

Abstract
We evaluated the therapeutic efficacy and mechanisms of action of both mouse and human B7-H4 Immunoglobulin fusion proteins (mB7-H4Ig; hB7-H4Ig) in treating EAE. The present data show that mB7-H4Ig both directly and indirectly (via increasing Treg function) inhibited CD4+ T-cell proliferation and differentiation in both Th1- and Th17-cell promoting conditions while inducing production of IL-10. B7-H4Ig treatment effectively ameliorated progression of both relapsing (R-EAE) and chronic EAE correlating with decreased numbers of activated CD4+ T-cells within the CNS and spleen, and a concurrent increase in number and function of Tregs. The functional requirement for Treg activation in treating EAE was demonstrated by a loss of therapeutic efficacy of hB7-H4Ig in R-EAE following inactivation of Treg function either by anti-CD25 treatment or blockade of IL-10. Significant to the eventual translation of this treatment into clinical practice, hB7-H4Ig similarly inhibited the in vitro differentiation of naïve human CD4+ T-cells in both Th1- and Th17-promoting conditions, while promoting the production of IL-10. B7-H4Ig thus regulates pro-inflammatory T-cell responses by a unique dual mechanism of action and demonstrates significant promise as a therapeutic for autoimmune diseases, including MS.
Keywords: Autoimmunity, B7-H4, CD4+ T-cell, Costimulatory/co-inhibitory molecule, EAE, Regulatory T-cells
1. Introduction
An important goal in autoimmune disease therapy is development of new treatments re-establishing durable tolerance in self-reactive CD4+ T-cells [1]. MS is an autoimmune disease characterized by Th1 (IFN-γ) and Th17 (IL-17/GM-CSF) CD4+ T-cell responses to epitopes on myelin basic protein (MBP), proteolipid protein (PLP), and/or myelin-oligodendrocyte glycoprotein (MOG) [2-5]. Experimental autoimmune encephalomyelitis (EAE) is a CD4+ T-cell mediated model of MS ideal for characterizing potential tolerance-based immunotherapies and their underlying mechanisms. Full activation of naïve T-cells (CD4+CD62LhiCD25−) requires interaction of the Ag-specific TCR (signal one) with peptide presented in the context of major histocompatibility complex II (MHC II) on the surface of antigen presenting cells (APCs), and delivery of positive co-stimulatory signals (signal two), i.e., ligation of CD28 on CD4+ T-cells by APC-expressed B7-family members B7-1 (CD80) and B7-2 (CD86) [6]. Costimulatory interactions serve as potential drug targets, e.g. CTLA-4Ig blocks CD80/CD86-CD28 interaction [1,7-9]. Additionally, both secreted cytokines and cell surface molecules are required for the differentiation of pro-inflammatory Th17 cells secreting IL-17, IL-6, IL-22, TNF-α, and GM-CSF [10] versus CD4+/FoxP3+ Tregs that are functionally dependent upon TGF-β and/or IL-10 [11-16]. TGF-β in combination with IL-2 is critical for differentiation of Tregs [17] while TGF-β in combination with IL-6 drives differentiation of pro-inflammatory Th17 cells [18].
Ligation of T-cell-expressed co-inhibitory receptors, e.g. PD-1 by its ligand PD-L1 (B7-H1), can effectively suppress T-cell responses. We have previously shown that PD-L1 deficient mice exhibit increased severity of MOG35–55-induced C-EAE, and treatment of wildtype mice with anti-PD-L1 blocking antibody enhances EAE by increasing the number of MOG35–55-specific CD4+ T-cells producing IFN-γ and IL-17 [19]. Similarly, B7-H4 is hypothesized to have immune modulatory function [20-23]. Tumor cell-expressed B7-H4 is suggested to be a mechanism by which these cells evade anti-tumor immune responses [24,25]. Therapeutically, B7-H4Ig treatment modulates the level of inflammatory CD4+ T-cell function in rheumatoid arthritis, the NOD model of T1D and in allogeneic islet cell transplantation [26-28]. Therefore, the goal of the present study was to determine the efficacy of B7-H4Ig treatment during EAE and more importantly to determine its mechanism(s) of action. We show that B7-H4Ig treatment ameliorates EAE progression via the unique dual mechanisms of inhibiting effector CD4± Th1/17-cell activity while concomitantly increasing the number and function of Tregs in both mouse and human T cells.
2. Materials and methods
2.1. Mice, CD4+ T-cell isolation and culture
Female SJL/JCrHsD (SJL/J), C57BL/6 (Harlan Labs; Indianapolis, IN), DO11.10, BALB/c (Jackson Laboratories; Bar harbor, ME); SJL Actin/GFP, SJL FoxP3/GFP, and C57BL/6 FoxP3/GFP (bred in-house) were housed under SPF conditions in the Northwestern University Center for Comparative Medicine. Naïve CD4+ T-cells (CD4+ l-selectinhi cells) were purified using AutoMacs Magnetic Bead cell separation technology (Miltenyi Biotech; Auburn, CA) from total LN cells isolated from unprimed mice with purity ranging from 98 to 99.9%. For in vitro activation, 3–5 × 105 naïve DO11.10 CD4+ T-cells were cultured with an equal number of irradiated BALB/c splenocytes plus OVA323–339 (10 μl/ml) in Th0 (200 U/ml IL-2); Th1-(200 U/ml IL-2, 10 ng/ml IL-12, 1 ug/ml anti-IL-4); Th2- (200 U/ml IL-2, 500 U/ml IL-4, 1 ug/ml anti-IFN-γ), Th17- (10 ng/ml TGF-β1, 50 ng/ml IL-6, 1 μg/ml anti-IFN-γ, 1 μg/ml anti-IL-4, 1 μg/ml anti-IL-2), or iTreg- (100 U/ml IL-2, 25 ng/ml TGF-β1, 100 nM retinoic acid) promoting conditions. Peptides (PLP139–151, PLP178–191, and OVA323–339) were purchased from Peptides International (Louisville, KY) and purified by HPLC (purity of 96–99%). After 3–7 d of culture, the T effector cells were isolated and culture supernatants collected and cytokine concentrations determined via multiplex Luminex Liqui-Chip (Millipore; Billerica, MA).
2.2. Preparation of mouse and human B7-H4Ig
mB7-H4Ig (mouse B7-H4 extracellular domain fused with murine IgG2a) and hB7-H4Ig (human B7-H4 extracellular domain fused with human IgG1) fusion proteins were produced in suspension culture in an animal protein-free-adapted CHOK1SV (Lonza Biologics, Allendale, NJ) cell line utilizing the glutamine synthetase gene expression system, and purified using Amplimmune’s protein purification process. The extracellular domain (ECD) of hB7-H4 is 90% identical to the murine sequence.
2.3. Human CD4+ T-cell isolation and culture
Naïve CD4+ T-cells were isolated from total PMBCs (LifeSource; Evanston, IL) of 10–12 healthy donors using AutoMacs cell separation technology. 5 × 105 cells/well were cultured with autologous irradiated total PBMCs (5 × 105 cells/well) and stimulated with anti-CD3 (clone OKT3; eBioscience) and anti-CD28 (Clone CD28.2; eBioscience) (0.5 μg/ml) plus Control-Ig or human B7-H4Ig (10 μg/ml). Cells were cultured in Th0- (100 U/ml IL-2, 1 μg/ml anti-IL-4, 1 μg/ml anti-IFN-γ), Th1- (100 U/ml IL-2, 10 ng/ml IL-12, and 1 μg/ml anti-IL-4), Th2- (100 U/ml IL-2, 10 ng/ml IL-4, 1 μg/ml anti-IFN-γ), Th17- (10 ng/ml TGF-β, 50 ng/ml IL-6, 10 ng/ml IL-1β, 1 μg/ml anti-IFN-γ, 1 μg/ml anti-IL-4), or iTreg- (100 U/ml IL-2, 25 ng/ml TGF-β1, and 100 nM retinoic acid) promoting conditions. After 3–7 d of culture, the T effector cells are isolated and culture supernatants collected and cytokine concentrations determined.
2.4. R-EAE, C-EAE, and PLP139–151 transfer R-EAE
6–7-wk-old female SJL/J or SJL-Actin/GFP mice were immunized s.c. with 100 μl of an emulsion containing 200 μg of M. tuberculosis H37Ra (BD Biosciences; San Jose, CA) and 50 μg of PLP139–151 distributed over three sites on the flank. C57BL/6 mice were immunized s.c. with 100 μl of an emulsion containing 200 μg of M. tuberculosis H37Ra and 200 μg of MOG35–55 distributed over three sites on the flank, and received pertussis toxin on d0 and d+2 post-disease induction via i.p. injection (200 μl of 1 μg/ml). To inactivate Tregs, mice received two injections of anti-CD25 [clone 7D4 at 500 μg/injection i.p. (Bio X Cell; West Lebanon, NH)] on the indicated days. To neutralize IL-10, mice received 6 injections of anti-IL-10 [clone JES5-2A5 at 100 μg/injection i.p. (Bio X Cell; West Lebanon, NH)] on the indicated days [29]. In transfer EAE experiments draining LNs were collected on day 8 post priming, and total draining LN cells were reactivated in the presence of 20 μg/ml of PLP139–151, at a cell density of 8 × 106 cells/ml for 72 h. After culture 3–5 × 106 blast cells were transferred to recipient SJL/J mice. Mice were treated with Control-Ig or B7-H4Ig either at the time of cell transfer or at the onset of remission (approximately day +15–20 for most animals). Individual animals were observed at the indicated time points and clinical scores assessed in a blinded fashion on a 0–5 scale: 0, no abnormality; 1, limp tail; 2, limp tail and hind limb weakness; 3, hind limb paralysis; 4, hind limb paralysis and forelimb weakness; and 5, moribund. Data are reported as the mean daily clinical score.
2.5. Delayed-type hypersensitivity (DTH) assay and ex vivo recall responses
On d+10 or d+35 post-disease induction, mice were assayed for DTH. Mice were anaesthetized by inhalation of isoflurane and ear thickness measured using a dial thickness gauge. 10 μg of PLP139–151 and PLP178–191 in 10 μl of PBS were injected into the left and right ears, respectively. The increase in ear thickness was determined after 24 h. Mice were then sacrificed and single cell suspension of spleens and draining LNs were cultured (5 × 105 cells/well) in the presence of medium alone, OVA323–339, PLP139–151 and PLP178–191 (20 μg/ml). 24 h post-culture initiation, wells were pulsed with 1 μCi of 3H-TdR and harvested at 72 h and 3H-TdR incorporation was detected using a Topcount Microplate Scintillation Counter. Results are expressed as the mean counts per minute (CPM) of triplicate cultures. Cytokine levels were determined via LiquiChip analysis.
2.6. Immunohistochemistry
Relapsing-remitting EAE was induced in SJL-FoxP/GFP mice with PLP139–151/CFA and mice received either Control-Ig or hB7-H4Ig was decreased above. On the day following the final treatment, mice were anesthetized and perfused with PBS followed by 1% paraformaldehyde, brains and spinal cords were removed and embedded with OCT compound (Sakura Finetek, Trrance, CA), frozen on dry-ice, and stored at −80 °C until processing and analysis. Eight-micrometer sections containing either the lumbar spinal cord or cerebellum were sectioned on the cryostat, sections were air dried at room temperature for 30 min, rinsed in DH2O to remove O.C.T., and fixed in −20 °C acetone for 10 min. Sections were dried at room temperature for 10 min, washed in PBS 3 × 5 min, blocked in 5% donkey serum in PBS + 0.1% triton x-100 (PBS+) for 60 min. prior to incubation with primary antibodies diluted in PBS + 0.1% Triton x-100 + 5% donkey serum overnight at 4 °C, i.e., anti-PLP antibody (clone plpc1) (AbD Serotec; Raleigh, NC) and anti-CD4 antibody (clone GK.15) (eBioscience). After washing in PBS 3 × 10 min, sections were incubated in secondary antibodies in PBS + for 1 h. at room temperature, i.e., Cy3 donkey anti-rat (Jackson ImmunoResearch; West Grove, PA) and FITC anti-rat IgG (Vector Labs; Burlingame, CA) respectively, washed in PBS 3 × 10 min, incubated in DAPI for nuclear staining 5 min, washed 3 × 5 min. in PBS then coverslipped using hard setting Vectamount (Vector Laboratories, Burlingame, CA). Images were taken on a Leica DM 5000B microscope using ImagePro software.
2.7. FACS analysis
FACS analysis was performed on spinal cord leukocytes isolated from individual mice (5 per group) perfused with 20 ml of PBS [30]. hB7-H4Ig (PE conjugated) was used for binding analysis of cells spleen and LN cells. Cells were stained with anti-CD4 (clone RM4-5), anti-FoxP3 (clone FJK-16s), anti-Helios (clone 22F6) (eBioscience; San Jose, CA), anti-CD25 (clone PC61), and anti-CD44 (clone IM7) (BD Bioscience; San Diego, CA). 5 × 105 viable cells were analyzed per individual sample using a BD Canto II cytometer (BD Bioscience), and the data were analyzed using FlowJo Version 9.5.2 software (Tree Star, Inc.; Ashland, OR).
2.8. Statistical analyses
Comparisons of the percentage of animals showing clinical disease were analyzed by X2 using Fisher’s exact probability, and two-way ANOVA with a Bonferroni post-test was used to determine statistical differences between mean clinical disease scores. Single comparisons of two means were analyzed by Student’s t-test.
3. Results
3.1. Efficacy of mouse and human B7-H4Ig treatment for regulating EAE induction and progression
The present studies were begun to determine the ability of B7-H4Ig to inhibit ongoing R-EAE. To do so, SJL/J mice were primed with PLP139–151/CFA and allowed to progress through the acute phase of disease, and treatment with either Control Ig or mB7-H4Ig was initiated during disease remission. The present data show that initiation of mB7-H4Ig treatment during disease remission significantly ameliorated further disease progression (Fig. 1A) decreasing the number of effector CD4+/CD44hi cells within the CNS, with a trend toward increasing the number of Tregs (Fig. 1B). Additionally, a significant increase in the number of Tregs was observed in the spleen (data not shown). These findings suggest that mB7-H4Ig treatment may allow for a more tolerogenic environment within the CNS since the ratio of Tregs to Teff-cells was dramatically increased (Control-Ig = 0.03 ± 0.0; 60 μg/dose B7-H4Ig = 2.89 ± 1.9; 300 μg/dose B7-H4Ig = 3.0 ± 1.0). While the mB7-H4Ig-induced increase in Tregs within the CNS did not achieve significance in the present experiment, in other experiments (Fig. 6A and B), significance was achieved in both the CNS and the spleen suggesting that the Tregs may be cycling between the spleen and the CNS during various stages of disease. We also demonstrated that treatment beginning on Day +20 with either a low dose (60 μg/dose) or high dose (300 μg/dose) of mB7-H4Ig or hB7-H4Ig significantly decreased disease progression of MOG35–55/CFA-induced C-EAE in C57BL/6 mice. Interestingly, no protection was seen in mice in which treatment was initiated on Day +60 (Fig. 1C and D) although ex vivo MOG35–55-specific CD4+ T-cell proliferation was inhibited in both treatment regimens (Fig. 1E).
Fig. 1.
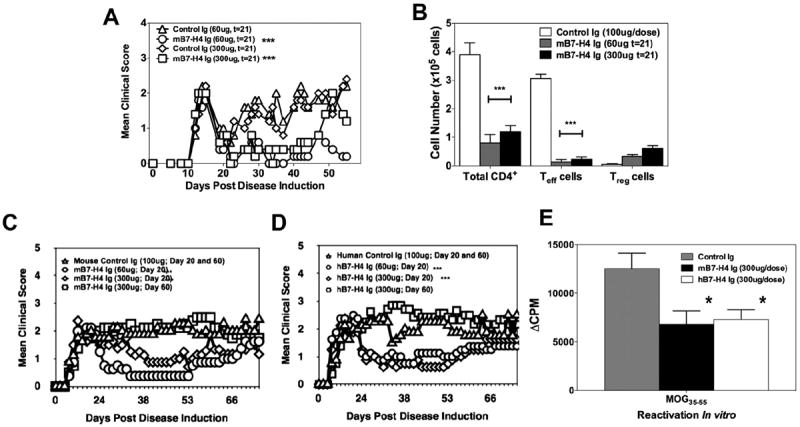
B7-H4Ig decreases EAE disease severity. SJL/J mice (n = 10/group) were primed with PLP139–151/CFA and (A) treated with 60 or 300 μg of species and isotype-matched Control-Ig or mB7-H4Ig three-times per week for four weeks beginning on Day +21 (t = 21) during disease remission. On Day +35 post-disease induction, (B) the CNS was collected from 3 representative mice per group and the numbers of total CD4+ T-cells, Teff cells (CD4+/CD44hi/FoxP3−), and Tregs (CD4+/CD25+/FoxP3+) were assessed via FACS analysis. C57BL/6 mice (n = 10/group) were primed with MOG35–55/CFA and treated beginning on Day +21 (t = 21) with 60 or 300 μg of species and isotype-matched Control-Ig or (C) mB7-H4Ig or (D) hB7-H4Ig three times per week for four weeks. The mice were followed for disease and the data are presented as the mean clinical score. Additionally, a subset of the Control-Ig treated mice from panels C and D were treated with mB7-B4Ig or hB7-H4Ig beginning on Day +60 post-disease induction and followed for disease severity. One representative experiment of 2–4 experiments is shown, and disease severity was found to be statistically significant when B7-H4Ig was administered at either the time of disease induction or at Day +20 post disease induction as compared to Control-Ig treated mice (p < 0.001). (E) On Day +75 post-priming total splenocytes were reactivated ex vivo with MOG35–55 (20 μg/ml), and the level of cellular proliferation was assessed via tritiated-thymidine incorporation. Asterisks (*, **, ***) indicate a statistically significant decrease in mean clinical score or in proliferation by cells from B7-H4Ig treated mice in comparison to cells collected from Control-Ig treated mice, p < 0.05, 0.01, 0.001 respectively.
Fig. 6.
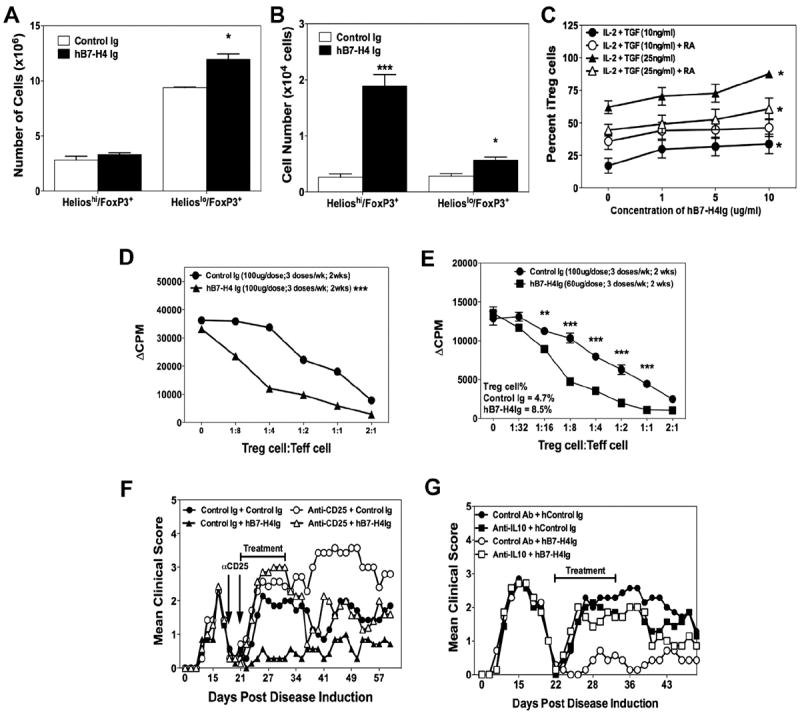
B7-H4Ig treatment induces an increase in the number and function of Tregs. SJL/J mice (n = 5/group) were primed with PLP139–151/CFA, and treated with Control-Ig or hB7-H4Ig beginning during disease remission. Following the last treatment (A) spleens and (B) CNS were collected and the number of CD4+/CD25+/FoxP3+/Helios+ versus CD4+/CD25+/FoxP3+/Helios− were enumerated from individual mice. (C) CD4+ T-cells were isolated from unprimed SJL/J mice and cultured (2.5 × 105 cells/well) with anti-CD3 + anti-CD28 (1 g/ml) plus an equal number of irradiated splenocytes in the presence of IL-2 (100 U/ml), TGF-β1 (10 or 25 ng/ml), and retinoic acid (100 nM) plus increasing concentrations of hB7-H4Ig. The cells were collected on Day 4 of culture and the percentage of CD4+/CD25+/FoxP3+ cells determined via FACS analysis. Asterisks (*) indicate a statistically significant increase in the number of cells in comparison to Control-Ig treated cells, p < 0.05. (D) Tregs were isolated from unprimed C57BL/6-FoxP3/GFP mice treated with either Control-Ig or hB7-H4Ig (100 μg/dose) via MoFlo sorting, co-cultured at various ratios with naïve CD4+ T-cell plus anti-CD3 (1 μg/ml), pulsed with 1 μCi tritiated thymidine at 24 h and cultures harvested at 72 h. (E) SJL-FoxP3/GFP mice were primed as above, and treated with Control-Ig or hB7-H4Ig beginning at disease remission. Following the final treatment splenic Tregs were sort purified and co-cultured at various ratios with 0.5 × 106 naïve CD4+ T-cells plus 0.5 × 106 irradiated APCs plus anti-CD3 (1 μg/ml). The cultures were pulsed with 1 μCi tritiated-thymidine at 24 h and harvested at 72 h. Asterisks (**, ***) indicate a statistically significant decrease in proliferation due to B7-H4Ig treatment as compared to Control-Ig treated cultures (p < 0.01 or 0.001, respectively). (F) SJL/J mice were primed with PLP139–151/CFA and treated with Control mAb or anti-CD25 (0.5 mg/dose) on Days +19 and 21 to inactivate Tregs, and then treated with Control-Ig or hB7-H4Ig beginning on Day +21. The mice were followed for disease, and the data is presented as the mean clinical score. Inactivation of Tregs reversed the normal hB7-H4Ig-induced decrease in disease during the period from Days +21–35. One representative experiment of three is shown. (G) To determine the functional relevance of the B7-H4Ig-induced increase in IL-10, SJL/J mice (n = 8 per treatment group) were primed with PLP139–151/CFA and treated with Control mAb or anti-IL-10 (0.1 mg/dose) in combination with either a species and isotype-matched Control-Ig or mB7-H4Ig three-times per week for two weeks beginning during disease remission (Day +22 post disease induction). The mice were followed for disease, and data from one experiment is presented as the mean clinical score.
3.2. B7-H4Ig treatment regulates CD4+ T-cell responses in vivo
To determine if mB7-H4Ig treatment decreases CD4+ T-cell responses during disease initiation, SJL/J mice were primed with PLP139–151/CFA and treated with Control-Ig or mB7-H4Ig beginning on the day of priming. Mouse B7-H4Ig treatment significantly inhibited the PLP139–151-specific DTH response as compared to Control-Ig-treated mice (Fig. 2A), and decreased the ex vivo PLP139–151-specific recall response (Fig. 2B). Relevant to disease therapy, splenocytes from mB7-H4Ig-treated mice exhibited decreased recall responses to both the disease-inducing PLP139–151 peptide and to PLP178–191, the dominant spread epitope, as determined by proliferation (Fig. 2C), and IL-17 (Fig. 2D) and IFN-γ secretion (Fig. 2E) demonstrating that mB7-H4Ig can both inhibit effector cytokine production by previously activated T-cells and priming of naïve T-cells.
Fig. 2.

B7-H4Ig decreases myelin-specific T-cell responses. SJL/J mice (n = 10/group) were primed with PLP139–151/CFA and treated with 60 or 300 μg of species and isotype-matched Control-Ig or mB7-H4Ig every other day until Day +10 when (A) PLP139–151 in vivo DTH responses and (B) ex vivo recall responses of total draining LN cells (5 × 105 cells/well) to increasing concentration of anti-CD3, OVA323–339, or PLP139–151 were determined. Cultures were pulsed with 1 μCi tritiated-thymidine at 24 h and cultures harvested at 72 h. SJL/J mice (n = 5/group) were primed with PLP139–151/CFA and treated with 60 or 300 μg of species and isotype-matched Control-Ig or mB7-H4Ig three-times per week for two weeks beginning during disease remission. On Day +35 post-disease induction, total splenocytes were collected from individual mice and activated ex vivo (5 × 105 cells/well) with PLP139–151 or PLP178–191 (20 μg/ml). Two replicate sets of wells were set up such that (C) the first was pulsed with 1 μCi of tritiated-thymidine at 24 h and proliferation and culture supernatants from the second were collected at 72 h and assayed for production of (D) IL-17 and (E) IFN-γ. One representative experiment of two-three is presented. Asterisks (*, **, ***) indicate a statistically significant decrease in proliferation and cytokine production by cells from B7-H4Ig treated mice in comparison to cells collected from Control-Ig treated mice, p < 0.05, 0.01, 0.001 respectively.
3.3. B7-H4Ig treatment inhibits in vitro differentiation of mouse Th1/Th17 cells
We next examined the ability of B7-H4Ig to modulate the differentiation of naïve DO11.10 CD4+ T-cells, either containing or depleted of CD25+ nTregs, and stimulated in Th1- vs. Th17-driving conditions. Mouse B7-H4Ig decreased the levels of IFN-γ (Fig. 3A) and IL-17 (Fig. 3C) produced in the absence of Tregs in a dose-dependent manner. Significantly, mB7-H4Ig-induced down-regulation of naïve T-cells containing CD25+ Tregs under both Th1 (Fig. 3B) and Th17 (Fig. 3D) driving conditions was more robust and associated with a dramatic increase in IL-10 production. Mouse B7-H4Ig-induced inhibition of Th1/Th17 cell differentiation was time-dependent. We next sought to determine if there was a temporal ability of mB7-H4Ig to modulate naïve CD4+ T-cell differentiation. To do so, naïve DO11.10 CD4+ T-cells depleted of CD25+ Tregs were cultured in Th1- or Th17-promoting conditions plus addition or wash out of Control-Ig or mB7-H4Ig at the time of culture initiation (t = 0) or 12, 24, 48, or 72 h post-culture initiation. Addition of mB7-H4Ig to the Th1 cell-promoting cultures at any time prior up to 72 h post-culture initiation inhibited IFN-γ production. Additionally, wash out of mB7-H4Ig in Th1 cell-promoting conditions before 24 h post-culture initiation, resulted in no decrease in IFN-γ production (Fig. 3E). Addition of mB7-H4Ig to the Th17 cell-promoting cultures at any time prior to 48 h post-culture initiation decreased IL-17 production. Wash out of mB7-H4Ig in Th17 cell-promoting cultures before 48 h post-culture initiation, resulted in no decrease in the level of IL-17 produced (Fig. 3F). This time frame is shifted to 12–24 h with regard to the ability of mB7-H4Ig to enhance the level of IL-10 produced by DO11.10 CD4+ T-cells activated in Th17-promoting conditions (Fig. 3G). Therefore, mB7-H4Ig alteration of naïve T-cell differentiation is time- and cell activation state-dependent.
Fig. 3.
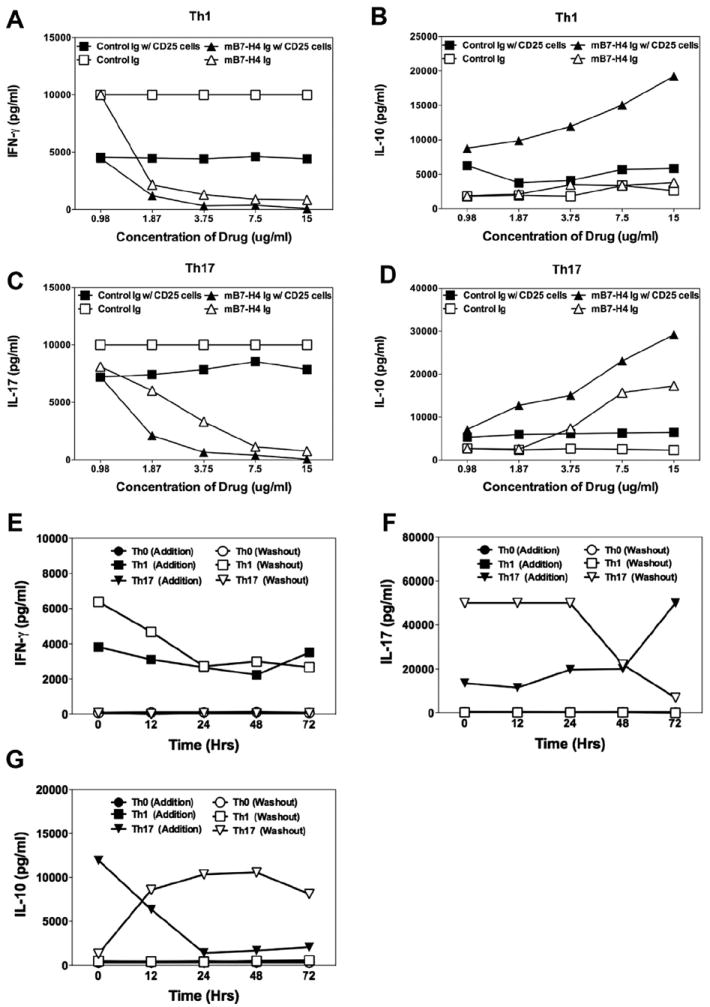
B7-H4Ig inhibits mouse Th1/Th17 cell differentiation. (A–D) CD4+ T-cells were isolated from naïve DO11.10 mice either specifically excluding CD25+ nTregs or not (following manufacturer protocol). Cells were cultured (5 × 105 cells + 5 × 105 irradiated BALB/c splenocytes + OVA323–339 (20 μg/ml) (A and B) in Th1- [IL-2 (100 U/ml), IL-12 (10 ng/ml), and anti-IL-4 (1 μg/ml)] or (C and D) Th17-promoting conditions [TGF-β (10 ng/ml), IL-6 (50 ng/ml), IL-23 (10 ng/ml), anti-IL-2, anti-IFN-γ, and anti-IL-4 (1 μg/ml)]) in the presence of increasing concentrations of species and isotype matched Control-Ig or mB7-H4Ig (0–15 μg/ml). (E–G) As above, CD4+ T-cells were isolated from naïve DO11.10 mice specifically excluding CD25+ cells and cultured in Th0 cell- [IL-2 (100 U/ml), anti-IL-4, and anti-IFN-γ (1 μg/ml)], Th1 cell-, or Th17 cell-promoting conditions. Control-Ig or mB7-H4Ig (10 μg/ml) was added to the cultures at the various times as indicated or washed out of the cultures at the various times after culture initiation and recultured for the remainder of the 96 h. The levels of IFN-γ, IL-17, and IL-10 were assayed via LiquiChip. One representative experiment of two is shown.
3.4. B7-H4Ig treatment suppresses adoptive transfer R-EAE by PLP139–151-specific T cells
The above data show that regulation of pro-inflammatory CD4+ T-cell function by B7-H4Ig is time-dependent. For the potential clinical development of B7-H4Ig as an autoimmune disease therapeutic, we chose to utilize hB7-H4Ig for the remainder of the studies. Given the ~90% amino acid homology between mouse and human B7-H4 [21], we reasoned that treatment of mice with hB7-H4Ig would be functional. After completing pilot studies to confirm hB7-H4Ig was functionally similar to mB7-H4Ig (data not shown), we next determined the time-dependence of B7-H4Ig binding to CD4+ T-cells in vivo, SJL/J mice were primed with PLP139–151/CFA, and binding of hB7-H4Ig to CD4+ T-cells from the draining inguinal lymph nodes (LNs), spleen, and non-draining cervical LNs was assessed at Days +3, +6 and +10. The data show an increase in hB7-H4Ig binding to T cells in the spleen and draining LNs 6 d post-priming (Fig. 4A). Additionally, hB7-H4Ig binds to an increased percentage of CD4+ T-cells from PLP139–151-primed mice following ex vivo peptide re-activation (Fig. 4B). Taking the time-dependency for mB7-H4Ig to modulate the level of cytokine produced with the above hB7-H4Ig binding data suggests that the putative B7-H4 receptor is induced on the surface of CD4+ T cells in an activation-induced manner.
Fig. 4.

B7-H4Ig decreases severity of adoptive transfer EAE. (A) Freshly isolated cells from spleens, inguinal LNs (Draining), and cervical LNs (Non-draining) were collected from SJL/J mice primed with PLP139–151/CFA on the indicated days and binding of hB7-H4Ig-PE to CD3+/CD4+ T-cells was assessed via FACS analysis. (B) Draining LN cells were collected from PLP139–151/CFA-primed SJL/J mice on Day +8, reactivated in vitro in the presence of PLP139–151 (20 μg/ml) for 3 days, and binding of hB7-H4Ig-PE on CD3+/CD4+ T-cells was assessed via FACS analysis. (C) SJL-actin/GFP mice were primed with PLP139–151/CFA, draining LNs were collected on Day +8, total LN cells were reactivated ex vivo with PLP139–151 (20 μg/ml), and on Day 3 of culture cell were collected and 5 × 106 blast cells were transferred i.v. to naïve recipient SJL/J mice (n = 15/group). Beginning on the day of cell transfer, mice received either three doses per week for two weeks of Control-Ig or hB7-H4Ig (60 μg/dose). The mice were followed for disease severity and the data is presented as the mean clinical score. On Day +10 post-cell transfer, (D) spleens and (E) CNS from the mice treated in Panel C were analyzed via FACS for the presence of transferred cells (n = 5/group). (F) Total cells isolated from individual CNS samples were reactivated ex vivo (1 × 106 cell/well) with PLP139–151, or OVA323–339 (20 μg/ml). Cultures were pulsed with 1 μCi tritiated-thymidine at 24 h and harvested at 72 h. One representative experiment of 3 is presented. (G) SJL/J mice were primed with PLP139–151/CFA, draining LNs were collected on Day +8, and total LN cells were reactivated ex vivo with PLP139–151 (20 μg/ml) plus Control-Ig or hB7-H4Ig (10 μg/ml). On day 3 of culture, cells were collected and supernatants were assessed for cytokine production. (H) 3 × 106 blast cells from Control-Ig-treated cultures, 3 × 106 blast cells from hB7-H4Ig-treated cultures, or 1.5 × 106 blast cells from Control-Igtreated cultures plus 1.5 × 106 blast cells from hB7-H4Ig-treated cultures were transferred i.v. to naïve recipient SJL/J mice (n = 10/group) and mice were followed for disease development. One representative experiment of 3 is presented. Asterisks (*, ***) indicate a statistically significant decrease in transferred cells, proliferation and cytokine production following B7-H4Ig treatment in comparison to Control-Ig treatment, p < 0.05, 0.01, 0.001 respectively.
We next determined if hB7-H4Ig treatment could inhibit adoptive transfer of R-EAE mediated by PLP139–151-specific Tcells derived from SJL-Actin/GFP Tg mice. Human B7-H4Ig treatment of wildtype SJL/J recipients beginning on the day of cell transfer dramatically decreased disease expression (2/10 mice affected with maximal clinical score of 1.0 each) as compared to Control-Ig-treated mice (10/10 affected) (Fig. 4C). The enhanced efficacy of hB7-H4Ig treatment in the adoptive transfer EAE model compared to active EAE (Fig. 1) likely represents the lack of continued T-cell activation via the peptide/CFA reservoir present in actively immunized mice. Human B7-H4Ig treatment did not result in a significant alteration in the number of donor GFP+ cells within the spleen (Fig. 4D), but significantly reduced the number of transferred cells within the CNS (Fig. 4E). Furthermore, PLP139–151-induced proliferative responses of CNS infiltrating T cells from hB7-H4Ig treated mice were significantly decreased as compared to CNS-infiltrating cells from Control-Ig treated mice (Fig. 4F). These findings indicate that hB7-H4Ig treatment inhibits the CNS transmigration of activated PLP139–151-specific CD4+ T-cells.
We next asked if adoptive R-EAE could be inhibited by addition of hB7-H4Ig to in vitro reactivation cultures of PLP139–151-sensitized total LN cells. Human B7-H4Ig significantly inhibited production of IFN-γ, IL-17, and GM-CSF, while enhancing the levels of IL-10 (Fig. 4G). Functionally, transfer of either 3 × 106 blast cells from hB7-H4Ig treated cultures or 1.5 × 106 blast cells from Control-Ig treated cultures plus 1.5 × 106 blast cells from hB7-H4Ig treated cultures resulted in decreased disease severity as compared to cells from Control-Ig-treated cultures (Fig. 4H). Therefore, hB7-H4Ig decreases the ability of PLP139–151 sensitized T-cells to induce adoptive R-EAE.
3.5. B7-H4Ig treatment decreases the number of effector CD4+ T cells and increases the number of Tregs in the CNS
The above data clearly show that treatment of mice with B7-H4Ig during both active and adoptive transfer EAE decreases the level of disease severity. The data also show that treatment with hB7-H4Ig decreases the ability of PLP139–151-sensitized cells from entering and/or being maintained within the CNS following transfer into an unprimed SJL/J recipients. Additionally, the data from Fig. 1C hinted at a B7-H4Ig treatment-induced decrease in the number of Teff-cells and concomitant increase in the number of Tregs. To further address this later possibility, immunohistochemical analysis was performed on SJL-FoxP3/GFP mice treated with B7-H4Ig during disease remission. Following the last treatment, mice were perfused and histological analysis of the lumbar spinal cord, cerebellum, and the spleen were performed. As shown in Fig. 5A and B, hB7-H4Ig treatment decreased the number of infiltrating CD4+ T cells present within the white matter tracks of the lumbar spinal cord. Within the cerebellum, lesions were found in both mice treated with Control-Ig and hB7-H4Ig treatment mice, however there was a decreased number of CD4+ T cells present within the lesions of hB7-H4Ig treated mice as compared to Control-Ig treated mice (Fig. 5C). Additionally, an increased number of FoxP3+ cells were found within the lesions of hB7-H4Ig treated mice (indicated by the white arrow in Fig. 5C). In further support of the hB7-H4Ig-induced increase in the number of Tregs, histological analysis of the spleen (Fig. 5D) also showed a significant increase in the number of FoxP3+ cells within the CD4+ T cell white pulp regions of the spleen of hB7-H4-Ig-treated mice. Taken together, this analysis shows that hB7-H4Ig treatment decreases the number of CD4+ T cells present within the CNS and increases the number of Tregs in the both spleen and the CNS.
Fig. 5.

B7-H4Ig treatment decreases the number of effector CD4+ T cells and increases the number of Tregs in the CNS. SJL-FoxP3/GFP mice (n = 5/group) were primed with PLP139–151/CFA, and treated with Control-Ig or hB7-H4Ig beginning during disease remission. Following the final treatment mice were perfused, and histological analysis of the lumbar spinal cord, cerebellum, and the spleen. Tissues were frozen in OCT, and 8 μm sections from the lumbar spinal cord were stained for (A) PLP (green) and nuclei (DAPI), and (B) CD4 (red), FoxP3 (green), and nuclei (DAPI). (C) For the sections taken from the cerebellum, similar analysis was completed, and a decrease it the number of CD4+ T cells was seem with a coordinate increase in the number of FoxP3+ cells within the lesions of hB7-H4Ig treated mice (white arrow). (D) Spleen sections were analyzed for the presence of CD4+ cells (red), FoxP3+ cells (green), and nuclei (DAPI).
Our results (Figs. 1C and 5C, and 5D) and others [27] suggest that B7-H4Ig treatment alters the number of Tregs within the target tissue of the ongoing autoimmune response. We thus asked if hB7-H4Ig treatment altered the number of natural Tregs (CD4+/CD25+/FoxP3+/Helioshi) versus induced Tregs (CD4+/CD25+/FoxP3+/Helioslo) present within the spleen and CNS. The present data show that hB7-H4Ig treatment induced an increase in the number Helioslo Tregs within the spleen and a significant increase in the number of both Helioshi and Helioslo Tregs within the CNS (Fig. 6A, B) arguably indicating drug-induced in vivo expansion of both nTregs and iTregs [31,32]. Supporting the ability of B7-H4Ig to induce iTreg expansion in vivo, hB7-H4Ig also induced a dose-dependent increase in the numbers of iTregs derived from naïve CD4+ T-cells stimulated in iTreg promoting cultures (Fig. 6C).
3.6. B7-H4Ig treatment increases the number and functional suppressive capacity of Tregs
We next determined if hB7-H4Ig treatment also altered Treg function. Tregs were isolated from unprimed C57BL/6-FoxP3/GFP mice following treatment with either Control-Ig or hB7-H4Ig and co-cultured at various ratios with naïve CD4+ T-cell plus anti-CD3 (Fig. 6D). FoxP3/GFP+ Tregs isolated from hB7-H4Ig-treated mice had significantly increased suppressor function, but not Treg number, as compared to Tregs isolated from Control-Ig-treated mice indicating that hB7-H4Ig directly enhances Treg suppressor function in the absence of an inflammatory immune response. To determine if antigen-stimulation is required for the hB7-H4Ig-induced increase in the number of Tregs previously observed (Fig. 6A, B), SJL-FoxP3/GFP mice were primed with PLP139–151 and treated with Control-Ig or hB7-H4Ig beginning at disease remission and Tregs were sort purified and assayed for suppressive function following the final treatment. Human B7-H4Ig treatment increased both the percentage and number of Tregs recovered and enhanced their in vitro suppressive function (Fig. 6E). Lastly, we determined the requirement for Tregs in hB7-H4Ig-mediated regulation of R-EAE in vivo. SJL/J mice were primed with PLP139–151/CFA and received either two treatments of Control mAb or anti-CD25 at disease remission to inactivate Tregs [33]. Beginning on the day of the second mAb treatment, half of each group was treated with either Control-Ig or hB7-H4Ig. Significantly, functional Tregs must be present at the time of hB7-H4Ig treatment for effective therapeutic regulation of R-EAE as anti-CD25 treatment reversed hB7-H4Ig-induced disease regulation.
We next determined if a mechanistic linkage between the B7-H4Ig-induced increase in the level of IL-10 production by both activated encephalitogenic T cells (Fig. 4G) and by naïve T cells stimulated in either Th1- or Th17-driving conditions (Fig. 3B, D) exists. Therefore, we asked if IL-10 was required for the disease modulating effects of B7-H4Ig treatment. SJL/J mice were primed with PLP139–151/CFA and treated with Control mAb or anti-IL-10 in combination with either Control-Ig or hB7-H4Ig beginning at disease remission. The present data show that neutralization of IL-10 reverses the therapeutic effect of hB7-H4Ig treatment clearly demonstrating the IL-10 dependence. Therefore, hB7-H4Ig treatment decreases the level of R-EAE severity in a functional Treg- and IL-10-dependent manner.
3.7. Human B7-H4Ig inhibits Th1/Th17 cell differentiation and enhances Th2 cell differentiation of human T cells
The above data show that both mB7-H4Ig and hB7-H4Ig modulate mouse CD4+ T-cell responses in vitro and in vivo. We next asked if hB7-H4Ig would similarly modulate human CD4+ T-cell responses. Naïve human CD4+ T-cells from 10 to 12 healthy donors were activated in Th0-, Th1-, Th2-, Th17-, or iTreg-promoting conditions as detailed in the Methods. The present data show that hB7-H4Ig treatment decreased cellular proliferation, the level of TNF-α secreted, and the level of lineage-associated cytokine produced in Th1- and Th17-promoting conditions, respectively. Similarly to mouse naïve CD4+ T cells activated in vitro, hB7-H4Ig treatment induced a trend toward increasing IL-10 production in Th0-, Th1-, Th2-, or iTreg-promoting conditions, with a statistically significant increase in the level of IL-10 produced by naïve human CD4+ T-cells activated in the presence of Th17-promoting conditions (Fig. 7). Thus, hB7-H4Ig modulates inflammatory cytokine production by human CD4+ T-cells replicating the mouse data. Most importantly, a significant increase in IL-10 production is noted, similar to the pattern seen with mouse cells, when human CD4+ T-cells are activated in Th17-promoting conditions. Additionally, hB7-H4Ig also significantly increased IL-4 production by human CD4+ T-cells activated in Th2-promoting conditions.
Fig. 7.
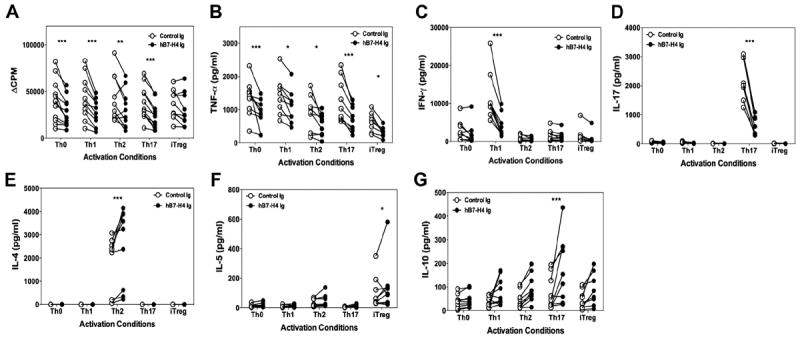
hB7-H4Ig decreases human Th1/Th17 cell differentiation and increases Th2 cell differentiation. Naive human CD4+ T-cells were isolated from total PMBCs collected from 10 to 12 healthy donors. The naive CD4+ T-cells (5 × 105 cells/well) were activated in the presence of autologous irradiated total PBMCs (5 × 105 cells/well) in the presence of anti-CD3 and anti-CD28 (0.5 μg/ml) plus Control-Ig or hB7-H4Ig (10 μg/ml). Cells were cultured in Th0- [IL-2 (100 U/ml), anti-IL-4, and anti-IFN-γ (1 μg/ml)], Th1- [IL-2 (100 U/ml), IL-12 (10 ng/ml), and anti-IL-4 (1 μg/ml)], Th2- [IL-2 (100 U/ml), IL-4 (10 ng/ml), and anti-IFN-γ (1 μg/ml)], Th17- [TGF-β (10 ng/ml), IL-6 (50 ng/ml), IL-1β (10 ng/ml), anti-IFN-γ, and anti-IL-4 (1 μg/ml)], or iTreg- [IL-2 (100 U/ml), TGF-β1 (25 ng/ml), and retinoic acid (100 nM)] promoting conditions. Two replicate sets of wells were set up such that one was (A) pulsed with 1 μCi of tritiated-thymidine at 24 h to determine cellular proliferation at 96 h, and for the second set the levels of (B) TNF-α, (C) IFN-γ, (D) IL-17, (E) IL-4, (F) IL-5, and (G) IL-10 were determined at 96 h post-culture initiation. Asterisks (*, **, ***) indicate a statistically significant alteration in the CPM values or cytokine level due to B7-H4Ig treatment as compared to Control-Ig treated cultures.
4. Discussion
Our results indicate that short-term treatment of either PLP139–151-induced R-EAE in SJL/J mice or MOG35–55-induced C-EAE in C57BL/6 mice with either mouse or human B7-H4Ig significantly inhibits disease progression by inhibiting pro-inflammatory Th1/Th17 cell activation/function via Treg activation. The present data are the first to show the functional linkage between the hB7-H4Ig-induced increase in the number of Tregs at the inflammatory site, the hB7-H4Ig-induced increase Treg suppressor function, and the requirement for both functional Tregs and IL-10 at the initiation of B7-H4Ig treatment. This clearly distinguishes the mechanism of B7-H4Ig therapy from the published mechanism of action for CTLA-4Ig treatment that blocks both CD80 and CD86, thereby inhibiting CD4+ T-cell activation and potentially Treg development [34]. Treg activation was required for regulation of EAE progression as depletion of CD25+ T-cells prior to B7-H4Ig therapy abrogated clinical protection. As IL-10 was also required for B7-H4Ig-induced regulation of EAE progression, it is likely that the Treg-mediated suppression was mediated via IL-10 production, which is an area of further research within the laboratory. In support of the hypothesis that mouse or human B7-H4Ig induces an inhibitory signal, as opposed to blocking its potential receptor, B7-H4−/− mice show increased EAE as compared to wildtype mice [35]. However in contrast to our present data and other studies using B7-H4Ig treatment, the B7-H4−/− (B7-X−/−) mice did not exhibit a decreased number of nTregs. This may be due to fundamental differences between the two experimental models, i.e., loss of a negative regulatory signal in B7-H4−/− mice vs. induction of a regulatory signal following ligation of the B7-H4 receptor following B7-H4Ig treatment. Collectively, our findings suggest that B7-H4Ig cross-links its receptor and induces a regulatory signal within activated target CD4+ T-cells. While the receptor for B7-H4 is still unknown, our data elucidates the in vivo function of B7-H4 on the cellular level, and the signaling pathways that may be involved in B7-H4 receptor-mediated CD4+ T-cell regulation.
To determine if B7-H4Ig treatment modulated the suppressive function of Tregs in vivo, we isolated Tregs from SJL-FoxP3/GFP mice following treatment with Control-Ig or B7-H4Ig during PLP139–151-induced R-EAE. Tregs isolated from B7-H4Ig-treated mice indeed showed significantly increased suppressive activity. Regarding the natural biology of B7-H4, published data has shown that alternatively activated macrophages (M2) are able to suppress EAE following transfer [36] and may possess neuroprotective activity [37]. As a potential innate regulatory effect of B7-H4 signaling, co-culture of IL-10/TGF-β treated M2 macrophages expressing B7-H4 decreases CD4+ T-cell proliferation and induces increased numbers of CD4+/FoxP3+ T-cells [38]. Collectively, our findings indicate that B7-H4Ig treatment induces a tolerogenic microenvironment by increasing the ratio of Tregs to effector CD4+ T-cells, especially within the CNS, which has been show to serve as a predictor of disease severity/resolution [39].
Other B7-CD28 family member proteins have been shown to modulate CD4+ T-cell function in vitro via a skewing of the effector response [40]. For example, activation of naïve CD4+ T-cells in the presence of beads coated with anti-CD3/28 and PD-L1Ig in Th1- or Th17-promoting conditions has been shown to decrease the number of resultant IFN-γ and IL-17 producing cells respectively, and to increase the number of CD25+/FoxP3+ Tregs [41]. Another B7-family receptor/ligand pair interaction, ICOS/ICOS-L, favors Th2 cell differentiation and IL-4 production [42]. Regarding the ability of an inhibitor molecule to modulate CD4+ T-cell function, the respective cell must express the correct receptor and the chromatin structure must allow for transcriptional regulation [43]. While the B7-H4 receptor is still unknown, our finding of a time-dependent ability of B7-H4Ig to modulate Th1 cell and Th17 cell differentiation in vitro is likely associated with transient B7-H4 receptor upregulation. Our data also show a time-dependent increase in B7-H4Ig binding to activated CD4+ T-cells within the draining LNs of PLP139–151/CFA primed mice. Furthermore, correlating with the time-dependence of B7-H4 receptor expression, is the time-dependency of the transcriptome related to differentiation of effector CD4+ Th1 and Th17 effector cells (V.K. Kuchroo, personal communication).
Our in vitro studies clearly show that mB7-H4Ig has a direct inhibitory effect on the differentiation of both Th1 and Th17 cells in the absence of nTregs, but did not detect any appreciable effect on differentiation of naïve mouse CD4+ T-cells toward the Th2 phenotype. Similarly, treatment of mice with mB7-H4Ig decreased both the level of cellular proliferation and inflammatory cytokine, i.e., IFN-γ and IL-17, by splenocytes reactivated ex vivo. Functionally, the decrease in the level of IL-17 produced by splenocytes reactivated with of PLP178–191 peptide supports that hypothesis that mB7-H4Ig decreases the level of epitope spreading that occurs following treatment. However, the data also show that the level of IL-17 secreted by the splenocytes isolated from mB7-H4Ig treated mice activated ex vivo with PLP139–151 peptide does not achieve that same level of significance when compared to the decrease in the PLP178–191 response. Potential explanations for this finding include the fact that B7-H4Ig treatment was initiated following PLP139–151 priming and that the bolus of PLP139–151/CFA present at the site of injection used to induce R-EAE is still present allowing for continued activation of PLP139–151-specific CD4+ T cells, which would not occur with the PLP178–191-specific CD4+ T cell population which is activated in response to small amounts of endogenously released myelin epitopes.
Significant to its translation to the clinic for autoimmune therapy, hB7-H4Ig similarly inhibited differentiation of naïve human CD4+ T-cells toward Th1 and Th17 phenotypes while concomitantly inducing IL-10 production perhaps suggesting induction of the recently-described regulatory Th17 phenotype [44]. There was also a trend for increased IL-10 production in iTreg-promoting conditions, and in contrast to the mouse data, hB7-H4Ig also induced increased production of IL-4 and IL-5 when human naïve CD4+ T-cells were activated in Th2-promoting conditions. Additionally, B7-H4Ig treatment enhanced IL-5 production by human naïve CD4+ T-cells activated in iTreg-promoting conditions. The later effect may be a consequence of species differences between mouse and human CD4+ T cells and the identity of the B7-H4 receptor(s), or former FoxP3 expressing cells that have begun to produce Th2-type of cytokines [45]. FoxP3 has been shown to repress inflammatory CD4+ T-cell-associated genes, while concurrently inducing the expression of genes associated with Treg function [46,47]. However, the ability to stably induce a Treg phenotype is dependent upon the level and stability of FoxP3 expression [48]. Human Tregs purified based on expression of CD4+CD25highCD127neg have been shown to be heterogeneous regarding their ability to maintain FoxP3 expression following in vitro culture [49]. Loss of FoxP3 expression is associated with a CD45RA− phenotype, while CD45RA+ Tregs maintain FoxP3 expression [50]. It has been recently shown that the CD4+CD25highCD127negCD45RA− Tregs that lose FoxP3 following culture upregulate Th2 cell-associated genes as well as secrete IL-4, IL-5, and IL-13, while secreting minimal levels of Th1 cell and Th17 cytokines [45]. To reconcile our present data with the above findings regarding FoxP3 expression stability, ongoing experiments are determining if B7-H4Ig treatment is able to induce a transient low level of FoxP3 expression when naïve CD4+ T-cells are active in the presence of Th2-promoting conditions.
While our results show that both mouse and human B7-H4Ig possess similar biological activity in EAE, we are concentrating on the use of hB7-H4Ig for potential clinical use. Several important points need to be considered in this context. First, the conformation of the Ig fusion protein needs to be assessed, i.e., whether the desired functional outcome may be increased by the utilization of monomeric, dimeric, or multimeric forms of the fusion protein. Secondly, the role of the fusion protein as an agonist versus an antagonist, for example is the functional activity of the fusion protein inhibited, unaffected, or enhanced by the its binding to host Fc receptors. Lastly, the serum half-life of the respective protein and the longevity of the treatment effect must be considered to optimize its desired efficacy while limiting undesired side effects. In conclusion, signaling via the B7-H4 receptor using B7-H4Ig down-regulates differentiation of mouse (and human) inflammatory Th1/17 cell subsets providing an effective therapy for pre-established EAE which may be highly relevant to the immunoregulation of the pathogenesis MS and other autoimmune diseases. Regulation is mediated by both a direct-cell-intrinsic pathway as well as by Treg-mediated indirect regulation of pro-inflammatory Th1/17 cells activity. Critically, our results show that B7-H4Ig therapy increases both the number and the functional capacity of Tregs and most importantly show the functional relevance of Treg induction to long-lived regulation the progression of relapsing and chronic EAE models.
Acknowledgments
We thank Dr. Daniel Getts for reviewing the manuscript.
Abbreviations
- B7-H4Ig
B7-H4-Immumoglobulin fusion protein
- hB7-H4Ig
human B7-H4Ig
- mB7-H4Ig
mouse B7-H4Ig
- C-EAE
chronic experimental autoimmune encephalomyelitis
- CNS
central nervous system
- CTLA-4
cytotoxic T lymphocyte associated antigen-4
- MBP
myelin basic protein
- MS
multiple sclerosis
- OVA
ovalbumin
- PLP
myelin proteolipid protein
- R-EAE
relapsing experimental autoimmune encephalomyelitis
- T1D
Type I diabetes
- TCR
T cell receptor
- CFA
complete Freund’s adjuvant
Footnotes
This work was supported by grants from the National Multiple Sclerosis Society Fast Forward Program and from Amplimmune, Inc.
References
- 1.Podojil JR, Miller SD. Molecular mechanisms of T-cell receptor and costimulatory molecule ligation/blockade in autoimmune disease therapy. Immunol Rev. 2009;229:337–55. doi: 10.1111/j.1600-065X.2009.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun J, Link H, Olsson T, Xiao BG, Andersson G, Ekre HP, et al. T and B cell responses to myelin-oligodendrocyte glycoprotein in multiple sclerosis. J Immunol. 1991;146:1490–5. [PubMed] [Google Scholar]
- 3.Zhang Y, Burger D, Saruhan G, Jeannet M, Steck AJ. The T-lymphocyte response against myelin-associated glycoprotein and myelin basic protein in patients with multiple sclerosis. Neurology. 1993;43:403–7. doi: 10.1212/wnl.43.2.403. [DOI] [PubMed] [Google Scholar]
- 4.Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. 2005;23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707. [DOI] [PubMed] [Google Scholar]
- 5.Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172:3893–904. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- 6.Lafferty KJ, Cunningham AJ. A new analysis of allogeneic interactions. Aust J Exp Biol Med Sci. 1975;53:27–42. doi: 10.1038/icb.1975.3. [DOI] [PubMed] [Google Scholar]
- 7.Miller SD, Vanderlugt CL, Lenschow DJ, Pope JG, Karandikar NJ, Dal Canto MC, et al. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–45. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 8.Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184:783–8. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karandikar NJ, Vanderlugt CL, Eagar TN, Tan L, Bluestone JA, Miller SD. Tissue-specific up-regulation of B7-1 expression and function during the course of murine relapsing experimental autoimmune encephalomyelitis. J Immunol. 1998;161:192–9. [PubMed] [Google Scholar]
- 10.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Garra A, Steinman L, Gijbels K. CD4+ T-cell subsets in autoimmunity. Curr Opin Immunol. 1997;9:872–83. doi: 10.1016/s0952-7915(97)80192-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Izikson L, Liu L, Weiner HL. Activation of CD25(+)CD4(+) regulatory T cells by oral antigen administration. J Immunol. 2001;167:4245–53. doi: 10.4049/jimmunol.167.8.4245. [DOI] [PubMed] [Google Scholar]
- 13.Zheng SG, Gray JD, Ohtsuka K, Yamagiwa S, Horwitz DA. Generation ex vivo of TGF-beta-producing regulatory T cells from CD4+CD25-precursors. J Immunol. 2002;169:4183–9. doi: 10.4049/jimmunol.169.8.4183. [DOI] [PubMed] [Google Scholar]
- 14.Fuss IJ, Boirivant M, Lacy B, Strober W. The interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J Immunol. 2002;168:900–8. doi: 10.4049/jimmunol.168.2.900. [DOI] [PubMed] [Google Scholar]
- 15.Pop SM, Wong CP, Culton DA, Clarke SH, Tisch R. Single cell analysis shows decreasing FoxP3 and TGFbeta1 coexpressing CD4+CD25+ regulatory T cells during autoimmune diabetes. J Exp Med. 2005;201:1333–46. doi: 10.1084/jem.20042398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 17.Zheng SG, Wang J, Wang P, Gray JD, Horwitz DA. IL-2 is essential for TGF-beta to convert naive CD4+CD25-cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J Immunol. 2007;178:2018–27. doi: 10.4049/jimmunol.178.4.2018. [DOI] [PubMed] [Google Scholar]
- 18.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–89. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Schreiner B, Bailey SL, Shin T, Chen L, Miller SD. PD-1 ligands expressed on myeloid-derived APC in the CNS regulate T-cell responses in EAE. Eur J Immunol. 2008:2706–17. doi: 10.1002/eji.200838137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi IH, Zhu G, Sica GL, Strome SE, Cheville JC, Lau JS, et al. Genomic organization and expression analysis of B7-H4, an immune inhibitory molecule of the B7 family. J Immunol. 2003;171:4650–4. doi: 10.4049/jimmunol.171.9.4650. [DOI] [PubMed] [Google Scholar]
- 21.Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–61. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 22.Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–73. doi: 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 23.Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A. 2003;100:10388–92. doi: 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichikawa M, Chen L. Role of B7-H1 and B7-H4 molecules in down-regulating effector phase of T-cell immunity: novel cancer escaping mechanisms. Front Biosci. 2005;10:2856–60. doi: 10.2741/1742. [DOI] [PubMed] [Google Scholar]
- 25.Simon I, Zhuo S, Corral L, Diamandis EP, Sarno MJ, Wolfert RL, et al. B7-h4 is a novel membrane-bound protein and a candidate serum and tissue biomarker for ovarian cancer. Cancer Res. 2006;66:1570–5. doi: 10.1158/0008-5472.CAN-04-3550. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Hao J, Metzger DL, Mui A, Ao Z, Akhoundsadegh N, et al. Early treatment of NOD mice with B7-H4 reduces the incidence of autoimmune diabetes. Diabetes. 2011;60:3246–55. doi: 10.2337/db11-0375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Hao J, Metzger DL, Mui A, Ao Z, Verchere CB, et al. B7-H4 induces donor-specific tolerance in mouse islet allografts. Cell Transplant. 2012;21:99–111. doi: 10.3727/096368911X582750. [DOI] [PubMed] [Google Scholar]
- 28.Azuma T, Zhu G, Xu H, Rietz AC, Drake CG, Matteson EL, et al. Potential role of decoy B7-H4 in the pathogenesis of rheumatoid arthritis: a mouse model informed by clinical data. PLoS Med. 2009;6:e1000166. doi: 10.1371/journal.pmed.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Getts DR, Turley DM, Smith CE, Harp CT, Feeney EM, Teague-Getts M, et al. Tolerance induced by apoptotic antigen-coupled leukocytes depends on PD-L1+, IL-10-producing splenic macrophages and induced Tregs. J Immunol. 2011;187:2405–17. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–9. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- 31.Thornton AM, Korty PE, Tran DQ, Wohlfert EA, Murray PE, Belkaid Y, et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J Immunol. 2010;184:3433–41. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akimova T, Beier UH, Wang L, Levine MH, Hancock WW. Helios expression is a marker of T cell activation and proliferation. PLoS One. 2011;6:e24226. doi: 10.1371/journal.pone.0024226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kohm AP, McMahon JS, Podojil JR, Smith Begolka W, DeGutes M, Kasprowicz DJ, et al. Cutting edge: anti-CD25 mAb injection results in the functional inactivation, not depletion of CD4+CD25+ Treg cells. J Immunol. 2006;176:3301–5. doi: 10.4049/jimmunol.176.6.3301. [DOI] [PubMed] [Google Scholar]
- 34.Paust S, Lu LR, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci U S A. 2004;101:10398–403. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei J, Loke P, Zang X, Allison JP. Tissue-specific expression of B7x protects from CD4 T cell-mediated autoimmunity. J Exp Med. 2011;208:1683–94. doi: 10.1084/jem.20100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mikita J, Dubourdieu-Cassagno N, Deloire MS, Vekris A, Biran M, Raffard G, et al. Altered M1/M2 activation patterns of monocytes in severe relapsing experimental rat model of multiple sclerosis. Amelioration of clinical status by M2 activated monocyte administration. Mult Scler. 2011;17:2–15. doi: 10.1177/1352458510379243. [DOI] [PubMed] [Google Scholar]
- 37.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao Q, Wang Y, Zheng D, Sun Y, Wang Y, Lee VW, et al. IL-10/TGF-beta-modified macrophages induce regulatory T cells and protect against adriamycin nephrosis. J Am Soc Nephrol. 2010;21:933–42. doi: 10.1681/ASN.2009060592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Korn T, Reddy J, Gao W, Bettelli E, Awasthi A, Petersen TR, et al. Myelin-specific regulatory T cells accumulate in the CNS but fail to control autoimmune inflammation. Nat Med. 2007;13:423–31. doi: 10.1038/nm1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 41.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol. 2000;165:5035–40. doi: 10.4049/jimmunol.165.9.5035. [DOI] [PubMed] [Google Scholar]
- 43.Avni O, Lee D, Macian F, Szabo SJ, Glimcher LH, Rao A. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol. 2002;3:643–51. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 44.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, et al. Pathogen-induced human TH17 cells produce IFN-gamma or IL-10 and are regulated by IL-1beta. Nature. 2012;484:514–8. doi: 10.1038/nature10957. [DOI] [PubMed] [Google Scholar]
- 45.Hansmann L, Schmidl C, Kett J, Steger L, Andreesen R, Hoffmann P, et al. Dominant Th2 differentiation of human regulatory T cells upon loss of FOXP3 expression. J Immunol. 2012;188:1275–82. doi: 10.4049/jimmunol.1102288. [DOI] [PubMed] [Google Scholar]
- 46.Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, et al. Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity. 2007;27:786–800. doi: 10.1016/j.immuni.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 47.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-dependent programme of regulatory T-cell differentiation. Nature. 2007;445:771–5. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 48.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–70. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 49.Edinger M. Regulatory T cells for the prevention of graft-versus-host disease: professionals defeat amateurs. Eur J Immunol. 2009;39:2966–8. doi: 10.1002/eji.200940030. [DOI] [PubMed] [Google Scholar]
- 50.Hoffmann P, Eder R, Boeld TJ, Doser K, Piseshka B, Andreesen R, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108:4260–7. doi: 10.1182/blood-2006-06-027409. [DOI] [PubMed] [Google Scholar]