell Proliferation assay
CXCR4-HaCaT cells (2×104) were cultured in a 6-well plate for 1 day followed by incubation with with recombinant human CXCL12 (Dr. Brian Volkman (Biochemistry, Medical College of Wisconsin; (Veldkamp et al, 2009) and/or recombinant human IL-22 for 2 days. Cells were trypsinized, washed with PBS, and resuspended in CellTiter-Glo™ Luminescent Cell Viability Assay (Promega) for measurement of luciferase activity.
siRNA transfection
siRNAs for SOCS3 (Silencer® Select Pre-designed siRNA, s17189) and non-specific control (Silencer® Select Negative Control #1 siRNA) were purchased from Ambion. They were used for transfection of CXCR4-HaCaT cells using Lipofectamine™ 2000 (Invitrogen) according to manufacturer’s protocol.
IL-23 injection model
Recombinant mouse IL-23 (500 ng/site in 20 μl PBS; eBioscience) was injected into ears of anesthetized mice every other day for 6 days as described (Hedrick et al, 2009). After harvesting mice ears, ventral skin sheets were incubated in PBS containing 0.5% trypsin (U.S. Biochemical) for 40 min at 37°C to separate epidermis from dermis. To obtain cell suspensions, epidermal sheets were repeatedly minced with scissors (Salgado et al, 1999). Cells were then filtered and washed with PBS prior to use.
Quantitative Real-Time RT-PCR
The extraction of RNA and cDNA synthesis from epidermal or dermal cell suspensions were performed using an RNeasy Kit (Qiagen) and High Capacity cDNA Reverse Transcription Kits (Applied Biosystems). RT-PCR was performed via StepOnePlus Real-Time PCR System (Applied Biosystems) using SYBR Green PCR master mix (Applied Biosystems). We confirmed PCR quality by performing melting curve analysis each time. The forward and reverse primers employed for real-time RT-PCR were shown in Supplementary Table S2 (Integrated DNA Technologies, Coralville, IA).
Western blotting
Total proteins were extracted using RIPA buffer (Sigma-Aldrich) containing protease inhibitor cocktail (Roche). After centrifugation, supernatants were collected and cell lysates were reduced, boiled, loaded onto 4-12% Bis-Tris gel (Invitrogen) and transferred to nitrocellulose membrane (Invitrogen). Membranes were blocked with 2% BSA (Sigma) for 1 hr and incubated with anti-phosphorylated STAT3 monoclonal antibody (Santa Cruz) at 1:200 dilutions for 1 hr or with anti STAT3 p92 polyclonal antibody (eBioscience) at 1:500 for overnight. Membranes were then washed three times and incubated with HRP-conjugated goat anti-mouse IgG (Santa Cruz), or with HRP-conjugated donkey anti-rabbit IgG (Santa Cruz) at 1: 500 dilutions for 1 hr. After washing, membranes were incubated with chemiluminescent solution (ECL Western Blotting Detection Reagents; GE Healthcare UK Ltd) for 5 min at room temperature. Images were acquired using ChemiDoc XRS+ system (Bio-Rad). Densitometry analysis was performed using ImageJ 1.45s software (NIH). When required, membranes were incubated in stripping buffer (Thermo Fisher Scientific) for 30 min at 37°C to remove bound antibodies.
Immunohistochemistry
Anti-CXCR4 (C-20) and anti-SOCS3 antibody (M-20; Santa Cruz) in combination with Alexa Fluor 568 donkey anti-goat IgG (H+L) secondary antibodies were used alone or with corresponding target peptides as specificity controls.
Mouse ears were excised, embedded in OCT compound without fixation, frozen, and then sectioned. CXCR4-HaCaT cells were cultured in Lab-Tek™II Chamber Slides (Thermo scientific) for 1 day followed serum deprivation for 1 day. Cells were then incubated with cytokines for 1 hr and were air-dried. For CXCR4 staining, tissue sections were fixed with ice-cold acetone for 10 min (CXCR4 staining) or with 4% paraformaldehyde PBS solution for 10 min followed by incubation with 95% ethanol for 20 min (SOCS3 staining), blocked for 1 hr at RT with 5% donkey serum, Fc-blocker (2.4G2; Bio X Cell), and donkey anti-goat IgG-HRP (Santa Cruz) in PBS containing 3% skim milk. Sections were incubated with primary antibody overnight at 4°C followed by incubation with secondary antibody for 30 min at RT and SlowFade Gold Antifade Reagent With DAPI (Invitrogen). Following image acquisition, ImageJ 1.45s software (NIH) was used for quantification. For measuring epidermal hyperplasia, we selected the thickest epidermal regions from each mouse ear sample and measured the epidermal thickness at four random points in a blinded fashion. We counted infiltrating cells to the dermis in a blinded fashion.
Statistical Analysis
All quantitative data were shown as the mean ± SD unless otherwise indicated. Simple comparisons of means and SD of data were made by using Student t test, and post hoc multiple comparisons (indicated by * next to p values) were made with one-way ANOVA and Scheffé’s test, using the Statcel statistical analysis software package (OMS Publishing Inc., Saitama, Japan).
Supplementary Material
Supplementary Figure S1. Genotyping of K14-CXCR4KO and control mice. Homozygous loxP flanked (floxed) CXCR4 mice (CXCR4f/f) were crossed with K14-Cre-mice as described in Materials and Methods. DNA was extracted from mouse tails and PCR for Cre and loxP flanked (floxed) CXCR4 genes was performed to confirm the presence of Cre, floxed CXCR4, and wildtype (wt) CXCR4 sequences. Two examples of genotyping of K14-CXCR4KO mice (CXCR4f/f-Cre+) are shown. CXCR4f/f-Cre− mice were used as positive controls and purified water was used as negative control.
Supplementary Figure S2. Incomplete CXCR4 knockout area of K14-CXCR4 KO treated with IL-23 showed thinner epidermis compared with complete CXCR4 knockout area. K14-CXCR4 KO ears were injected with IL-23 every other day for 5 days and collected at day 5. CXCR4 staining was performed and incomplete CXCR4 knockout area and complete CXCR4 knockout area are shown. The superior dotted lines represent the approximate level of the cornified layer while the inferior dotted lines represent the approximate level of the epidermal basement membrane. White dotted square areas of left upper panel, right upper panel, and right lower-middle panel are shown at panels shown just blow each panel.
Supplementary Figure S3. Immunostaining for SOCS3 in CXCR4-HaCaT cells cultured with IL-22 and/or CXCL12. CXCR4-HaCaT cells were cultured with IL-22 and/or CXCL12 for 1 hr and then stained using anti-SOCS3 Ab. A representative image from each treatment group is shown. Scale bars, 100 μm (A). Four HPFs, which contained similar numbers of cells by DAPI staining, were selected for measurement of SOCS3-positive area using ImageJ software (B). *Indicates significance using Scheffé’s test for post hoc multiple comparison.
Supplementary Figure S4. STAT3 and pSTAT3 protein expression in CXCR4-HaCaT cells cultured with IL-22 and/ or CXCL12. STAT3 protein levels and pSTAT3 protein levels in CXCR4-HaCaT cells cultured with IL-22 and/or CXCL12 were evaluated by Western blot [full-length blot shown, and cropped blot is shown in Fig 5D]. The same membrane was used for second antibody staining after stripping the first antibody prior to restaining.
Supplementary Figure S5. RNA interference for SOCS3 in CXCR4-HaCaT cells. siRNA for S0CS3 was transduced to CXCR4-HaCaT cells. Cells were cultured in slide chamber with CXCL12 and IL-23 for 1 hr and then stained using anti-S0CS3 antibody. Four HPFs, which contained SOCS3 expressing cells, were selected and SOCS3-positive area was measured using ImageJ software. *lndicates significance using Scheffé’s test for post hoc multiple comparison.
Supplementary Figure S6. CXCR4 and SOCS3 expression in human psoriatic skin. Three human psoriasis skin samples containing the junctional zone between lesional and peri-lesional regions were stained using anti-CXCR4 and anti-SOCS3 antibodies (Patients’ information is shown in Supplementary Table S1.). CXCR4 staining of patient 1′s sample is shown. White dotted square area of left-upper panel is shown at left-lower panel (A). SOCS3 staining of patient 3′s sample is shown (B). Scale bars, 100 μm.
Supplementary Table S1: Patients’ information of Figure 6
Supplementary Table S2: Primer sequences
Acknowledgments
We thank Dr. Joshua J. Ziarek (Biochemistry, MCW), Ms. Jenny Grewal (Microbiology and Molecular Genetics, MCW), Mr. Nathan Duncan, Brian C. Schulte (Dermatology, MCW), Ms. Tamami Kaga (Dermatology, Univ. of Tokyo), and Dr. Uimook Choi (Laboratory of Host Defenses, NIH) for their assistance. These studies were supported by Advancing Healthier Wisconsin Research Funds (to STH), a National Psoriasis Foundation Translational Grant (to STH), and NIDDK grant DK062066 (to MBD).
Abbreviations List
- KC
keratinocyte
- CXCR
chemokine receptor
- K14
keratin 14
- STAT3
signal transducer and activator of transcription 3
- SOCS3
suppressor of cytokine signaling 3
Footnotes
Conflict of Interest The authors state no conflict of interest.
References
- Avniel S, Arik Z, Maly A, Sagie A, Basst HB, Yahana MD, et al. Involvement of the CXCL12/CXCR4 pathway in the recovery of skin following burns. J Invest Dermatol. 2006;126:468–476. doi: 10.1038/sj.jid.5700069. [DOI] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–833. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- Boniface K, Bernard FX, Garcia M, Gurney AL, Lecron JC, Morel F. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J Immunol. 2005;174:3695–3702. doi: 10.4049/jimmunol.174.6.3695. [DOI] [PubMed] [Google Scholar]
- Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley EC, Benveniste EN. Signal transducer and activator of transcription-3: A molecular hub for signaling pathways in gliomas. Mol Cancer Res. 2008;6:675–684. doi: 10.1158/1541-7786.MCR-07-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2577–2587. doi: 10.1084/jem.20060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’Agnola C, Biragyn A. Clinical utilization of chemokines to combat cancer: The double-edged sword. Expert Rev Vaccines. 2007;6:267–283. doi: 10.1586/14760584.6.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Lee VC, Cha E, Zhang H, Hwang ST. CCR7 regulates B16 murine melanoma cell tumorigenesis in skin. J Leukoc Biol. 2008;84:965–972. doi: 10.1189/jlb.1107776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch E, Harper E, Skorcheva I, Kurtz SE, Blauvelt A. Pathophysiology of psoriasis: Recent advances on IL-23 and Th17 cytokines. Curr Rheumatol Rep. 2007;9:461–7. doi: 10.1007/s11926-007-0075-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R, Soriano SF, Rodriguez-Frade JM, Gomez L, Martin de Ana A, Sanchez-Gomez M, et al. CXCR4-mediated suppressor of cytokine signaling up-regulation inactivates growth hormone function. J Biol Chem. 2004;279:44460–44466. doi: 10.1074/jbc.M408010200. [DOI] [PubMed] [Google Scholar]
- Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, Singh SP, et al. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. 2009;119:2317–2329. doi: 10.1172/JCI37378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi Y, Bae SJ, Katayama I. Overexpression of the suppressor of cytokine signalling 3 (SOCS3) in severe atopic dermatitis. Clin Exp Dermatol. 2006;31:100–104. doi: 10.1111/j.1365-2230.2005.01979.x. [DOI] [PubMed] [Google Scholar]
- Kreis S, Philippidou D, Margue C, Rolvering C, Haan C, Dumoutier L, et al. Recombinant interleukin-24 lacks apoptosis-inducing properties in melanoma cells. PLoS One. 2007;2:e1300. doi: 10.1371/journal.pone.0001300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke A, Goren I, Bosl MR, Pfeilschifter J, Frank S. The suppressor of cytokine signaling (SOCS)-3 determines keratinocyte proliferative and migratory potential during skin repair. J Invest Dermatol. 2010;130:876–885. doi: 10.1038/jid.2009.344. [DOI] [PubMed] [Google Scholar]
- Lonsdorf AS, Hwang ST, Enk AH. Chemokine receptors in T-cell-mediated diseases of the skin. J Invest Dermatol. 2009;129:2552–66. doi: 10.1038/jid.2009.122. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Chang TW, Quinter S, Hwang ST. Chemokine receptors in the pathogenesis and therapy of psoriasis. J Dermatol Sci. 2012;65:4–11. doi: 10.1016/j.jdermsci.2011.11.007. [DOI] [PubMed] [Google Scholar]
- Mabuchi T, Takekoshi T, Hwang ST. Epidermal CCR6+ gammadelta T cells are major producers of IL-22 and IL-17 in a murine model of psoriasiform dermatitis. J Immunol. 2011;187:5026–5031. doi: 10.4049/jimmunol.1101817. [DOI] [PubMed] [Google Scholar]
- Mansbridge JN, Knapp AM. Changes in keratinocyte maturation during wound healing. J Invest Dermatol. 1987;89:253–263. doi: 10.1111/1523-1747.ep12471216. [DOI] [PubMed] [Google Scholar]
- Murakami T, Cardones AR, Finkelstein SE, Restifo NP, Klaunberg BA, Nestle FO, et al. Immune evasion by murine melanoma mediated through CC chemokine receptor-10. Journal of Experimental Medicine. 2003;198:1337–47. doi: 10.1084/jem.20030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO, et al. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–7334. [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: The universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Nakagawa R, Yoshida H, Asakawa M, Tamiya T, Inoue N, Morita R, et al. Pyridone 6, a pan-JAK inhibitor, ameliorates allergic skin inflammation of NC/Nga mice via suppression of Th2 and enhancement of Th17. J Immunol. 2011;187:4611–4620. doi: 10.4049/jimmunol.1100649. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- Petit I, Szyper-Kravitz M, Nagler A, Lahav M, Peled A, Habler L, et al. G-CSF induces stem cell mobilization by decreasing bone marrow SDF-1 and up-regulating CXCR4. Nat Immunol. 2002;3:687–694. doi: 10.1038/ni813. [DOI] [PubMed] [Google Scholar]
- Romanowska M, Reilly L, Palmer CN, Gustafsson MC, Foerster J. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PLoS One. 2010;5:e9701. doi: 10.1371/journal.pone.0009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger C, Solovan C, Rosenberger AD, Jinping L, Treudler R, Frei U, et al. Upregulation of hypoxia-inducible factors in normal and psoriatic skin. J Invest Dermatol. 2007;127:2445–2452. doi: 10.1038/sj.jid.5700874. [DOI] [PubMed] [Google Scholar]
- Salgado CG, Nakamura K, Sugaya M, Tada Y, Asahina A, Fukuda S, et al. Differential effects of cytokines and immunosuppressive drugs on CD40, B7-1, and B7-2 expression on purified epidermal langerhans cells1. J Invest Dermatol. 1999;113:1021–1027. doi: 10.1046/j.1523-1747.1999.00785.x. [DOI] [PubMed] [Google Scholar]
- Schon MP, Boehncke WH. Psoriasis. N Engl J Med. 2005;352:1899–1912. doi: 10.1056/NEJMra041320. [DOI] [PubMed] [Google Scholar]
- Sestito R, Madonna S, Scarponi C, Cianfarani F, Failla CM, Cavani A, et al. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 2011;25:916–927. doi: 10.1096/fj.10-172288. [DOI] [PubMed] [Google Scholar]
- Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von hippel-lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- Urbich C, Dimmeler S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ Res. 2004;95:343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- Veldkamp CT, Ziarek JJ, Su J, Basnet H, Lennertz R, Weiner JJ, et al. Monomeric structure of the cardioprotective chemokine SDF-1/CXCL12. Protein Sci. 2009;18:1359–1369. doi: 10.1002/pro.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss G, Shemer A, Trau H. The koebner phenomenon: Review of the literature. J Eur Acad Dermatol Venereol. 2002;16:241–248. doi: 10.1046/j.1473-2165.2002.00406.x. [DOI] [PubMed] [Google Scholar]
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–51. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zimmerman NP, Vongsa RA, Faherty SL, Salzman NH, Dwinell MB. Targeted intestinal epithelial deletion of the chemokine receptor CXCR4 reveals important roles for extracellular-regulated kinase-1/2 in restitution. Lab Invest. 2011;91:1040–1055. doi: 10.1038/labinvest.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Genotyping of K14-CXCR4KO and control mice. Homozygous loxP flanked (floxed) CXCR4 mice (CXCR4f/f) were crossed with K14-Cre-mice as described in Materials and Methods. DNA was extracted from mouse tails and PCR for Cre and loxP flanked (floxed) CXCR4 genes was performed to confirm the presence of Cre, floxed CXCR4, and wildtype (wt) CXCR4 sequences. Two examples of genotyping of K14-CXCR4KO mice (CXCR4f/f-Cre+) are shown. CXCR4f/f-Cre− mice were used as positive controls and purified water was used as negative control.
Supplementary Figure S2. Incomplete CXCR4 knockout area of K14-CXCR4 KO treated with IL-23 showed thinner epidermis compared with complete CXCR4 knockout area. K14-CXCR4 KO ears were injected with IL-23 every other day for 5 days and collected at day 5. CXCR4 staining was performed and incomplete CXCR4 knockout area and complete CXCR4 knockout area are shown. The superior dotted lines represent the approximate level of the cornified layer while the inferior dotted lines represent the approximate level of the epidermal basement membrane. White dotted square areas of left upper panel, right upper panel, and right lower-middle panel are shown at panels shown just blow each panel.
Supplementary Figure S3. Immunostaining for SOCS3 in CXCR4-HaCaT cells cultured with IL-22 and/or CXCL12. CXCR4-HaCaT cells were cultured with IL-22 and/or CXCL12 for 1 hr and then stained using anti-SOCS3 Ab. A representative image from each treatment group is shown. Scale bars, 100 μm (A). Four HPFs, which contained similar numbers of cells by DAPI staining, were selected for measurement of SOCS3-positive area using ImageJ software (B). *Indicates significance using Scheffé’s test for post hoc multiple comparison.
Supplementary Figure S4. STAT3 and pSTAT3 protein expression in CXCR4-HaCaT cells cultured with IL-22 and/ or CXCL12. STAT3 protein levels and pSTAT3 protein levels in CXCR4-HaCaT cells cultured with IL-22 and/or CXCL12 were evaluated by Western blot [full-length blot shown, and cropped blot is shown in Fig 5D]. The same membrane was used for second antibody staining after stripping the first antibody prior to restaining.
Supplementary Figure S5. RNA interference for SOCS3 in CXCR4-HaCaT cells. siRNA for S0CS3 was transduced to CXCR4-HaCaT cells. Cells were cultured in slide chamber with CXCL12 and IL-23 for 1 hr and then stained using anti-S0CS3 antibody. Four HPFs, which contained SOCS3 expressing cells, were selected and SOCS3-positive area was measured using ImageJ software. *lndicates significance using Scheffé’s test for post hoc multiple comparison.
Supplementary Figure S6. CXCR4 and SOCS3 expression in human psoriatic skin. Three human psoriasis skin samples containing the junctional zone between lesional and peri-lesional regions were stained using anti-CXCR4 and anti-SOCS3 antibodies (Patients’ information is shown in Supplementary Table S1.). CXCR4 staining of patient 1′s sample is shown. White dotted square area of left-upper panel is shown at left-lower panel (A). SOCS3 staining of patient 3′s sample is shown (B). Scale bars, 100 μm.
Supplementary Table S1: Patients’ information of Figure 6
Supplementary Table S2: Primer sequences