

Abstract
BACKGROUND AND PURPOSE
Recovery from stroke requires neuroplasticity within surviving adjacent cortex. The axon initial segment (AIS) is the site of action potential initiation and a focal point for tuning of neuronal excitability. Remodeling of the AIS may be important to neuroplasticity after stroke.
METHODS
Focal cortical stroke in forelimb motor cortex was induced by photothrombosis and compared to sham controls. White matter stroke was produced through stereotactic injection of a vasoconstrictor together with biotinylated dextran amine to retrogradely label injured cortical neurons. AIS length, morphology and number were measured using immunofluorescence and confocal microscopy two weeks after stroke.
RESULTS
Within the peri-infarct cortex and after white matter stroke, AIS length decreases. This shortening is accompanied by altered AIS morphology. In peri-infarct cortex, the decrease in AIS length after stroke occurs from the distal end of the AIS resulting in a loss of NaV1.6 channels. GABAR-α2 subunit staining at axoaxonic synapses along the AIS is significantly decreased. In addition, a significant increase in small, immature initial segments is present in layers 2/3 of peri-infarct cortex, reflecting maturation of axonal sprouting and new initial segments from surviving neurons.
CONCLUSIONS
Stroke alters the compartmental morphology of surviving adjacent neurons in peri-infarct cortex and in neurons whose distal axons are injured by white matter stroke. With a key role in modulation of neuronal excitability, these changes at the AIS may contribute to altered neuronal excitability after injury and prove crucial to increasing neuroplasticity in surviving tissue affected by stroke.
Indexing Terms: axon initial segment, stroke, white matter, plasticity
Introduction
Stroke is a devastating neurologic illness with limited functional recovery. The adjacent surviving cortex plays a pivotal role in the limited neuroplasticity and recovery seen after stroke. Regulation of neuronal excitability is an important feature of recovery after stroke. Tonic GABA inhibition is increased in peri-infarct cortex, increasing overall basal inhibition and lowering neuronal excitability 1. Pharmacologic blockade of tonic GABA signaling improves functional recovery after stroke 1. Stimulation of glutamate signaling through the AMPA receptor promotes neuronal excitability in peri-infarct cortex, stimulating local BDNF production and enhancing recovery 2. Thus, stroke alters peri-infarct neuronal excitability and these alterations play a role in the recovery of function.
The axon initial segment (AIS) regulates neuronal excitability by serving as a gatekeeper for action potential initiation. The AIS regulates the threshold for action potential initiation through voltage-gated sodium (NaV) channel clustering, neurotransmission, and axoaxonic synaptic connections 3. Recently, the molecular makeup of the AIS has been clarified and appears dependent on ankyrinG (ankG) 4–6. Beta-IV spectrin follows ankG to the initial segment and serves to anchor the NaV channel-ankG complex to the axonal cytoskeleton creating a clustered zone of high density depolarizing membrane ion channels 7, 8. Initiation and propagation of the action potential is dependent on depolarization of a defined length of axon referred to as the spike trigger zone (TZ) 9, 10, thus the length of NaV channel clustering at the distal AIS determines the trigger zone and participates in fine tuning of neuronal excitability.
As a gatekeeper of action potential initiation, the axon initial segment is uniquely positioned to play an important role in brain plasticity after injury 11. Disruption of the presynaptic input of the avian brainstem auditory system lengthens the AIS and lowers the threshold for excitation by increasing the distribution of NaV channel12, while direct optogenetic stimulation of hippocampal neurons in culture alters AIS position (relative to the soma) over time 13. Mutation of alpha-II spectrin, similar to a microdeletion observed in a cohort of patients with early-onset West syndrome, alters clustering of the NaV channel-ankG complex and increases the threshold required for action potential firing 14. In a rat model of temporal lobe epilepsy, pyramidal cells from the entorhinal cortex display increased action potential firing correlating with increases in NaV1.6 staining at the AIS 15. In the AbetaPP/PS1 transgenic mouse model of Alzheimer’s disease, GABAergic axoaxonic synapses present along the AIS are lost from cells in proximity to amyloid plaques, suggesting an explanation for the altered neuronal excitability of cells adjacent to plaques 16. Thus, alterations in AIS molecular organization are relevant to a variety of neurologic injuries.
Here, we show that molecular remodeling of the AIS occurs after stroke within surviving peri-infarct cortex in focal cortical stroke and in cortical neurons injured by white matter stroke. We also demonstrate new AIS formation within peri-infarct cortex suggesting that post-stroke axonal sprouting may result in new functional axons. Together these data suggest that plasticity in the length and morphology of the AIS may contribute to post-stroke changes in neuronal excitability and provide a potential morphologic neuronal target to enhance recovery from stroke.
Methods
Photothrombotic stroke model
Three separate cohorts (sham n=12, stroke = 15) of adult wild type C57/Bl6 male mice (Jackson Laboratory) 2–4 months of age were subjected to either a sham or photothrombotic stroke over the left forelimb motor cortex according to previously published methods 1. For studies of specific Layer 5 cortical neurons, male Thy-1-eYFP-H homozygous transgenic mice 17 at 2–4 months of age were used (sham n=4, stroke n=7). Animal maintenance and surgical procedures were consistent with UCLA Animal Research Council Guidelines. In all animal cohorts, tissue processing and analyses were performed 14 days after stroke.
White matter stroke model
Focal ischemic lesions with retrograde neuronal tracing were produced in five adult wild type C57/Bl6 male mice (Jackson Laboratory) at 2 months of age through stereotactic pneumatic injection of 150 nL of a 54 mg/mL solution of N5-(1-iminoethyl)-L-ornithine, dihydrochloride (L-Nio, Calbiochem) mixed 1:1 with a 20% solution of 10,000 M.W. biotinylated dextran amine (BDA) (Life Technologies, Inc.) (final concentration of L-Nio 27 mg/mL and 10% BDA), at each of three stereotactic coordinates using a previously described approach 18.
Tissue processing and immunofluorescence
Mice with focal cortical or white matter stroke were processed for tissue section immunofluorescent visualization of various AIS markers and GABAR-α2 subunit 19 (Supplemental Methods, please see http://stroke.ahajournals.org).
Quantitation of axon initial segment morphology
From each animal, three 40 μm cryostat sections from a 160 μm series were immunostained for ankG and beta-IV spectrin. Stitched 0.5 mm x 0.7 mm 100X confocal z-stacks corresponding to 7.5 μm optical sections were obtained from peri-infarct cortex or sham motor cortex (Nikon C2 laser scanning confocal microscope). The 200μm peri-lesional area adjacent to the stroke was cropped, rotated, and a 100 μm2 square grid overlayed. Five 100 μm2 areas were selected at random for initial segment measurement from each section and independently for each immunolabel. Axon initial segment length was measured in pixels (NeuronJ:Fiji). All linear structures within each measured field in which the beginning and end could be determined were measured, excluding nodes of Ranvier. This included a fraction of (short-appearing) initial segments not completely within the actual or optical section but this fraction did not differ between control and stroke.
Quantitation of NaV1.6 and ankG length was performed by measuring the pixel length staining for each immunolabel in 5–10 layer 5 cortical neurons in five, 100X fields/animal in motor (sham) and peri-infarct cortex as above.
In YFP-positive cells, AIS length was measured by tracing the labeled AIS of YFP-positive cells using the multipoint line feature of NIS Elements software (Nikon). The peri-infarct region of 200 μm lateral to the stroke edge was imaged from two sections from a 120 μm series. AIS length in 4–15 YFP-positive cells/animal was measured compared to a corresponding region of motor cortex from sham animals.
Axon initial segment number was quantified using the same 0.5 mm x 0.7 mm stitched confocal images used for length measurements. These images included the upper boundary of cortical layer 2 to the lower boundary of layer 6 and included the lateral edge of the stroke. The region of interest for AIS number was defined as the region between 100 and 200 μm lateral to the stroke edge. Within this ROI, all beta-IV spectrin-positive structures were counted that had a typical AIS appearance: more than 5 μm in length with a wide base and tapered end. AIS number was binned according to 100 μm squares.
For AIS length measurement after white matter stroke, three 50 μm sections from a 150μm series in each of five animals were imaged using a 0.5 mm x 0.5 mm stitched 100X confocal z-stack of the motor cortex overlying the stroke. BDA-positive cells were counted and their AIS length measured as above (NeuronJ:Fiji). Percent change in AIS length was calculated by measuring AIS length from unlabeled cells within the same region.
For length measurements in both focal cortical and white matter stroke, statistical significance was determined using Welch’s t-test assuming unequal variance with a p-value <0.05. For AIS number analysis, a Bonferroni correction was applied for multiple comparisons and established statistical significance at p<0.007 (Microsoft Excel).
Results
Peri-infarct changes in axon initial segment length
Two weeks after photothrombotic stroke in the motor cortex, mice were sacrificed and double-immunolabeled for ankyrinG and beta-IV spectrin, two key cytoskeletal scaffolds that position NaV channels to the AIS. In the peri-infarct cortex immediately lateral to the stroke (within 200 μm), axon initial segments showed altered morphology and length compared to sham (Figure 1A–B). Focal cortical stroke produces near complete cell death in the stroke core and results in the loss of all AIS labeling within the area of focal ischemia (Figure 1B), consistent with prior reports 11. In sham controls, axon initial segments labeled for ankG (left panel) or beta-IV spectrin (right panel) have a regular, linear morphology and project inferiorly (Figure 1C). Within peri-infarct cortex, axon initial segments with non-linear projection patterns were common (Figure 1D). The length of the initial segment is proposed as a key mechanism for the regulation of excitability 20. Therefore, we measured ankG, beta-IV spectrin, and NaV1.6-positive axon initial segment length within peri-infarct cortex compared to sham controls. Two weeks after stroke, ankG and beta-IV spectrin AIS length decreases by 14.0% (+/− 0.99%) and 15.4% (+/− 1.04%), respectively, corresponding to a ~3 μm absolute change in length (p<0.0001) (Table S1, please see http://stroke.ahajournals.org). Immunolabeling and quantification of the ankG to NaV1.6 length ratio shows that this ratio is unchanged from sham (76%) to stroke (77%; p=0.37, n.s.) (Figure S1). Thus, stroke reduces the length of the AIS by altering its distal end, thereby decreasing the overall number of NaV1.6 channels at this region.
Figure 1.

Axon initial segment morphology and length are altered in surviving neurons in peri-infarct cortex after photothrombotic stroke over the motor cortex. Schematic showing the region of interest and appearance of AIS labeling in motor cortex from sham controls (A). Two weeks after focal cortical stroke, AIS labeling is lost within the stroke (B) and the adjacent peri-lesional region is highlighted (B). Cortical neurons labeled for ankG (left) and beta-IV spectrin (right) in sham control (C) or peri-infarct regions (D) two weeks after stroke. Axon initial segment morphology is altered in peri-infarct cortex and overall length decreases by 14.0–15.4% on average. 100X digital images; scale bar in C, D = 20 μm.
The previous data show that the AIS shortens in peri-infarct cortex in cells distributed throughout all cortical layers. However, we reasoned that subcortical projecting layer 5 cortical neurons in peri-infarct sensorimotor cortex might play a disproportionate role in post-stroke plasticity and recovery given their role in motor function. Therefore, we utilized the Thy-1-eYFP-H transgenic mouse line to assess AIS length and morphology specifically in this cell population within peri-infarct cortex compared to sham control two weeks after stroke. This analysis demonstrates that YFP-positive layer 5 neurons show a similar alteration in AIS morphology as observed in other cortical layers (Figure 2). In peri-infarct cortex, the AIS of YFP-positive cells often showed a blunt distal end (arrows, Figure 2B–C) whereas in sham control, the AIS tapers off in the distal end (Figure 2A). Using beta-IV spectrin immunostaining (red), quantification of AIS length in this specific cell population demonstrates a similar 14.2% (3.71 μm) decrease in peri-infarct cortex compared to sham (p<0.00001; Table S1).
Figure 2.

Stroke alters the AIS in YFP-positive layer 5 cortical neurons. Beta-IV spectrin labeling of the initial segment in Thy1-eYFP-H transgenic mice in sham (A) versus stroke animals (B–D) two weeks after stroke. AIS length (brackets) in this specific cell population decreases by 14.1% (Table S1). The distal end of the AIS often shows a blunt appearance (arrows in B, C) compared to sham. Post-stroke, axonal branching from the AIS was noted with early expression of beta-IV spectrin in the new axon (arrow, D). 100X digital images; scale bar = 5 μm.
GABAR-α2 axoaxonic synpases in peri-infarct cortex
In cortical pyramidal neurons, chandelier cells form axoaxonic synaptic connections along the AIS. These synapses are exclusively GABAergic and specifically involve the alpha-2 subunit of the GABA receptor. These axoaxonic synapses function at the AIS to further control the fidelity of neural circuitry. Thus, to demonstrate that post-stroke plasticity at the AIS includes changes in its circuitry in addition to changes in its structure, we labeled GABAR-α2 positive subunits in both sham and peri-infarct tissue two weeks after stroke. Within peri-infarct cortex, GABAR-α2 positive synaptic boutons along the AIS are significantly decreased compared to sham control (Figure 3A–B). The absolute number of GABAR-α2 positive boutons per AIS in peri-infarct is decreased (sham = 9.13, stroke = 4.41, p<0.005). When controlled for ankG-positive AIS length, the average length of initial segment between GABAR-α2 positive boutons increases modestly but significantly in peri-infarct tissue (Figure 3C). Notably, there was substantial variability in the number of axoaxonic synapses in cortical neurons in both sham and stroke conditions. Thus, stroke causes a decrease in GABA innervation of the AIS by synaptic pruning of GABAergic synapses.
Figure 3.

GABAR-α2 subunit synaptic bouton labeling along the axon initial segment in sham and peri-infarct cortical neurons two weeks after stroke. Multiple GABAR-α2-positive boutons (arrows) are visible along the length of the ankG-positive initial segment in sham animals (A) while in peri-infarct cortex, most of these axoaxonic boutons are lost (B). On the left axis, the average length of the initial segment between GABAR-α2-positive boutons (■) is plotted for both sham and stroke, demonstrating a small but statistically significant increase, consistent with a loss of GABAR-α2 boutons in peri-infarct cortex. On the right axis, the mean number of boutons per measured AIS (X) is plotted (p<0.005).
Peri-infarct changes in axon initial segment number
While assessing AIS length and morphology differences in peri-infarct cortex, we observed an apparent increase in small, irregular appearing initial segments in upper cortical layers. Because of the randomized way AIS length was assessed in Figure 1, this increase is not responsible for the shorter AIS length observed throughout peri-infarct cortex, however it was intriguing as axonal sprouting is known to occur within peri-infarct cortex. Therefore, we examined AIS number according to relative cortical depth within lateral peri-infarct cortex two weeks after stroke (Figure 4A–B). The number of beta-IV spectrin-positive AIS increases significantly between 47–109% greater than sham animals at cortical depths consistent with layers 2/3 (Figure 4C). Small, irregular appearing initial segments (arrows, Figure 4B) were largely responsible for the increase in AIS number after stroke. Rarely, occasional neurons within peri-infarct cortex could be identified with supernumerary AIS (Figures 4D and S2), perhaps accounting for some of the increase in total AIS number. When present, these new initial segments typically arose from the most proximal portion of beta-IV spectrin labeling and were oriented laterally rather than the typical inferiorly projecting orientation for cortical motor neurons.
Figure 4.
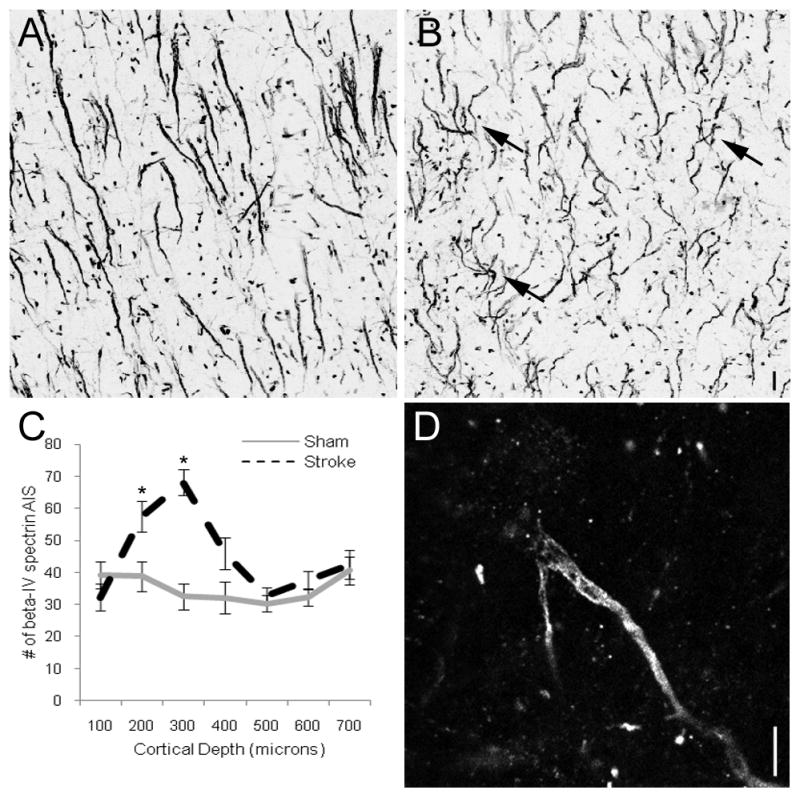
AIS number increases in upper cortical layers within peri-infarct cortex two weeks after stroke. In sham animals, beta-IV spectrin-positive initial segments show an even distribution throughout cortical depth (A). In peri-infarct cortex, approximately 100 μm from the lateral edge of the stroke, there is an increased density in AIS in upper cortical layers at depths consistent with layers 2/3 (B). The increased density results from multiple small, immature axon initial segments (arrows, B). AIS number plotted against relative cortical depth reveals a statistically significant increase in AIS number in peri-infarct cortex at depths consistent with layers 2/3 (C). Single 0.25 μm confocal optical slice showing a supernumerary AIS arising from the proximal portion of the AIS of a single cortical neuron (D). Scale bars = 5 μm.
White matter stroke alter AIS morphology and length
To further support that AIS remodeling is part of post-stroke plasticity, we utilized a mouse model of focal white matter stroke combined with retrograde labeling 18. This method produces precise localization of stroke in the white matter underlying forelimb motor cortex (Figure 5A). Combined with retrograde labeling, this technique allows visualization of individual cortical neurons that have undergone distal ischemic axotomy, as uninjured axons do not readily pick up the retrograde label (data not shown). As established in the previous study, only a minority (~10%) of neurons within overlying motor cortex project through the focal ischemic lesion and pick up the tracer (red, Figure 5B), thus allowing unlabeled cells in the adjacent cortex to serve as internal controls, as they lie adjacent to neurons that have injured axons. Co-immunolabeling for AIS markers allows identification and measurement of the AIS of individual cortical neurons injured by the stroke (Figure 5C). Two weeks after white matter stroke, BDA-positive cells (arrows, 5C) have both altered morphology and length of their AIS (green) when compared to adjacent, unlabeled cells (arrowheads, 5C) within the same region of motor cortex. Quantification of beta-IV spectrin-positive AIS length in labeled versus neighboring unlabeled cells, demonstrated a 33.0% decrease in AIS length two weeks after stroke (p<0.0001; Table S2). In labeled cells, decreases in AIS length were often accompanied by altered morphology, particularly a decrease in the caliber or width of the AIS both at its proximal base and distal end, as well as a relative decrease in the beta-IV spectrin (arrows, left panel 5C) and ankG (data not shown) immunoreactivity. Thus, white matter stroke produces remodeling of the AIS in neurons far from the stroke site. Figure 5D summarizes how cortical and white matter stroke alters the AIS in surviving neurons.
Figure 5.

Focal white matter stroke alters the AIS. White matter stroke in the YFP-H transgenic mouse produces an area of focal axonal loss (*) in the white matter underlying motor cortex (A). Neurons with axons projecting through and injured by stroke (*) are retrogradely labeled with BDA (red, B). High magnification of BDA-labeled cells (red) immunolabeled for beta-IV spectrin (green) shows decreased AIS length and altered morphology (arrows, C) compared to neighboring unlabeled cells with intact AIS (arrowheads) two weeks after stroke. Schematic showing the three major effects cortical and white matter stroke have on the AIS (red) (D). Scale bar in A and B = 250 μm, C = 5 μm.
Discussion
Recovery from stroke depends in large part on plasticity within peri-infarct cortex. Understanding the regulation of surviving cortical networks after injury is essential to designing neural repair therapies. Modulation of neuronal excitability in peri-infarct cortex can benefit recovery from stroke 1, 2. Post-stroke changes in peri-infarct dendritic arborization are known to occur and thereby alter the morphology of surviving neurons after stroke 21. The axon initial segment (AIS) provides a structural platform for the regulation of neuronal excitability through dense localization of NaV channels and integration of axoaxonic input. Here we examined AIS length and morphology in surviving neurons within peri-infarct cortex with structurally intact but stunned circuitry. Two weeks after stroke, cell death and peri-infarct inflammation are waning, while axonal sprouting, gene expression changes, and dendritic remodeling are robust 22–28, suggesting that post-stroke neuroplasticity is at a first peak. Within peri-infarct cortex, neurons are characterized by a decrease in the length of the AIS, occurring from the distal end of the initial segment, and lose axoaxonic synapses along the length of the AIS. These changes potentially alter the spike trigger zone and may negatively impact peri-infarct excitability. We also show that the absolute number of axon initial segments increases within peri-infarct cortex, perhaps reflecting maturation of post-stroke axonal sprouting into functional axons. Finally, we demonstrate that AIS remodeling occurs in a model of white matter stroke, thus supporting the concept that this axonal microdomain is sensitive to neuronal injury and essential for neuroplasticity after varied types of stroke.
Functioning as the integrative site of neuronal excitability, the AIS is a finite cellular compartment that plays a key role in neuronal plasticity. Several studies suggest the primary mechanism for AIS regulation of cellular excitability is through control of its length. For example, de-afferentation of the chick brainstem auditory nucleus resulted in an increased AIS length with a greater distribution of NaV channels and a lower threshold for excitation 12. In a mouse model of Angelman syndrome, the AIS lengthened in CA1 and CA3 areas by similar percentages as seen here 29. In this model, the action potential amplitude and rate of rise of the action potential increased while the threshold potential was lower, correlating with increased expression of AIS proteins, ankG and NaV1.6. Of these, ankG appears to be the driver of AIS formation and maintenance. In its absence, axonal compartments revert to dendritic molecular makeup 30. In direct neuronal ischemia, proteolysis of ankG is mediated by calpain-dependent proteolysis and partially reversed by calpain-inhibition 11. Calpain activity increases in peri-infarct cortex 31, therefore AIS remodeling in peri-infarct cortex may be dependent on calpain activation.
While the post-stroke change in AIS length is small, alterations within this critical cell compartment are likely to have an impact on cellular excitability. The observed decrease in the length of NaV1.6 staining at the AIS in peri-infarct cortex suggests that fewer NaV channels are present at this location. Stroke directly alters the gene expression of sodium channels in sprouting neurons adjacent to the infarct 28 and in human peri-infarct tissue after intracerebral hemorrhage 32. Electrophysiological techniques to measure the excitability effects of sodium channel changes at the AIS are difficult. Only recently have voltage-sensitive dyes and imaging techniques improved enough to allow observations of current at the AIS 9, 33 and these techniques still have resolution limits that preclude their application to this model system 9. However, localization of the action potential spike trigger zone to the distal half of the AIS 9, where we observed the greatest degree of change, suggests the peri-infarct decrease in AIS length from its distal end would decrease the ability of neurons to fire action potentials with the same fidelity as unaffected cells, thereby altering cortical circuitry post-stroke.
Axoaxonic synapses present at the AIS introduce an additional regulatory element to the initiation site of action potentials. Chandelier cells that provide axoaxonic input are predominantly GABAergic interneurons whose role in cortical networks is thought to be inhibitory at the AIS 34. However, recent data has suggested that axoaxonic synapses may have depolarizing effects at rest despite their exclusive use of GABA 35–37. Loss of these synapses after stroke provides evidence that remodeling at the AIS is not limited to intrinsic pyramidal cell regulation of AIS length but includes synaptic remodeling. The actual electrophysiological meaning of GABAergic synapse loss at the post-stroke AIS remains to be clarified as the field determines whether these are inhibitory or excitatory synapses. However, the data presented here supports the idea that AIS remodeling changes not just the intrinsic excitability of peri-infarct pyramidal neurons but also their local circuitry.
In rodent models of stroke, axonal sprouting occurs in the peri-infarct region resulting in reorganization of topographic maps of adjacent somatosensory cortex 38–40 and stimulating a sprouting transcriptome that varies with age 28. The extent to which axonal sprouting occurs from the neuronal cell body or from more distal axon collaterals is unknown. Regardless, the functional maturation of these new axons requires the molecular clustering of NaV channels at either nodes of Ranvier or AIS to facilitate conduction. Here, we show that in upper layers of cortex adjacent to stroke, the absolute number of initial segments increases. The significance of this increase in AIS is not clear but prior data has demonstrated that there is not significant neurogenesis within this region of peri-infarct cortex 41, 42. Thus, we believe that the increased number of initial segments seen here reflects supernumerary initial segments arising from neurons in this region as a consequence of axonal sprouting, evidenced by the example provided in Figure 6. Axonal sprouting from the cell body has been observed in models of spinal cord injury, where these supernumerary axons from axotomized spinal motor neurons were shown to be cholinergic, GAP-43-positive 43, and form synapses 44. Here, we demonstrate evidence for supernumerary initial segments arising from the existing AIS in adult cortical neurons in vivo after stroke and show that these axons begin the molecular organization that begets function.
The loss of AIS integrity after white matter stroke heralds a new concept in our understanding of the widespread effect of ischemic lesions deep within the brain. The retrograde effect of white matter stroke on the neuronal cell body is poorly understood but here we show that its ability to fire action potentials is likely jeopardized through remodeling of the AIS. In this disease model of white matter stroke, the neuronal cell body is protected from direct injury and thus only the effect of distal ischemic axotomy on the neuron is observed. This data expands on the observations by Schafer et al. (2009) that acute neuronal injury produces alterations in the AIS cytoskeleton by showing that distal ischemic axonal injury in an in vivo animal model of stroke can induce similar changes. Though the retrograde axon-to-neuron signal that initiates AIS remodeling is likely different after white matter stroke, the molecular mechanisms that execute AIS remodeling are likely the same.
Stroke alters many cortical networks, simultaneously de-afferenting both excitatory and inhibitory neurons, and producing stunned neuronal circuitry. Extrapolating from a comparatively simple sensory network, in which de-afferentation increases the excitability of central auditory neurons through elongation of the AIS 12, to the complex multicellular network present in neocortex, is difficult. However, the observed decrease in AIS length in neurons surviving stroke may reflect a common injury response; a final common effector pathway of peri-infarct hypoexcitability serving an endogenous neuroprotective role by limiting excitotoxic damage 45. Likewise, the effect of pharmacologic manipulation of post-stroke excitability may further alter AIS morphology/length in ways that suggest this morphologic compartment of the cell holds key molecular signals for neuroplasticity.
Supplementary Material
Acknowledgments
We thank Nathan Rabanzia for assistance in tissue processing.
Sources of Funding:
This work was supported by NIH - NS071481 (STC), NS044916 (MNR), NS065723 (JDH) and the Adelson Medical Research Foundation.
Footnotes
Conflicts of Interest: None
References
- 1.Clarkson AN, Huang BS, Macisaac SE, Mody I, Carmichael ST. Reducing excessive gaba-mediated tonic inhibition promotes functional recovery after stroke. Nature. 2010;468:305–309. doi: 10.1038/nature09511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clarkson AN, Overman JJ, Zhong S, Mueller R, Lynch G, Carmichael ST. Ampa receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci. 2011;31:3766–3775. doi: 10.1523/JNEUROSCI.5780-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kole MH, Stuart GJ. Signal processing in the axon initial segment. Neuron. 2012;73:235–247. doi: 10.1016/j.neuron.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. Ankyring is required for clustering of voltage-gated na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jenkins SM, Bennett V. Ankyrin-g coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and l1 cams at purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, et al. A common ankyrin-g-based mechanism retains kcnq and nav channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. Betaiv spectrin is recruited to axon initial segments and nodes of ranvier by ankyring. J Cell Biol. 2007;176:509–519. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, et al. Betaivsigma1 spectrin stabilizes the nodes of ranvier and axon initial segments. J Cell Biol. 2004;166:983–990. doi: 10.1083/jcb.200408007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Popovic MA, Foust AJ, McCormick DA, Zecevic D. The spatio-temporal characteristics of action potential initiation in layer 5 pyramidal neurons: A voltage imaging study. J Physiol. 2011;589:4167–4187. doi: 10.1113/jphysiol.2011.209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meeks JP, Mennerick S. Action potential initiation and propagation in ca3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- 11.Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuba H, Oichi Y, Ohmori H. Presynaptic activity regulates na(+) channel distribution at the axon initial segment. Nature. 2010;465:1075–1078. doi: 10.1038/nature09087. [DOI] [PubMed] [Google Scholar]
- 13.Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saitsu H, Tohyama J, Kumada T, Egawa K, Hamada K, Okada I, et al. Dominant-negative mutations in alpha-ii spectrin cause west syndrome with severe cerebral hypomyelination, spastic quadriplegia, and developmental delay. Am J Hum Genet. 2010;86:881–891. doi: 10.1016/j.ajhg.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hargus NJ, Merrick EC, Nigam A, Kalmar CL, Baheti AR, Bertram EH, 3rd, et al. Temporal lobe epilepsy induces intrinsic alterations in na channel gating in layer ii medial entorhinal cortex neurons. Neurobiol Dis. 2011;41:361–376. doi: 10.1016/j.nbd.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon-Espinosa G, Defelipe J, Munoz A. Effects of amyloid-beta plaque proximity on the axon initial segment of pyramidal cells. J Alzheimers Dis. 2012;29:841–852. doi: 10.3233/JAD-2012-112036. [DOI] [PubMed] [Google Scholar]
- 17.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, et al. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of gfp. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 18.Sozmen EG, Kolekar A, Havton LA, Carmichael ST. A white matter stroke model in the mouse: Axonal damage, progenitor responses and mri correlates. J Neurosci Methods. 2009;180:261–272. doi: 10.1016/j.jneumeth.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider Gasser EM, Straub CJ, Panzanelli P, Weinmann O, Sassoe-Pognetto M, Fritschy JM. Immunofluorescence in brain sections: Simultaneous detection of presynaptic and postsynaptic proteins in identified neurons. Nat Protoc. 2006;1:1887–1897. doi: 10.1038/nprot.2006.265. [DOI] [PubMed] [Google Scholar]
- 20.Grubb MS, Shu Y, Kuba H, Rasband MN, Wimmer VC, Bender KJ. Short- and long-term plasticity at the axon initial segment. J Neurosci. 2011;31:16049–16055. doi: 10.1523/JNEUROSCI.4064-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown CE, Boyd JD, Murphy TH. Longitudinal in vivo imaging reveals balanced and branch-specific remodeling of mature cortical pyramidal dendritic arbors after stroke. J Cereb Blood Flow Metab. 2010;30:783–791. doi: 10.1038/jcbfm.2009.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael ST. Plasticity of cortical projections after stroke. The Neuroscientist. 2003;9:64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- 23.Carmichael ST. Rodent models of focal stroke: Size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ng SC, de la Monte SM, Conboy GL, Karns LR, Fishman MC. Cloning of human gap-43: Growth association and ischemic resurgence. Neuron. 1988;1:133–139. doi: 10.1016/0896-6273(88)90197-3. [DOI] [PubMed] [Google Scholar]
- 25.Stroemer RP, Kent TA, Hulsebosch CE. Neocortical neural sprouting, synaptogenesis, and behavioral recovery after neocortical infarction in rats. Stroke. 1995;26:2135–2144. doi: 10.1161/01.str.26.11.2135. [DOI] [PubMed] [Google Scholar]
- 26.Brown CE, Li P, Boyd JD, Delaney KR, Murphy TH. Extensive turnover of dendritic spines and vascular remodeling in cortical tissues recovering from stroke. J Neurosci. 2007;27:4101–4109. doi: 10.1523/JNEUROSCI.4295-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Overman JJ, Clarkson AN, Wanner IB, Overman WT, Eckstein I, Maguire JL, et al. A role for ephrin-a5 in axonal sprouting, recovery, and activity-dependent plasticity after stroke. Proc Natl Acad Sci U S A. 2012;109:E2230–2239. doi: 10.1073/pnas.1204386109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Overman JJ, Katsman D, Kozlov SV, Donnelly CJ, Twiss JL, et al. An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat Neurosci. 2010;13:1496–1504. doi: 10.1038/nn.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of angelman syndrome. J Neurosci. 2011;31:17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hedstrom KL, Ogawa Y, Rasband MN. Ankyring is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kambe A, Yokota M, Saido TC, Satokata I, Fujikawa H, Tabuchi S, et al. Spatial resolution of calpain-catalyzed proteolysis in focal cerebral ischemia. Brain Res. 2005;1040:36–43. doi: 10.1016/j.brainres.2005.01.080. [DOI] [PubMed] [Google Scholar]
- 32.Carmichael ST, Vespa PM, Saver JL, Coppola G, Geschwind DH, Starkman S, et al. Genomic profiles of damage and protection in human intracerebral hemorrhage. J Cereb Blood Flow Metab. 2008;28:1860–1875. doi: 10.1038/jcbfm.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradley J, Luo R, Otis TS, DiGregorio DA. Submillisecond optical reporting of membrane potential in situ using a neuronal tracer dye. J Neurosci. 2009;29:9197–9209. doi: 10.1523/JNEUROSCI.1240-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woodruff AR, Anderson SA, Yuste R. The enigmatic function of chandelier cells. Front Neurosci. 2010;4:201. doi: 10.3389/fnins.2010.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of gabaergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- 36.Molnar G, Olah S, Komlosi G, Fule M, Szabadics J, Varga C, et al. Complex events initiated by individual spikes in the human cerebral cortex. PLoS Biol. 2008;6:e222. doi: 10.1371/journal.pbio.0060222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodruff A, Xu Q, Anderson SA, Yuste R. Depolarizing effect of neocortical chandelier neurons. Front Neural Circuits. 2009;3:15. doi: 10.3389/neuro.04.015.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dancause N, Barbay S, Frost SB, Plautz EJ, Chen D, Zoubina EV, et al. Extensive cortical rewiring after brain injury. J Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown CE, Aminoltejari K, Erb H, Winship IR, Murphy TH. In vivo voltage-sensitive dye imaging in adult mice reveals that somatosensory maps lost to stroke are replaced over weeks by new structural and functional circuits with prolonged modes of activation within both the peri-infarct zone and distant sites. J Neurosci. 2009;29:1719–1734. doi: 10.1523/JNEUROSCI.4249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carmichael ST, Wei L, Rovainen CM, Woolsey TA. New patterns of intracortical projections after focal cortical stroke. Neurobiol Dis. 2001;8:910–922. doi: 10.1006/nbdi.2001.0425. [DOI] [PubMed] [Google Scholar]
- 41.Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J, et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. J Neurosci. 2006;26:1269–1274. doi: 10.1523/JNEUROSCI.4480-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoang TX, Nieto JH, Havton LA. Regenerating supernumerary axons are cholinergic and emerge from both autonomic and motor neurons in the rat spinal cord. Neuroscience. 2005;136:417–423. doi: 10.1016/j.neuroscience.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 44.Havton L, Kellerth JO. Regeneration by supernumerary axons with synaptic terminals in spinal motoneurons of cats. Nature. 1987;325:711–714. doi: 10.1038/325711a0. [DOI] [PubMed] [Google Scholar]
- 45.Carmichael ST. Brain excitability in stroke: The yin and yang of stroke progression. Arch Neurol. 2012;69:161–167. doi: 10.1001/archneurol.2011.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.