

Abstract
Nrf2 is a transcription factor that protects against inflammatory diseases, but the underlying mechanism of this effect remains unclear. Here, we report that Nrf2 uses lipocalin–prostaglandin D synthase (L-PGDS) as a mechanism for suppressing inflammation. Exogenously added prostaglandin D2 (PGD2) induced L-PGDS expression in bone-marrow-derived macrophages (BMDMs), suggesting a positive feedback loop between L-PGDS expression and PGD2. Unlike lipopolysaccharide (LPS)-induced L-PGDS expression, PGD2-mediated expression was independent of MAPK, PU.1, or TLR4. Sequence analysis located a putative Nrf2 binding site in the murine L-PGDS promoter, to which Nrf2 bound when treated with PGD2. Chemical activation, or overexpression, of Nrf2 was sufficient to induce L-PGDS expression in macrophages, BMDMs, or lungs of Nrf2-knockout (KO) mice, but treatment with PGD2 failed to do so, suggesting a pivotal role for Nrf2 in the expression of L-PGDS. Consistent with this, expression of Nrf2 in the lungs of Nrf2-KO mice was sufficient to induce the expression of L-PGDS and to reduce neutrophilic lung inflammation elicited by LPS. Furthermore, expression of L-PGDS in mouse lungs decreased neutrophilic infiltration, ameliorating lung inflammation in mice. Together, our results show that Nrf2, activated by PGD2, induced L-PGDS expression, resulting in decreased inflammation. We suggest that the positive feedback induction of L-PGDS by PGD2 is part of the mechanism by which Nrf2 regulates inflammation.
Keywords: Nrf2, Lung inflammation, Lipocalin-prostaglandin D synthase, Prostaglandin D2, Gene expression, Free radicals
Prostaglandin D2 (PGD2), a major prostanoid produced by macrophages and mast cells [1,2], affects inflammation. How PGD2 influences inflammation has been controversial, though. This is because PGD2 either promotes or suppresses inflammation depending on the experimental setting. For example, PGD2 challenge induces eosinophil infiltration and thus exacerbates asthma [3–5], suggesting that PGD2 promotes inflammation. By contrast, PGD2 suppresses inflammation in lungs of animals challenged with bleomycin or monosodium urate monohydrate crystal [6–8]. PGD2 also facilitates resolution of acute inflammation [9], functioning as a proresolving lipid mediator along with resolvins, protectins, lipoxins, and aspirin-triggered lipoxins [10,11]. PGD2 is bound to generate 15-deoxy-Δ12,14-PGJ2 via nonenzymatic processes, and it has been well documented that 15-deoxy-Δ12,14-PGJ2 suppresses inflammation by activating peroxisome proliferator-activated receptor (PPAR)-γ [12,13] and NF-E2-related factor 2 (Nrf2) [14]. Therefore, these studies show that PGD2, directly or through 15-deoxy-Δ12,14-PGJ2, can suppress inflammation, contingent on the inflammatory milieu.
Lipopolysaccharide (LPS), a cell wall component of gram-negative bacteria, binds to Toll-like receptor 4 (TLR4) on macrophages and activates mitogen-activated protein kinases (MAPKs) and NF-κB [15], resulting in the production of proinflammatory mediators including cyclooxygenase (COX)-2 [16]. Along with the constitutively expressed isozyme COX-1, COX-2 synthesizes prostaglandin H2 (PGH2), which is used as a common substrate for other prostanoids, including PGD2 [17]. Two distinctive enzymes convert PGH2 to PGD2: hematopoietic-type (H-PGDS) and lipocalin-type prostaglandin D synthase (L-PGDS). H-PGDS is constitutively expressed mainly in hematopoietic cells, whereas L-PGDS is mainly detected in the central nervous system and related organs [18]. Given the key role of hematopoietic cells in inflammation, H-PGDS has been known to account for the production of PGD2 in inflammation [9,19]. It was, however, reported that LPS induces the expression of L-PGDS in macrophages and in mouse lungs, which contributes to at least half of the PGD2 produced by macrophages [20]. In addition, L-PGDS expression is detected in macrophages that have infiltrated atherosclerotic plaques [21]. These studies suggest that L-PGDS also plays a role in regulating inflammation. However, the significance and mechanism of L-PGDS expression in inflammation remain less understood.
Reactive oxygen species (ROS) generated by activated macrophages elicit oxidative stress and inflict damage to tissues while helping to remove invading pathogens [22]. In response to ROS, cells express various proteins to counter the deleterious effects of ROS by activating Nrf2, a member of the cap’n’collar family of basic leucine-zipper transcription factors [14]. Under unstimulated conditions, Nrf2 is constantly degraded by Keap1 in the cytoplasm, which keeps the level of Nrf2 low in the cytoplasm [23]. However, oxidative stress inhibits the function of Keap1 against Nrf2, which allows Nrf2 to accumulate in the nucleus, resulting in expression of phase II detoxification and antioxidant enzymes such as glutamate–cysteine ligase catalytic subunit (GCLC), NAD(P)H:quinine oxidoreductase-1 (NQO1), and heme oxygenase-1 (HO-1) [24,25]. Studies have shown that Nrf2 is protective against various inflammatory lung diseases in animal models of acute lung injury, asthma, and smoke-induced emphysema [26–28]. Although nullifying the deleterious effects of ROS probably contributes to suppression of inflammation, the mechanisms by which Nrf2 suppresses inflammation remain less understood.
In this study, we sought to understand how Nrf2 protects from inflammatory diseases. We found that Nrf2 induced the expression of L-PGDS, which was associated with decreased inflammation. Nrf2 bound to the promoter of L-PGDS and induced the expression of the enzyme when treated with PGD2. Whereas genetic ablation of nrf2 failed to induce L-PGDS expression, supplementation of Nrf2 to Nrf2-knockout (KO) mice induced L-PGDS expression, which was associated with reduced neutrophilic lung inflammation. Similarly, expression of L-PGDS in mouse lungs was sufficient to relieve lung inflammation. Given that Nrf2 and PGD2 are involved in suppressing inflammation and that L-PGDS was sufficient for suppressing lung inflammation, we suggest that a positive feedback regulation of L-PGDS expression by PGD2 is a part of the mechanism of the anti-inflammatory activity of Nrf2.
Material and methods
Reagents
PGD2, 15-deoxy-Δ12,14-PGJ2, PGE2, PPARγ agonist rosiglitazone, PPARγ inhibitor GW9146, and COX-1 inhibitor SC-560 were purchased from Cayman Chemical (Ann Arbor, MI, USA). TLR4-specific Escherichia coli LPS was obtained from Enzo Biochemical (Farmingdale, NY, USA). Sulforaphane was from Sigma Chemical Co. (St. Louis, MO, USA). MAPK inhibitors including SB 220025 (p38 inhibitor), JNK inhibitor II, and U0126 (ERK1/2 inhibitor) were purchased from EMD Millipore Chemicals (Darmstadt, Germany).
Macrophages and culture
A murine macrophage cell line, RAW 264.7 cells (ATCC, Rock-ville, MD, USA), was cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Grand Island, NY, USA) containing 10% fetal bovine serum (FBS; Invitrogen). Bone-marrow-derived macrophages (BMDMs) were prepared as described elsewhere [29]. In short, cellular material from mouse femurs was flushed out with phosphate-buffered saline (PBS) and cultured in DMEM containing 10% FBS and 10% L929 cell culture medium for 5 days before experiments.
Animal model
C57BL/6, TLR4 mutant, and COX-2-knockout mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Along with these mice, Nrf2-knockout mice [26] were housed in certified, standard laboratory cages and fed with food and water ad libitum before experiments. Mice weighing 20–28 g were used for experiments. All experimental procedures were in accordance with the NIH of Korea Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Pusan National University, Pusan, Korea (Approval No. PNU-2010-00028). Mice were anesthetized with Zoletil (Virbac Korea, Seoul, Korea) and received a single dose of PGD2 (0.03 μg/kg body wt) in 25 μl of PBS, which was loaded in a MicroSprayer Aerosolizer-Model IA-1C (Penn-Century, Wyndmoor, PA, USA) and delivered in aerosol to the lung via the trachea under visual guidance. After incubation with EC10, a liposome that carries DNA effectively to the lung [30], a plasmid (1.5 mg/kg body wt) encoding L-PGDS [18] or Nrf2 [31] was similarly loaded in a MicroSprayer and delivered in aerosol to the lung, 24 h before LPS instillation. Mice received a single dose of 10 mg E. coli LPS (serotype 055:B5; Sigma)/kg body wt intranasally. At 24 h after administration of LPS, the mice were euthanized with CO2 gas. The trachea was exposed through a midline incision and cannulated with a sterile 24-gauge intravascular catheter. Bilateral bronchoalveolar lavage (BAL) was performed by two consecutive instillations of 1.0 ml of PBS. Total cell numbers in BAL fluid were counted with a hemocytometer, and the cells in BAL fluid were prepared by cytospin and stained for the differentiation of macrophages, lymphocytes, or neutrophils by Hemacolor (Merck, Darmstadt, Germany). Three hundred cells in total were counted, and 100 of the cells in each microscopic field were scored. The mean number of cells per field was reported.
For collecting lung tissue, mice were perfused with saline and the whole lung was inflated with fixatives. After paraffin embedding, 5-μm sections were cut and placed on charged slides and stained with hematoxylin and eosin (H&E). Three separate H&E-stained sections were evaluated in 200 microscopic magnifications per mouse.
Plasmid, siRNA, and transfection
Murine Nrf2 [31] and L-PGDS [18] were subcloned into pcDNA3.1(+) (Invitrogen), the fidelity of which was verified by sequencing. DNA was transfected using Lipofectamine 2000 and the manufacturer’s protocol (Invitrogen). Scrambled and siRNA specific for PU.1 was purchased and transfected per the manufacturer’s manual (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Isolation of total RNA and semiquantitative RT-PCR
Total RNA from cells or tissue was isolated using Trizol reagent and the manufacturer’s instructions (Invitrogen). After the concentration of RNA was determined by spectrophotometer, 2 μg of RNA was reverse-transcribed by M-MLV reverse transcriptase (Promega, Madison, WI, USA) to generate cDNA. The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNA level from each sample was used to normalize the samples for differences in PCR efficiency. L-PGDS mRNA quantity was determined by using end-point dilution PCR, including three serial 1 to 5 dilutions (1:1, 1:5, 1:25, and 1:125) of RT products for PCR amplification. To eliminate genomic DNA contamination, equal amounts of total RNA from each sample were PCR amplified without RT reaction. A portion of the cDNA was amplified with 0.5 U of Taq polymerase (PerkinElmer, Waltham, MA, USA) and appropriate oligonucleotides at 94 °C for 40 s, 60 °C for 30 s, and 72 °C for 40 s for 35 cycles, with an initial 4-min denaturation at 95 °C and final 10-min extension at 72 °C. The primers used in this study were as follows: the primers for L-PGDS were 5′-GGTTCCGGGAGAAGAAAGCT-3′ and 5′-CACTGACACGGAGTGGATGC-3′; the primers for murine Nrf2 were 5′-AAAGCACAGCCAGCACATTC-3′ and 5′-GACCAGGACTCACGGGAACT-3′; the primers for NQO1 were 5′-GCAGTGCTTTCCATCACCAC-3′ and 5′-TGGAGTGTGCCCAATGCTAT-3′; the primers for HO-1 were 5′-TGAAGGAGGCCACCAAGGAGG-3′ and 5′-AGAGGTCACCCAGGTAGCGGG-3′; the primers for GCLC were 5′-CACTGC-CAGAACACAGACCC-3′ and 5′-ATGGTCTGGCTGAGAAGCCT-3′; and those for GAPDH were 5′-GGAGCCAAAAGGGTCATCAT-3′ and 5′-GTGATGGCATGGACTGTGGT-3′.
Protein isolation and Western blot
Total cell lysate was prepared with radioimmunoprecipitation assay cell lysis buffer (50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% sodium orthovanadate, 1% Triton X-100, 0.5% deoxycholate, 0.1% sodium dodecyl sulfate) supplemented with protease inhibitors (Roche Korea, Seoul, Korea). Nuclear proteins were isolated from the lung tissue of mice using a Thermo Scientific NE-PER nuclear extraction kit and the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, USA). Protein content was quantified by the Bradford assay (Bio-Rad, Hercules, CA, USA) as specified by the manufacturer. Equal amounts of proteins were fractionated by SDS–PAGE and then transferred to PVDF membrane (Bio-Rad). Blots were blocked for at least 1 h with 5% nonfat dry milk before incubation with appropriate antibodies at 4 °C overnight. Immune complex was detected by enhanced chemiluminescence (ECL Plus; GE Healthcare Biosciences, Pittsburgh, PA, USA). All antibodies used in this study were purchased from Santa Cruz Biotechnology.
Chromatin immunoprecipitation (ChIP) assay
Reagents were obtained from Upstate Biotechnology (Lake Placid, NY, USA), and the assay was performed as described previously [32]. One microgram of anti-Nrf2 antibody (Santa Cruz Biotechnology) was used for precipitation of Nrf2 bound to DNA, along with isotypic IgG (Santa Cruz Biotechnology) to exclude nonspecific precipitation. PCR was performed as follows: 94 °C for 240 s; 30–32 cycles at 94 °C for 40 s, 54 °C for 40 s, and 72 °C for 60 s; and final elongation at 72 °C for 10 min. PCR for the input was performed with 100 ng of genomic DNA. The primers used for the PCR were 5′-ATGGAGCTGAGTGTTCTGGG-3′ and 5′-TCTGGGGCACAGGATCATTT-3′. This primer set covers the murine L-PGDS promoter segment from −945 to −776 nt, which contains the putative Nrf2 binding site.
Statistical analysis
Data are presented as 7SEM (standard error of the mean) of at least three separate experiments. For comparison among groups, paired or unpaired t tests and one-way analysis of variance were used (with the assistance of InStat, GraphPad Software, San Diego, CA, USA). P values less than 0.05 were considered statistically significant.
Results
A positive feedback induction of L-PGDS by PGD2 in macrophages
Because the level of PGD2 culminates and then dwindles during inflammation [9,20,33], we began the study by hypothesizing that PGD2 is involved in regulating the production of itself. To test this hypothesis, we first examined the possibility that PGD2 regulates its own production by suppressing the expression of H-PGDS or L-PGDS, given that these enzymes specifically synthesize PGD2. BMDMs were prepared from femurs of C57BL/6 mice and treated with PGD2 (0.1 μM) or LPS (0.1 μg/ml). At 16 h after treatment, total cell lysates were prepared from the treated cells and analyzed by Western blotting for H-PGDS and L-PGDS. Consistent with the previous study [20], LPS treatment induced the expression of L-PGDS without affecting constitutive expression of H-PGDS (Fig. 1A). Similar to LPS, PGD2 treatment also induced the expression of L-PGDS without affecting the level of H-PGDS. Given that L-PGDS produces PGD2, these results suggest the possibility that there is a novel, self-regulatory mechanism for the expression of L-PGDS.
Fig. 1.

PGD2 induces the expression of L-PGDS. (A) One million BMDMs were treated with LPS (0.1 μg/ml), PGD2 (0.1 μM), or vehicle (DMSO) for 16 h. 50 μg of proteins in total cell lysate was fractionated by SDS–PAGE and analyzed by Western blotting for L-PGDS (top) and H-PGDS (bottom). (B) Similar to (A), 1,000,000 BMDMs were treated with LPS or PGD2 and analyzed by Western blotting for COX-2 (top) and L-PGDS (middle). (C) BMDMs prepared from cox-2−/− mice were treated with 0.1 mM (recommended by manufacturer) or 1 mM SC-560 for 24 h before PGD2 treatment. At 16 h after PGD2 (0.1 μM) treatment, 50 μg of total proteins was analyzed by Western blotting for L-PGDS. The membrane was stripped and blotted again for β-actin. All the experiments were repeated at least three times.
Next, we determined whether PGD2 induces L-PGDS expression directly or indirectly via other prostanoids. To do this, we examined whether L-PGDS expression by PGD2 accompanied the expression of COX-2. As shown in Fig. 1B, treatment of BMDMs with PGD2 for 16 h did not induce COX-2 expression. Similarly, no induction was detected when BMDMs were treated with PGD2 for 4 or 8 h (data not shown). Because COX-1 is constitutively expressed to produce a basal level of prostanoids, we tested whether the prostanoids derived from COX-1 affect the induction of L-PGDS expression. BMDMs were prepared from cox-2−/− mice, pretreated with two different doses of a specific COX-1 inhibitor, SC-560, for 24 h, and then treated with PGD2 for 16 h before Western blot analysis of L-PGDS. As shown in Fig. 1C, the COX-1-specific inhibitor did not significantly affect the expression of L-PGDS induced by PGD2. Together, these results indicate that PGD2 induces L-PGDS expression without involvement of other prostanoids.
15-Deoxy-Δ12,14-PGJ2 induces L-PGDS expression independent of PPAR-γ
Because PGD2 is bound to generate 15-deoxy-Δ12,14-PGJ2, we examined whether 15-deoxy-Δ12,14-PGJ2 is also capable of inducing L-PGDS expression. Given that a high concentration of 15-deoxy-Δ12,14-PGJ2 is required to execute its effect [34], we treated BMDMs with 10 μM 15-deoxy- Δ12,14-PGJ2. At 16 h after treatment, total proteins were extracted from the treated cells and analyzed by Western blotting for L-PGDS. As shown in Fig. 2A, 15-deoxy-Δ12,14-PGJ2 induced L-PGDS expression, albeit less than PGD2.
Fig. 2.

15-Deoxy-Δ12,14-PGJ2 induces L-PGDS expression independent of PPAR-γ. (A) One million BMDMs were treated with 15-deoxy-Δ12,14-PGJ2 (10 μM), LPS (0.1 μg/ml), PGD2 (0.1 μM), or PGE2 (10 μM) for 16 h. Total cell lysate was quantitated and analyzed by Western blotting for L-PGDS (top) and internal controls (bottom). (B) One million RAW 264.7 cells were treated with the indicated amounts of rosiglitazone (lanes 4 and 5) or GW 9662 along with 15-deoxy-Δ12,14-PGJ2 (10 μM) (lanes 6 and7). Western blot was performed for L-PGDS (top) and internal controls (bottom). All the experiments were repeated at least three times.
Because 15-deoxy-Δ12,14-PGJ2 is known as a ligand for PPAR-γ [12,13], we examined whether L-PGDS expression induced by 15-deoxy-Δ12,14-PGJ2 is mediated by PPAR-γ. We hypothesized that if PPAR-γ were involved in inducing L-PGDS, pharmacologic ligands for PPAR-γ should induce the expression of L-PGDS; conversely, inhibitors against PPAR-γ should suppress the expression of L-PGDS induced by 15-deoxy-Δ12,14-PGJ2. RAW 264.7 cells were treated with an agonist, rosiglitazone, for 16 h, and total cell lysate was prepared and analyzed by Western blotting for L-PGDS. As shown in Fig. 2B, no L-PGDS expression was detected (lanes 4 and 5). When RAW 264.7 cells were treated with an antagonist, GW 9662, for 2 h and subsequently with 15-deoxy-Δ12,14-PGJ2 for 16 h, expression of L-PGDS induced by 15-deoxy-Δ12,14-PGJ2 was not blocked (lanes 6 and 7). Taken together, these results suggest that 15-deoxy-Δ12,14-PGJ2 also induces the expression of L-PGDS, without involvement of PPAR-γ.
Induction of L-PGDS expression by PGD2 is distinct from that by LPS
In previous studies, we demonstrated that LPS induces the expression of L-PGDS via MAPKs, such as JNK and p38 [20,29]. Thus, we examined whether PGD2 induces L-PGDS expression in a similar fashion. RAW 264.7 cells, pretreated with inhibitors of JNK, p38, and ERK1/2 for 2 h, were treated with LPS (1 μg/ml) or PGD2 (1 μM). At 16 h after treatment, the expression of L-PGDS was analyzed by Western blot. As shown in Fig. 3A, unlike the LPS-induced L-PGDS expression, which was inhibited by JNK or p38 inhibitors (lanes 3 and 4), those inhibitors did not affect L-PGDS expression induced by PGD2 (lanes 7 and 8).
Fig. 3.
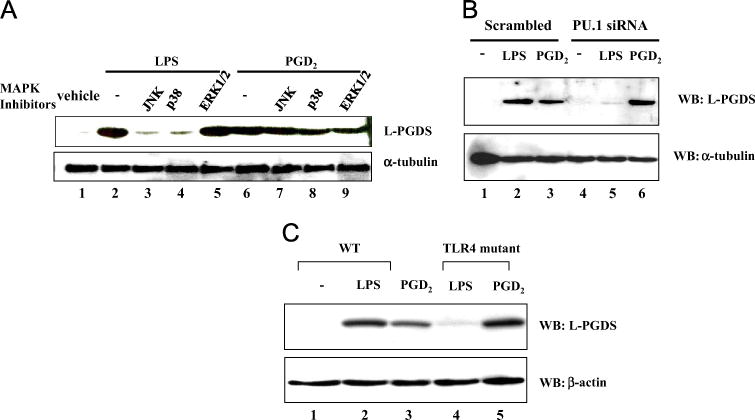
Induction of L-PGDS by PGD2 is distinct from that by LPS. (A) One million RAW 264.7 cells were treated with a final 10 μM MAPK inhibitors against JNK, p38, or ERK1/2 for 2 h and subsequently with LPS (lanes 2 to 5) or PGD2 (lanes 6 to 9). At 16 h after treatment, L-PGDS expression was determined by Western blotting for L-PGDS (top). The membrane was stripped and blotted again for α-tubulin to ensure equal loading. (B) RAW 264.7 cells were transfected with scrambled (lanes 1 to 3) or PU.1 siRNA (lanes 4 to 6) before LPS or PGD2 treatment. Western blot was performed for L-PGDS (top) and internal controls (bottom). (C) BMDMs were prepared from wild-type littermate (WT) or TLR4-defective mice. One million BMDMs were treated with LPS or PGD2. Western blot was performed for L-PGDS (top) and internal controls (bottom). All the experiments were repeated at least three times.
Because PU.1 is also involved in the induction of L-PGDS expression in LPS-treated macrophages [29], we examined whether PGD2 induces L-PGDS expression via PU.1. RAW 264.7 cells were transfected with either scrambled or PU.1-specific siRNA. The transfected cells were treated with LPS (0.1 μg/ml) or PGD2 (0.1 μM) for 16 h and then analyzed by Western blotting for L-PGDS. As shown in Fig. 3B, whereas LPS treatment failed to induce L-PGDS expression when PU.1 was silenced (lanes 2 and 5), PGD2 successfully induced it (lanes 3 and 6), suggesting that PU.1 is not involved in L-PGDS expression induced by PGD2. Furthermore, whereas functional TLR4 was necessary for LPS to induce L-PGDS expression (lanes 2 and 4, Fig. 3C), PGD2 robustly induced it without functional TLR4 (lanes 3 and 5). Together, these results demonstrate that induction of L-PGDS by PGD2 uses a pathway distinct from that of LPS.
Nrf2 binds to a putative site in the promoter of murine L-PGDS upon PGD2 treatment
To find the mechanism by which PGD2 induced L-PGDS expression, we analyzed the promoter of murine L-PGDS by using the TFSEARCH program (version 1.3, Tokyo University, Tokyo, Japan). Sequence analysis located a putative Nrf2 binding site, which was positioned in reverse from −868 to −878 nt upstream of the transcription initiation site of the gene (Fig. 4A). Given that 15-deoxy-Δ12,14-PGJ2, a by-product of PGD2, activates Nrf2 [14], we tested the possibility that PGD2 induces the binding of Nrf2 to the putative binding site on the promoter. To this end, we performed a ChIP assay. RAW 264.7 cells were treated with 1 μM PGD2 for various periods. DNA bound to Nrf2 was precipitated with anti-Nrf2 antibody and amplified by PCR with a set of specific primers flanking the putative Nrf2 binding site in the promoter. As shown in Fig. 4B, at 12 h after PGD2 treatment, Nrf2 bound to the putative site on the promoter of L-PGDS. In a similar experiment with isotypic IgG, no Nrf2 binding was detected. These results suggest that Nrf2, activated by PGD2, binds to the putative cognate site in the promoter of L-PGDS.
Fig. 4.

PGD2 induces the binding of Nrf2 to a putative cognate site in the promoter of murine L-PGDS. (A) A putative Nrf2 binding site in the murine L-PGDS promoter is shown in the box. The sequences used for ChIP assay are underlined. (B) One million RAW 264.7 cells were treated with PGD2 (1 μM) for the indicated periods. DNA fragments bound to Nrf2 were precipitated with anti-Nrf2 antibody and amplified by PCR. A similar experiment was performed with isotypic IgG to exclude nonspecific precipitation.
Chemical activation, or overexpression, of Nrf2 was sufficient to induce L-PGDS expression
Next, we tested whether activation of Nrf2 is sufficient for the induction of L-PGDS expression. First, RAW 264.7 cells were treated with sulforaphane (5 μM), a well-documented activator of Nrf2. At 16 h after treatment, total cell lysate was prepared and analyzed by Western blotting for L-PGDS. As shown in Fig. 5A, sulforaphane treatment was sufficient to induce the expression of L-PGDS (lane 4). Western blot analysis of nuclear proteins shows that induction of L-PGDS was correlated with nuclear localization of Nrf2, indicative of activated Nrf2 (Fig. 5B). To confirm that Nrf2 sufficed for the induction of L-PGDS expression, we examined whether overexpression of Nrf2 results in L-PGDS expression. RAW 264.7 cells were transfected with a plasmid encoding Nrf2. At 48 after transfection, total cell lysate was prepared and analyzed by Western blotting for L-PGDS. As shown in Fig. 5C, forced expression of Nrf2 was sufficient to support L-PGDS expression, although the level of L-PGDS was low because of low transfection efficiency (approximately 5%). Nevertheless, these results indicate that Nrf2 activation was sufficient for the induction of L-PGDS expression.
Fig. 5.

Activation of Nrf2 is sufficient to induce L-PGDS expression. (A) One million BMDMs were treated with LPS (0.1 μg/ml), PGD2 (0.1 μM), or sulforaphane (1 μM), an activator of Nrf2, for 16 h. Total cell lysate was prepared and analyzed by Western blotting for L-PGDS (top) and for β-actin as an internal control (bottom). (B) Cells were treated as for (A), and nuclear proteins were isolated and analyzed by Western blotting for nuclear-localized Nrf2, indicative of activated Nrf2 (top), and lamin A/C as an internal nuclear protein control (bottom). (C) RAW 264.7 cells were transfected with a host vector (lane 1) or an Nrf2-expressing plasmid (lane 2). At 48 h after transfection, total cell lysates of the transfected cells were quantitated, and 50 μg of the cell lysate was analyzed by Western blotting for L-PGDS (top) and β-actin as an internal control (bottom).
Nrf2 is essential for the expression of L-PGDS induced by PGD2
To determine the impact of Nrf2 on the expression of L-PGDS, we prepared BMDMs from nrf2−/− mice and treated them with LPS or PGD2. At 16 h after treatment, Western blot was performed to measure the expression of L-PGDS. As shown in Fig. 6A, PGD2 treatment failed to induce the expression of L-PGDS in the absence of Nrf2, as opposed to LPS treatment, which induced L-PGDS expression despite the absence of Nrf2. We further tested whether this is also the case in mouse lungs. Wild-type (WT) littermate or Nrf2-KO (nrf2−/−) mice (n = 5 per group) received an intratracheal (i.t.) spray of 0.3 μg/kg body wt PGD2 or vehicle (dimethyl sulfoxide (DMSO)). At 16 h after treatment, mouse lungs were harvested, from which total RNA was extracted and analyzed by semiquantitative RT-PCR for the expression of L-PGDS. Consistent with the result shown in Fig. 6A, PGD2 treatment induced L-PGDS expression in the lungs of WT, but not Nrf2-KO, mice (Fig. 6B). These results demonstrate that Nrf2 is an essential factor for PGD2-mediated expression of L-PGDS.
Fig. 6.
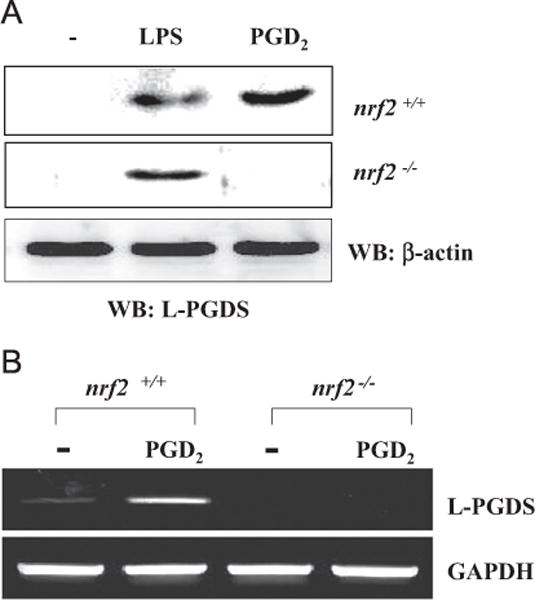
Nrf2 is essential for PGD2-mediated L-PGDS expression. (A) BMDMs of wild-type littermate (top) and nrf2−/− mice (bottom) were treated with LPS (0.1 μg/ml) or PGD2 (0.1 μM) for 16 h. Total cell lysates of variously treated cells were quantitated, and 50 μg of cell lysate was analyzed by Western blotting for L-PGDS. (B) Wild-type littermate and nrf2−/− mice (n = 5 per group) received an intratracheal injection of vehicle (DMSO) or PGD2 (0.03 μg/kg body wt). At 16 h after the injection, the lungs of the mice were harvested, and total RNA was analyzed by semiquantitative RT-PCR for L-PGDS and GAPDH.
Reintroduction of Nrf2 induces L-PGDS expression and reduces lung inflammation in Nrf2-KO mice
Given that Nrf2 suppresses lung inflammation [26–28], we tested whether Nrf2 performs this function by inducing the expression of L-PGDS in the lung. First, we tested whether reintroduction of nrf2 in Nrf2-KO mice results in induction of L-PGDS expression in the lung. Nrf2-KO mice (n=5 per group) received in an aerosol either a host vector (pcDNA3.1) or a plasmid expressing Nrf2 (1.5 mg/kg body wt) via the trachea. At 48 h after DNA treatment, mouse lungs were harvested, and total RNA was extracted for semiquantitative RT-PCR analysis of L-PGDS. As shown in Fig. 7A, expression of Nrf2 in the lungs of Nrf2-KO mice was sufficient to induce the expression of L-PGDS, along with that of Nrf2-dependent genes, such as NQO1, GCLC, and HO-1.
Fig. 7.

Nrf2 induces L-PGDS expression and suppresses neutrophilic inflammation in the lung. (A) Nrf2-KO (nrf2−/−) mice (n = 5 per group) received an i.t. spray of a host vector or an Nrf2-expressing vector, which was mixed with EC10 liposomes (see Material and methods). Total RNA was extracted from the lungs, and the expression of L-PGDS and Nrf2-dependent genes in the lungs of the treated mice was determined by semiquantitative RT-PCR. Shown are representatives of five mouse lungs per group. (B) Differential cell counting was performed to score inflammatory cell infiltrates in the lung. Nrf2-KO mice received an i.t. spray of a host vector (first and second columns) or an Nrf2-expressing plasmid (third and fourth columns). At 48 h after DNA treatment, the mice received intranasal LPS (10 mg/kg body wt), along with PGD2 (3 μg/kg body wt) (second and fourth columns). BAL was performed, from which total cells and neutrophils in the lung were counted. Data are represented as the mean 7 SEM (n = 5; *P < 0.05 compared with the LPS-treated; **P < 0.05 compared with other groups).
Next, we examined whether restoring Nrf2 in the lungs of Nrf2-KO mice results in reduced lung inflammation. As in Fig. 7A, Nrf2-KO mice received either the host vector or the Nrf2-expressing plasmid in aerosol. At 48 h after DNA treatment, Nrf2-KO mice (n = 5 per group) were treated with intranasal LPS (10 mg/kg body wt) for the induction of lung inflammation 2 h before an i.t. spray of PGD2 (0.3 μg/kg body wt) or vehicle. At 24 h after LPS treatment, BAL was performed, and lung infiltrates were scored by differential cell counting. As shown in Fig. 7B, LPS induced neutrophil infiltration into the lungs, which was slightly but significantly suppressed by subsequent PGD2 treatment (first and second closed columns), suggesting that PGD2 is able to suppress lung inflammation to a degree without Nrf2. On the other hand, single delivery of Nrf2 to the lungs of the Nrf2-KO mice was sufficient to reduce neutrophil infiltration (third closed column); this suppressive effect was significantly enhanced by PGD2 (fourth closed column). Together, these results suggest that Nrf2 is sufficient to induce L-PGDS in the lung and contributes to the decrease in neutrophilic lung inflammation.
L-PGDS expression is sufficient for reducing lung inflammation in mice
Finally, given our results showing that Nrf2 induced L-PGDS expression and suppressed inflammation in the lungs, we tested whether L-PGDS is also involved in suppressing lung inflammation. To do this, we performed experiments similar to those described for Fig. 7. C57BL/6 mice (n = 5 per group) received an i.t. spray of 1.5 mg/kg body wt of a host vector (pcDNA3.1) or a plasmid expressing L-PGDS. At 48 h after treatment, the mice received intranasal LPS (2 mg/kg body wt) for the induction of lung inflammation. At 24 h after LPS treatment, bronchoalveolar lavage was performed, and the lung sections of the mice were analyzed to determine the effect of L-PGDS on lung inflammation. As shown in Fig. 8A, histologic analysis revealed that the expression of L-PGDS in the lung significantly reduced lung inflammation. Concordantly, expression of L-PGDS significantly reduced cellular (Fig. 8B) and neutrophil infiltration into the lung (Fig. 8C). These results show that L-PGDS contributed to the suppression of neutrophilic lung inflammation. Together, these results show that Nrf2 induced L-PGDS expression, which was associated with suppression of lung inflammation. Our findings suggest that Nrf2 uses L-PGDS as part of a mechanism for suppression of lung inflammation.
Fig. 8.

The expression of L-PGDS ameliorates lung inflammation. (A) C57BL/6 mice (n = 5 per group) received an i.t. spray of a host vector (images a and b) or a plasmid encoding murine L-PGDS (image c). At 48 h after DNA spraying, the mice received intranasal delivery of PBS (image a) or LPS (images b and c). At 24 h after LPS treatment, the lungs of the mice were perfused and analyzed by histological examination (original magnification 200×). Shown are representatives of at least three different areas of H&E-stained lung sections. BAL was performed before lung harvest, and (B) total cells and (C) neutrophils in the lung were counted. Data represent the mean 7 SEM of five mice (*P < 0.05, compared to the LPS-treated).
Discussion
In this study, we sought to understand how Nrf2 exerts its anti-inflammatory function, as Nrf2 is known to protect from various inflammatory diseases in animal models. We found that Nrf2 used L-PGDS as part of an anti-inflammatory mechanism. When treated with PGD2, Nrf2 was activated, which resulted in L-PGDS expression. Whereas genetic ablation of nrf2 abolished the induction of L-PGDS expression, expression of Nrf2 in the lung of Nrf2-KO mice restored the expression of L-PGDS, suggesting that Nrf2 was essential for the induction of L-PGDS expression. Expression of Nrf2 in Nrf2-KO mice reduced neutrophilic lung inflammation elicited by LPS. Similarly, expression of L-PGDS alone was sufficient to ameliorate lung inflammation in mice. Therefore, our results show that PGD2 activated Nrf2, resulting in L-PGDS expression, which was associated with suppression of lung inflammation. Our findings suggest the possibility that Nrf2 wields L-PGDS expression as a part of the mechanism to suppress lung inflammation.
We designed a study around our hypothesis that PGD2 is involved in the downregulation of its own production via a feedback regulatory mechanism because the levels of PGD2 culminate and dwindle during the inflammatory response. If this were the case, one could expect that PGD2 treatment would lower the expression of L-PGDS and/or H-PGDS. However, our results showed otherwise: PGD2 induced L-PGDS expression without affecting the constitutive expression of H-PGDS. Because L-PGDS expression probably results in increased production of PGD2 [23], our results suggest that PGD2 promotes its own production by inducing L-PGDS expression, contributing to maximizing the level of PGD2 and thereby the anti-inflammatory effect of PGD2. In theory, this positive feedback regulation of L-PGDS expression by PGD2 could perpetuate the production of PGD2. However, this is unlikely because the level of PGD2 should be dependent on the availability of its precursor, PGH2, and of COX-1 and COX-2 that generate PGH2 [35]. Although there is a report showing that prostanoids, such as PGE2, 15-deoxy-Δ12,14-PGJ2, and 6-keto-PGF2, induce COX-2 expression [36], our results show that exogenously added PGD2 did not induce COX-2 expression (Fig. 1B). In addition, induction of L-PGDS expression by PGD2 was not affected by other prostanoids that COX-1 and COX-2 produce (Fig. 1C). Therefore, it is conceivable that L-PGDS, along with H-PGDS, converts available PGH2 to PGD2 rapidly in the inflammatory milieu, which in turn induces the expression of L-PGDS, maximizing the level of PGD2 contributing to suppression of inflammation. During inflammation, this positive feedback induction of L-PGDS expression depletes PGH2, resulting in a decrease in PGD2.
It is well documented that PGD2 is bound to yield 15-deoxy-Δ12,14-PGJ2 via nonenzymatic processes [34,37]. Our results indicate that not only PGD2 but also 15-deoxy-Δ12,14-PGJ2 induced L-PGDS expression (Fig. 3A). Although 15-deoxy-Δ12,14-PGJ2 is known as a ligand of PPAR-γ [12,13], our pharmacologic experiments, at least, excluded the possibility that PPAR-γ activated by15-deoxy-Δ12,14-PGJ2 induces L-PGDS expression (Fig. 3B). Because 15-deoxy-Δ12,14-PGJ2 activates Nrf2 [14], it is possible that activation of Nrf2 by PGD2 is mainly due to 15-deoxy-Δ12,14-PGJ2 generated from PGD2. Although it is unclear, at this moment, whether PGD2 alone is sufficient to induce L-PGDS expression without involvement of 15-deoxy-Δ12,14-PGJ2, our results showing that the degree of L-PGDS expression by PGD2 was more robust than that by 15-deoxy-Δ12,14-PGJ2 (Fig. 3A) suggest the possibility that PGD2 can activate Nrf2 independent of 15-deoxy-Δ12,14-PGJ2. The effect of PGD2 vs 15-deoxy-Δ12,14-PGJ2 on L-PGDS expression is under investigation in our laboratory.
In previous studies, we reported that LPS induces L-PGDS expression in macrophages, in which JNK and p38 MAPK and PU.1 play key roles [20,29]. In this study, we found that PGD2 was also a potent inducer of L-PGDS expression. However, unlike the LPS-mediated expression, neither MAPKs nor PU.1 was involved in the PGD2-induced L-PGDS expression. Rather, our results show that Nrf2 was critical because genetic ablation of nrf2 completely blocked the induction of L-PGDS expression by PGD2 but not by LPS (Fig. 6). These results strongly suggest that there are at least two distinctive pathways to induce L-PGDS expression in inflammation. It is conceivable that invading bacteria or LPS triggered induction of COX-2 expression and thereby abundant production of PGH2. Constitutively expressed H-PGDS plays an early role in converting PGH2 to PGD2 [9]. Meanwhile, LPS activated MAPK and PU.1 to induce L-PGDS expression, which contributes to the conversion of PGH2 to PGD2. Accumulating PGD2, in turn, activated Nrf2, resulting in L-PGDS expression. Together with H-PGDS, L-PGDS, which was now maximally produced through TLR4- and Nrf2-mediated pathways, would contribute to maximizing the level of PGD2. An inflammatory response often accompanies the production of ROS [38]. Because LPS induces the production of ROS, a potent activator of Nrf2 [39], ROS could contribute to the production of L-PGDS and PGD2 by activating Nrf2. Nevertheless, our results suggest the possibility that TLR4- and Nrf2-dependent pathways work together to accentuate the expression of L-PGDS and thereby the production of PGD2 in the inflammatory milieu.
Genetic ablation of nrf2 shows that Nrf2 was the essential factor for PGD2 to induce L-PGDS expression, positioning Nrf2 in the positive feedback loop of L-PGDS and PGD2. Because Nrf2 is involved in suppressing inflammation and PGD2 regulates inflammation, the functional association between Nrf2 and PGD2 shown in this study suggests that PGD2 executes an anti-inflammatory activity via Nrf2. Conversely, Nrf2 executes its anti-inflammatory activity via PGD2. In addition to this, our results show that PGD2 activated Nrf2 to induce L-PGDS expression and that L-PGDS alone might have an anti-inflammatory activity. These results suggest that Nrf2 or PGD2 exerts its anti-inflammatory activity via L-PGDS expression and that Nrf2 uses L-PGDS as an effector molecule for its anti-inflammatory function. Therefore, it is conceivable that Nrf2 executes its anti-inflammatory function by galvanizing the positive feedback loop of L-PGDS expression and PGD2 production. It is unknown how L-PGDS contributes to suppression of inflammation. L-PGDS expression may simply contribute to the increase in PGD2 by converting PGH2 that is likely to be abundant in the inflammatory milieu. Given that L-PGDS is a secretory protein with lipophilic, small-molecule binding properties [40], it is also possible that this nonenzymatic property of L-PGDS is associated with the immunosuppressive effect. Regardless of the underlying mechanism, our results suggest that L-PGDS expression contributes to suppression of inflammation.
Conclusions
In summary, we report that when activated by PGD2, Nrf2 induced L-PGDS expression, which was associated with decreased neutrophilic lung inflammation. In line with this, L-PGDS expression alone was sufficient to suppress neutrophilic lung infiltration in mice. Given the well-documented anti-inflammatory function of Nrf2, we propose that this novel, positive feedback loop of PGD2 and L-PGDS expression is a means for Nrf2 to wield its anti-inflammatory activity.
Acknowledgments
We thank Mr. Minjae Kwon at Vanderbilt University School of Medicine for his technical help. This study was support in part by the NRF of Korea (20110018985 to M.J.).
References
- 1.Brown GP, Monick MM, Hunninghake GW. Human alveolar macrophage arachidonic acid metabolism. Am J Physiol. 1988;254:C809–815. doi: 10.1152/ajpcell.1988.254.6.C809. [DOI] [PubMed] [Google Scholar]
- 2.Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ., 2nd Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982;129:1627–1631. [PubMed] [Google Scholar]
- 3.Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, Sugimoto Y, Kobayashi T, Ushikubi F, Aze Y, Eguchi N, Urade Y, Yoshida N, Kimura K, Mizoguchi A, Honda Y, Nagai H, Narumiya S. Prostaglandin D2 as a mediator of allergic asthma 1. Science. 2000;287:2013–2017. doi: 10.1126/science.287.5460.2013. [DOI] [PubMed] [Google Scholar]
- 4.Fujitani Y, Kanaoka Y, Aritake K, Uodome N, Okazaki-Hatake K, Urade Y. Pronounced eosinophilic lung inflammation and Th2 cytokine release in human lipocalin-type prostaglandin D synthase transgenic mice. J Immunol. 2002;168:443–449. doi: 10.4049/jimmunol.168.1.443. [DOI] [PubMed] [Google Scholar]
- 5.Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, Trottein F, Dombrowicz D. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174:3703–3708. doi: 10.4049/jimmunol.174.6.3703. [DOI] [PubMed] [Google Scholar]
- 6.Ando M, Murakami Y, Kojima F, Endo H, Kitasato H, Hashimoto A, Kobayashi H, Majima M, Inoue M, Kondo H, Kawai S, Hayashi I. Retrovirally introduced prostaglandin D2 synthase suppresses lung injury induced by bleomycin 8. Am J Respir Cell Mol Biol. 2003;28:582–591. doi: 10.1165/rcmb.2002-0162OC. [DOI] [PubMed] [Google Scholar]
- 7.Ianaro A, Ialenti A, Maffia P, Pisano B, Di Rosa M. Role of cyclopentenone prostaglandins in rat carrageenan pleurisy. FEBS Lett. 2001;508:61–66. doi: 10.1016/s0014-5793(01)03035-6. [DOI] [PubMed] [Google Scholar]
- 8.Murakami Y, Akahoshi T, Hayashi I, Endo H, Hashimoto A, Kono S, Kondo H, Kawai S, Inoue M, Kitasato H. Inhibition of monosodium urate monohydrate crystal-induced acute inflammation by retrovirally transfected prostaglandin D synthase. Arthritis Rheum. 2003;48:2931–2941. doi: 10.1002/art.11271. [DOI] [PubMed] [Google Scholar]
- 9.Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW. Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci USA. 2007;104:20979–20984. doi: 10.1073/pnas.0707394104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12,14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma 1. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- 13.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation 1. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- 14.Mochizuki M, Ishii Y, Itoh K, Iizuka T, Morishima Y, Kimura T, Kiwamoto T, Matsuno Y, Hegab AE, Nomura A, Sakamoto T, Uchida K, Yamamoto M, Sekizawa K. Role of 15-deoxy delta(12,14) prostaglandin J2 and Nrf2 pathways in protection against acute lung injury. Am J Respir Crit Care Med. 2005;171:1260–1266. doi: 10.1164/rccm.200406-755OC. [DOI] [PubMed] [Google Scholar]
- 15.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signalling. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 16.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol. 2003;3:169–176. doi: 10.1038/nri1004. [DOI] [PubMed] [Google Scholar]
- 17.FitzGerald GA. COX-2 and beyond: approaches to prostaglandin inhibition in human disease 1. Nat Rev Drug Discovery. 2003;2:879–890. doi: 10.1038/nrd1225. [DOI] [PubMed] [Google Scholar]
- 18.Urade Y, Eguchi N. Lipocalin-type and hematopoietic prostaglandin D synthases as a novel example of functional convergence. Prostaglandins Other Lipid Mediators. 2002;68–69:375–382. doi: 10.1016/s0090-6980(02)00042-4. [DOI] [PubMed] [Google Scholar]
- 19.Joo M, Sadikot RT. PGD synthase and PGD2 in immune response. Mediators Inflammation. 2012;2012:503128. doi: 10.1155/2012/503128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joo M, Kwon M, Sadikot RT, Kingsley PJ, Marnett LJ, Blackwell TS, Peebles RS, Jr, Urade Y, Christman JW. Induction and function of lipocalin prostaglandin D synthase in host immunity. J Immunol. 2007;179:2565–2575. doi: 10.4049/jimmunol.179.4.2565. [DOI] [PubMed] [Google Scholar]
- 21.Cipollone F, Fazia M, Iezzi A, Ciabattoni G, Pini B, Cuccurullo C, Ucchino S, Spigonardo F, De Luca M, Prontera C, Chiarelli F, Cuccurullo F, Mezzetti A. Balance between PGD synthase and PGE synthase is a major determinant of atherosclerotic plaque instability in humans. Arterioscler Thromb Vasc Biol. 2004;24:1259–1265. doi: 10.1161/01.ATV.0000133192.39901.be. [DOI] [PubMed] [Google Scholar]
- 22.Cathcart MK. Regulation of superoxide anion production by NADPH oxidase in monocytes/macrophages: contributions to atherosclerosis 3. Arterioscler Thromb Vasc Biol. 2004;24:23–28. doi: 10.1161/01.ATV.0000097769.47306.12. [DOI] [PubMed] [Google Scholar]
- 23.Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 24.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 25.Jaiswal AK. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med. 2004;36:1199–1207. doi: 10.1016/j.freeradbiomed.2004.02.074. [DOI] [PubMed] [Google Scholar]
- 26.Chan K, Kan YW. Nrf2 is essential for protection against acute pulmonary injury in mice. Proc Natl Acad Sci USA. 1999;96:12731–12736. doi: 10.1073/pnas.96.22.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joo M, Kwon M, Cho YJ, Hu N, Pedchenko TV, Sadikot RT, Blackwell TS, Christman JW. Lipopolysaccharide-dependent interaction between PU.1 and c-Jun determines production of lipocalin-type prostaglandin D synthase and prostaglandin D2 in macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L771–779. doi: 10.1152/ajplung.90320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shim G, Choi HW, Lee S, Choi J, Yu YH, Park DE, Choi Y, Kim CW, Oh YK. Enhanced intrapulmonary delivery of anticancer siRNA for lung cancer therapy using cationic ethylphosphocholine-based nanolipoplexes. Mol Ther. 2013;21:816–824. doi: 10.1038/mt.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sekhar KR, Yan XX, Freeman ML. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene. 2002;21:6829–6834. doi: 10.1038/sj.onc.1205905. [DOI] [PubMed] [Google Scholar]
- 32.Joo M, Park GY, Wright JG, Blackwell TS, Atchison ML, Christman JW. Transcriptional regulation of the cyclooxygenase-2 gene in macrophages by PU.1 3. J Biol Chem. 2004;279:6658–6665. doi: 10.1074/jbc.M306267200. [DOI] [PubMed] [Google Scholar]
- 33.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, Serhan CN. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. 2005;174:4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 34.Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114:100–109. doi: 10.1016/j.clim.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- 36.Vichai V, Suyarnsesthakorn C, Pittayakhajonwut D, Sriklung K, Kirtikara K. Positive feedback regulation of COX-2 expression by prostaglandin metabolites. Inflammation Res. 2005;54:163–172. doi: 10.1007/s00011-004-1338-1. [DOI] [PubMed] [Google Scholar]
- 37.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swindle EJ, Hunt JA, Coleman JW. A comparison of reactive oxygen species generation by rat peritoneal macrophages and mast cells using the highly sensitive real-time chemiluminescent probe pholasin: inhibition of antigen-induced mast cell degranulation by macrophage-derived hydrogen peroxide. J Immunol. 2002;169:5866–5873. doi: 10.4049/jimmunol.169.10.5866. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2–Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signaling. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- 40.Urade Y, Hayaishi O. Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase 2. Biochim Biophys Acta. 2000;1482:259–271. doi: 10.1016/s0167-4838(00)00161-8. [DOI] [PubMed] [Google Scholar]