

Abstract
Inflammatory monocyte-derived effector cells play an important role in the pathogenesis of numerous inflammatory diseases. However, no treatment option exists that is capable of modulating these cells specifically. We show that infused negatively charged, immune-modifying microparticles (IMPs), derived from polystyrene, microdiamonds, or biodegradable poly(lactic-co-glycolic) acid, were taken up by inflammatory monocytes, in an opsonin-independent fashion, via the macrophage receptor with collagenous structure (MARCO). Subsequently, these monocytes no longer trafficked to sites of inflammation; rather, IMP infusion caused their sequestration in the spleen through apoptotic cell clearance mechanisms and, ultimately, caspase-3–mediated apoptosis. Administration of IMPs in mouse models of myocardial infarction, experimental autoimmune encephalomyelitis, dextran sodium sulfate–induced colitis, thioglycollate-induced peritonitis, and lethal flavivirus encephalitis markedly reduced monocyte accumulation at inflammatory foci, reduced disease symptoms, and promoted tissue repair. Together, these data highlight the intricate interplay between scavenger receptors, the spleen, and inflammatory monocyte function and support the translation of IMPs for therapeutic use in diseases caused or potentiated by inflammatory monocytes.
INTRODUCTION
Inflammatory monocytes (ΦIM) play an important role in a wide range of immune-mediated diseases, including autoimmune disorders, immunopathological viral infections, and ischemia-reperfusion injury (1-11). In both humans and mice, ΦIM express chemokine receptor 2 (CCR2), the chemokine ligand 2 (CCL2) receptor principally responsible for their recruitment to inflammatory sites (12-14). CCR2+ monocyte-derived macrophages accumulate and participate in the immune pathology observed in a number of diseases including human rheumatoid arthritis, HIV encephalitis, chronic liver disease, amyotrophic lateral sclerosis, atherosclerosis, and cardiac infarction (8, 12, 14-16). Upon entry into inflamed tissues, ΦIM differentiate into tissue macrophages or dendritic cells (DCs) that may secrete numerous proinflammatory cytokines, proteases, and other mediators including nitric oxide, culminating in tissue damage, scarring, and even death (2, 4-6, 10, 11, 17). To date, there is no safe or effective therapy for specifically targeting ΦIM, with physicians relying on broad-acting steroids, nonsteroidal anti-inflammatory agents, or antibodies that briefly neutralize components of the ΦIM response. Experimentally, CCR2 small interfering RNAs (siRNAs) delivered in liposomes can reduce ΦIM trafficking in mouse models of atherosclerosis (18). However, siRNA technology has proven highly complex, with multiple targeting and toxicity issues culminating in the termination of several therapeutic siRNA programs (19). In addition, a number of CCL2 antagonists and antibodies have also been tested with mixed results (20). Mixed outcomes are likely the result of ubiquitous CCR2 expression by B cells, T cells, and numerous human monocyte subsets including myeloid suppressor cells (21, 22). Together, these findings highlight the need for safer and more targeted therapies capable of modulating monocytes.
The therapeutic accessibility of ΦIM in the bloodstream, their inherent propensity to engulf particulate material, and our experience from cell trafficking experimentation (2) suggested that microparticle-based therapeutics might provide a readily translatable solution. To date, most micromedical approaches have focused on imaging as well as drug delivery and targeting, with an emphasis on developing methods to avoid immune-mediated clearance (23). Little investigation into how microparticle biophysical surface properties may be harnessed for therapeutic use has been conducted. Here, we show that highly negatively charged immune-modifying microparticles (IMPs) bind to circulating ΦIM via a scavenger receptor, the macrophage receptor with a collagenous structure (MARCO). This results in the induction of apoptosis and sequestration of ΦIM in the spleen, culminating in reduced immune pathology at peripheral sites of inflammation. Finally, IMPs derived from poly(lactic-co-glycolic acid) (PLGA-IMP) show excellent efficacy. Given that PLGA-IMP is a commonly used biodegradable clinical polymer, IMPs composed of this polymer may be a new tool for inhibiting ΦIM in numerous inflammatory disease indications.
RESULTS
IMPs reduce inflammatory monocyte immigration and increase survival in a mouse model of West Nile virus encephalitis
Previously, we have shown that morbidity associated with West Nile virus (WNV) brain infection is the direct result of ΦIM trafficking into the brain (2, 11, 24). In this model, weight loss of ≥5% is a biomarker that correlates with ΦIM trafficking into the central nervous system (CNS) and subsequent lethality (2, 11). At least 50% of infected animals will lose ≥5% weight, and of these, 100% will succumb to infection within 72 hours of initial weight loss. Animals that do not lose weight do not show any other symptoms of encephalitis but will develop lifelong sterilizing immunity (Fig. 1A). To monitor monocyte trafficking instead of infusing neutral polystyrene microparticles (PS-NPs) at initial weight loss (day 6), we unintentionally used 500-nm-diameter, negatively charged, carboxylated polystyrene particles (PS-IMPs). Surprisingly, PS-IMP infusion almost immediately reduced symptoms such as ruffled fur, malaise, and seizures, uniformly associated with clinical WNV infection of the CNS (24). This reduction correlated with a reduction in CNS and systemic proinflammatory cytokines and chemokines (fig. S1). Furthermore, daily infusion of PS-IMPs for up to 5 days resulted in 60% survival of mice that would otherwise have succumbed to infection (Fig. 1A). In these mice, body weight typically stabilized and returned to control levels within 5 to 6 days. PS-NP– or vehicle-treated mice showed continued weight loss, with ≤10% survival. Rechallenge with a lethal virus dose, 30 days after recovery, indicated that survivors had developed sterilizing immunity. Consistent with our previous observations (2, 11, 24), survival correlated with a significant reduction in ΦIM-derived macrophages (SSClo/FSClo/CD11b+/CD45hi/Ly6C+/hi) in the brains of PS-IMP–treated mice (P < 0.001) compared to phosphate-buffered saline (PBS; vehicle)–or PS-NP–treated control animals (Fig. 1B).
Fig. 1. PS-IMPs ameliorate WNV encephalitis in mice.
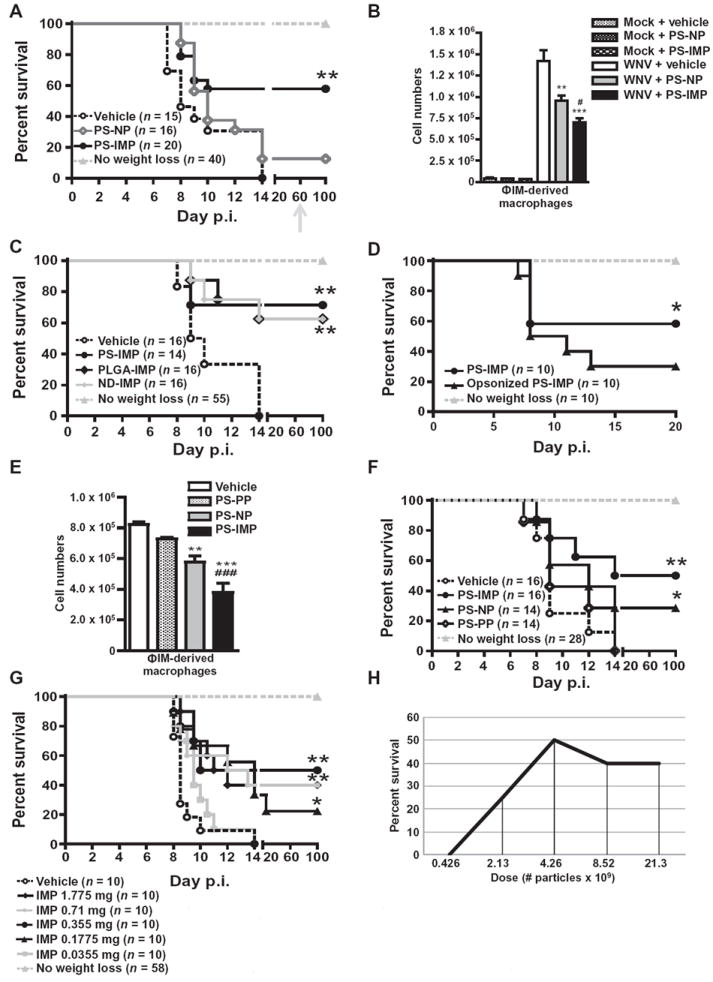
(A) Survival of WNV-infected mice treated with PS-IMPs, PS-NPs (neutral particle control), or vehicle control. Surviving mice were rechallenged with lethal 3 × 108 plaque-forming units (PFU) of WNV on day 60 (gray arrow). (B) Numbers of ΦIM-derived macrophages (SSClo/FSClo/CD45hi/CD11b+/Ly6C+/hi) in the brains of PS-IMP, PS-NP, or vehicle control–treated mock or WNV-infected mice 24 hours after therapy (day 7 after infection). (C) Survival of WNV-infected mice treated with PLGA-IMP, PS-NP, ND-IMP, or vehicle. (D) Survival of WNV-infected mice treated with PS-IMP, opsonized PS-IMP, or vehicle control. (E) Numbers of ΦIM-derived macrophages in the brains of PS-IMP, PS-NP, PS-PP (positively charged particles), or vehicle control–treated or WNV-infected mice 24 hours after therapy (day 7 after infection). (F) Survival of WNV-infected mice treated with PS-IMP, PS-NP, PS-PP, or vehicle control. (G and H) Survival of WNV-infected mice treated with increasing doses of PS-IMP or vehicle control (G) per dose (mg) or (H) as particle number. Survival data represent three separate experiments with 10 to 20 mice per group. Statistical analysis was conducted with the Mantel-Haenszel log-rank test. Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Statistical analysis was conducted with one-way analysis of variance (ANOVA) and Tukey-Kramer post-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing PS-IMP and NP groups to vehicle control groups; #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.001, comparing PS-IMP to NP groups.
IMPs must be negatively charged
To determine the critical physical characteristics of IMPs for mediating this protective effect, we infused 500-nm carboxylated microdiamond IMPs (ND-IMPs) or carboxylated poly(lactic-co-glycolic acid) IMPs (PLGA-IMPs) (table S1) using the same administration criteria as for PS-IMPs. Survival statistics for PLGA-IMPs were similar to those seen in animals treated with PS-IMPs (Fig. 1C), indicating that the particle core is independent of the property responsible for the therapeutic effect of IMPs. Because opsonization is purported to be a potential mechanism by which circulating microparticles are cleared (23), we determined the plasma proteins that bind to PS-IMPs by mass spectrometry. Of the 114 proteins found to associate with IMPs, 96 were from the normal plasma proteome, including opsonins known to enhance particle uptake, such as complement and vitronectin (25-27) (table S2). However, other studies have shown that uptake of negative particles, like IMPs, may occur in anopsonin-independent fashion (28, 29). Incubating particles with mouse plasma to preopsonize IMPs reduced the therapeutic efficacy, compared to non-opsonized particles in WNV-infected animals, which tended toward significance (P = 0.06) (Fig. 1D).
Initial pilot studies used non-opsonized IMPs with a ζ potential of less than −50 mV (table S1). We compared PS microparticles with ζ potentials of −50 mV, ~−0.5 [neutral; polystyrene neutral particles (PS-NPs)], or +40 mV [aminated particles; polystyrene-positive particles (PS-PPs)]. PS-IMPs (−50 mV) caused the greatest reduction in ΦIM trafficking to the brain (Fig. 1E). In contrast, PS-PPs did not decrease infiltration of inflammatory monocyte-derived macrophages (Fig. 1E), nor did they increase survival (Fig. 1F). Furthermore, IMPs coated with mouse plasma had a reduced negative charge, with the ζ potential changing from −50 mV (non-opsonized IMPs) to −16 mV (opsonized IMPs), correlating with reduced therapeutic efficacy (Fig. 1D). Together, the data suggest that negative charge plays a role in IMP therapeutic efficacy. The dose response for 500-nm-diameter PS-IMPs in mice with WNV encephalitis showed the most effective dose to be ~4 × 109 IMP particles or 0.355 mg of particles per mouse (Fig. 1, G and H). The finding that doses higher than 0.355 mg have no added advantage suggests that potential saturation occurs at this dose. In the context of a potential human equivalent dose, using a body surface area equation [as defined in (30)], this represents a dose of 1.4 mg/kg or ~107 mg per 75-kg patient.
IMP treatment results in reduced ΦIM in the brain and increased ΦIM in the spleen
After infusion, fluorescein isothiocyanate (FITC)–labeled PS-IMPs (FITC–PS-IMPs) were predominantly localized with Ly6Chi monocytes and disappeared from the blood within 24 hours (fig. S2, A and B). In the context of organ distribution, FITC-labeled PS-IMPs predominantly localized to the lungs, spleen, and liver (Fig. 2A), with no FITC+ particles in the brain (fig. S2, C and D). In the spleen, PLGA-IMPs were almost undetectable by fluorescence histology 1 hour after infusion (Fig. 2B). Flow cytometry revealed some IMP+ Ly6G+ neutrophils, CD11c+ DCs, CD3+ T cells, NK1.1+ natural killer (NK) cells, and B cells (Fig. 2C). However, IMP+ Ly6C+ ΦIM were between two- and fourfold increased in numbers relative to any other population in WNV-infected mice, suggesting that these cells are the primary target of IMPs (Fig. 2C; P < 0.05). Furthermore, spleens from infected mice treated with FITC–PS-IMPs had significantly more Ly6Chi ΦIM than did those treated with PS-NPs or vehicle control (Fig. 2, C to F; P < 0.001), closely corresponding to a decrease in circulating ΦIM in the peripheral blood in these mice (Fig. 2G). As we have previously shown that morbidity in WNV encephalitis is caused by ΦIM (2, 11), we hypothesized that IMPs mediate their therapeutic activity by binding to ΦIM, abrogating their migration to the inflamed brain, and instead diverting them to the spleen. To confirm this, we sorted Ly6Chi ΦIM from the bone marrow of WNV-infected mice on day 6 after infection, labeled them with membrane-inserting dye (PKH26), and transferred them intravenously into mock- or WNV-infected recipients on day 6 after infection (Fig. 3A). This was followed immediately by separate intravenous infusion of PS-IMPs, PS-NPs, or vehicle only. As previously described (2), in vehicle-treated mice, adoptively transferred PKH26-labeled Ly6Chi ΦIM migrated into the WNV-infected brain, differentiating into macrophages (Fig. 3B). This migration was significantly reduced by PS-IMP treatment (Fig. 3, B and D; P < 0.001), with both fewer adoptively transferred PKH26+ cells and host ΦIM migrating into the WNV-infected brain (Fig. 3, B and D; P < 0.001). The reduced trafficking of adoptively transferred cells into the infected brain by PS-IMP treatment correlated with a significant increase in PKH26-labeled ΦIM in the spleen (Fig. 3, C and E). Furthermore, greater than 30% of these adoptively transferred cells had taken up FITC+ PS-IMPs (Fig. 3C). Together, the data clearly show that in the absence of IMPs, adoptively transferred ΦIM migrate to the WNV-infected brain, as we have previously described (2). The introduction of IMPs to the circulation prevents this migration, instead resulting in accumulation of adoptively transferred ΦIM in the spleen.
Fig. 2. PS-IMP treatment redirects inflammatory monocytes to mouse spleen.
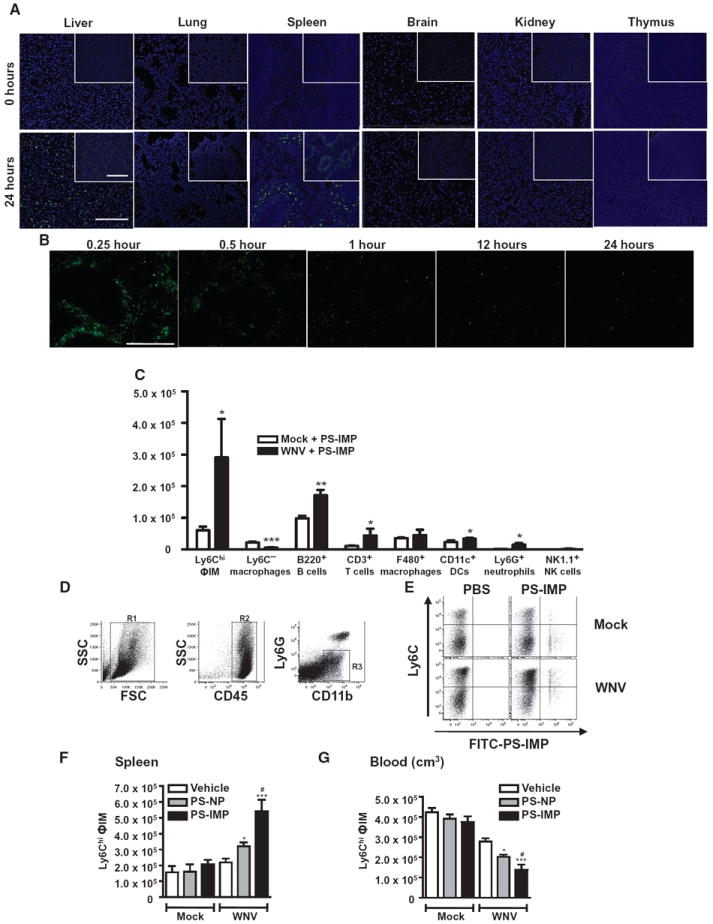
(A and B) Distribution of FITC–PS-IMPs (green) in the brain, kidney, lung, thymus, spleen, and liver. (B) After intravenous injection of FITC–PLGA-IMPs (green), mouse spleens were processed for fluorescent immunohistology at the time points indicated. (C) Mock- and WNV-infected mice were injected with FITC–PS-IMPs intravenously on day 6 after infection, and spleens were processed for flow cytometry on day 7 after infection to determine localization of PS-IMPs in CD11b+ Ly6C+ inflammatory monocytes (ΦIM), Ly6C− macrophages, ΦIM, B220+ B cells, CD3+ T cells, F4/80+ macrophages, CD11c+ DCs, Ly6G+ neutrophils, and NK1.1+ NK cells. (D to G) With flow cytometry, CD45+ Ly6G− Ly6Chi monocytes were gated progressively and enumerated in the spleens (F) and blood (G) of mock- and WNV-infected mice 24 hours after FITC–PS-IMP infusion. Immunohistochemistry data represent three separate experiments with three mice per group. Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Statistical analysis was conducted with one-way ANOVA and Tukey-Kramer post-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing PS-IMP and NP to vehicle control groups; #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.001, comparing PS-IMP to NP groups. (A and B) Scale bars, 200 μm (main photomicrograph) and 50 μm (inset).
Fig. 3. PS-IMP treatment reduces migration of inflammatory monocytes into the mouse CNS.
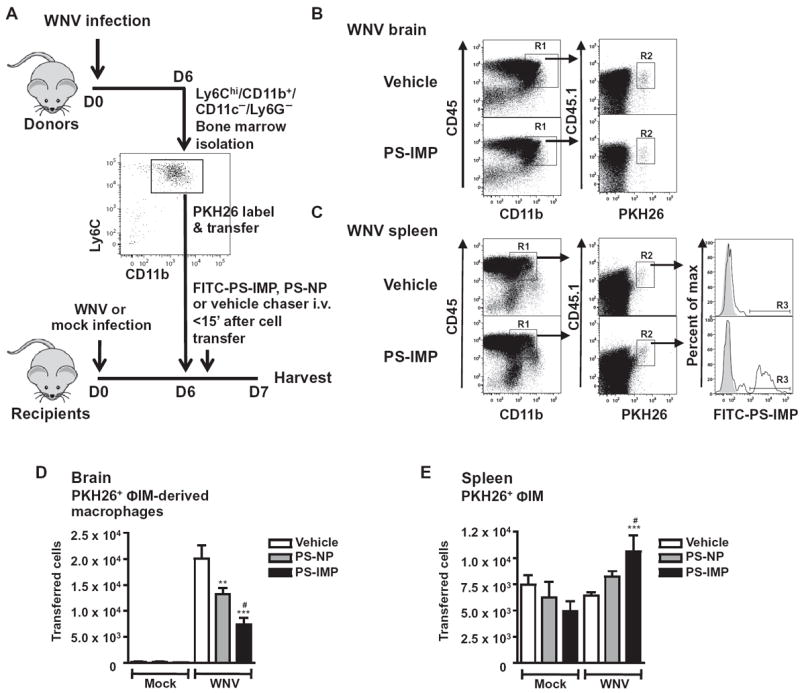
(A) SSClo/FSClo/CD11b+/CD11c−/Ly6Chi inflammatory monocytes (ΦIM) were isolated from the bone marrow of mock- and WNV-infected mouse donors on day 6 after infection, labeled with PKH26 fluorescent dye, and injected into matched recipients on day 6 after infection, immediately followed by a separate injection of vehicle, FITC–PS-NP, or FITC–PS-IMP. (B to E) Brains (B and D) and spleens (C and E) were processed on day 7 after infection for flow cytometry. (B and D) SSClo/FSClo/CD45hi/CD11b+ macrophages (R1) and adoptively transferred PKH26 (R2) populations from brain tissue were progressively gated (B), and transferred cells were enumerated (D). In the spleen (C and E), CD11c−/CD45hi/CD11b+ macrophages (R1), adoptively transferred PKH26 (R2), and IMP+ macrophage populations (R3) were progressively gated for examination (B), with transferred cells enumerated in (E). Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Statistical analysis was conducted with one-way ANOVA and Tukey-Kramer post-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing PS-IMP and NP to vehicle control groups; #P ≤ 0.05, ##P ≤ 0.01, ###P ≤ 0.001, comparing PS-IMP and NP groups.
IMPs inhibit peritoneal inflammation
Viral infection results in a complex array of immune responses involving numerous immune elements. To address the specificity of IMPs in a nonviral setting, we used a sodium thioglycollate (TG) peritoneal inflammation mouse model (Fig. 4A). Immigration of leukocytes into the TG-inflamed peritoneum follows a stereotypical pattern, with neutrophils attracted within the first 4 to 18 hours, followed by the CCR2- dependent accumulation of ΦIM-derived macrophages from about 12 hours onward (31). Infusion of either PS-IMPs (Fig. 4B) or PLGA-IMPs, 24 hours after intraperitoneal TG injection, significantly reduced ΦIM trafficking into the site of inflammation (P < 0.001; Fig. 4B and fig. S2, E and F). Similar to observations in WNV-infected animals, IMP treatment correlated with accumulation of ΦIM in the spleen of animals suffering TG-induced peritonitis (Fig. 4G). The role of the spleen in IMP efficacy was addressed in splenectomized mice. Surprisingly, removal of the spleen resulted in complete abrogation of IMP efficacy in both WNV encephalitis and TG peritonitis mouse models, as determined by the similar number of ΦIM at the site of inflammation in splenectomized IMP-treated mice and splenectomized untreated control mice (Fig. 4, C and D).
Fig. 4. The scavenger receptor MARCO mediates IMP activity.
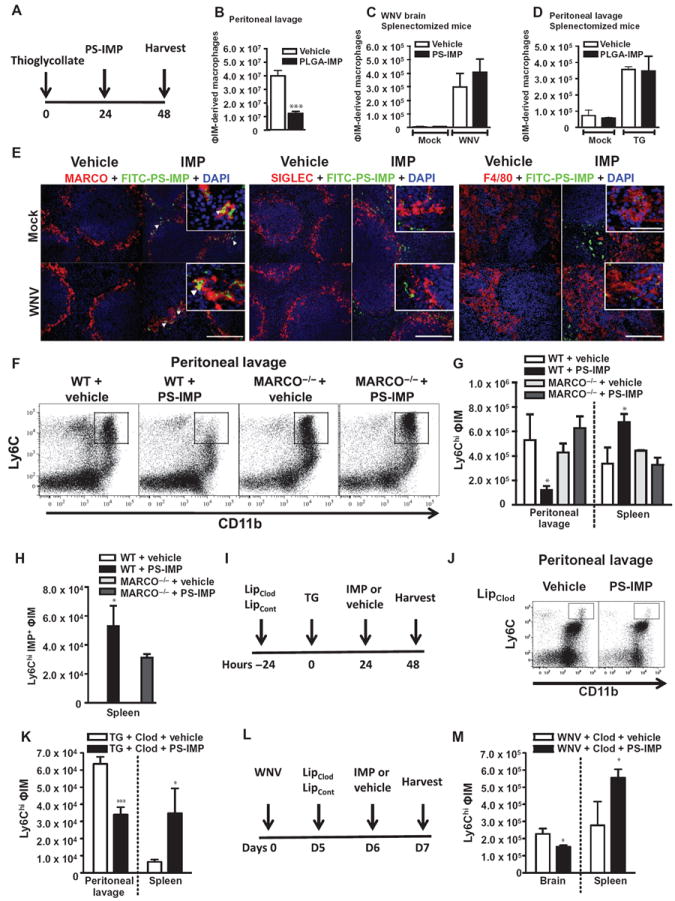
(A and B) PLGA-IMP or vehicle (A) was injected 24 hours after TG injection, with the number of peritoneal CD11b+/Ly6Chi/Ly6G− inflammatory monocytes (ΦIM) enumerated by flow cytometry (B). (C and D) The importance of the spleen for IMP efficacy was addressed in splenectomized mice, with ΦIM numbers in the brains of WNV-infected mice (C) or the peritoneal cavity (D) of mice with TG-induced peritonitis compared between vehicle and IMP treatment. (E) FITC–PS-IMPs (green) or vehicle control were infused on day 6 after WNV or mock infection and processed 24 hours later for expression of MARCO, SIGLEC, or F4/80 immunohistology with 4′,6-diamidino-2-phenylindole (DAPI) (blue) counterstaining. White arrowheads indicate co-localization between MARCO+ cells and FITC–PS-IMP. (F and G) PS-IMP or vehicle was injected intravenously into wild-type (WT) or MARCO−/− mice 24 hours after TG injection, with total peritoneal inflammation examined by flow cytometry (F) and the total number of ΦIM-derived macrophages determined (G). (H) In addition, both total (G) and PS-IMP+ (H) Ly6Chi ΦIM macrophages were enumerated in the spleen. (I to M) Deletion of marginal zone macrophages was performed with clodronate liposomes, and IMP efficacy in mice with TG-induced peritonitis (I to K) or WNV encephalitis (L and M) was examined. In TG-treated animals (I), ΦIM-derived macrophages in the peritoneal cavity and spleen were enumerated (J and K) by flow cytometry. In WNV-infected mice (L), Ly6Chi ΦIM-derived macrophages in the brain and spleen were examined by flow cytometry on day 7 after infection (M). Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Statistical analysis was conducted with one-way ANOVA and Tukey-Kramer post-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing the WT PS-IMP groups to all other groups. Immunohistochemistry data represent three separate experiments with three mice per group. (E) Scale bars, 200 μm (main photomicrograph) and 50 μm (inset).
IMP efficacy is dependent on MARCO expression by ΦIM
It has been shown that the spleen is an important organ for the removal of systemic debris, microparticles, and apoptotic cells via a scavenger receptor expressed by DCs, F4/80+ red pulp macrophages, sialic acid–binding immunoglobulin-type lectin-expressing (SIGLEC+) metallophilic macrophages, and MARCO+ marginal zone macrophages (32-35). With FITC–PS-IMPs, few IMPs were found to colocalize with CD11c+ DCs (Fig. 1C and fig. S3A), F4/80-expressing red pulp macrophages, or SIGLEC-expressing metallophilic macrophages (Fig. 4E). Instead, colocalization was found predominantly to occur with cell populations that expressed MARCO (indicated by the yellow color when green IMP staining and red MARCO staining are overlaid; Fig. 4E). MARCO has been implicated in the binding of negatively charged microparticles and polystyrene nanoparticles to cells expressing this scavenger receptor (29, 35). Using the TG peritonitis model and MARCO-deficient mice (MARCO−/−), we further examined the role of this scavenger receptor. Intraperitoneal TG injection resulted in the accumulation of similar numbers of peritoneal macrophages in wild-type and MARCO−/− mice at 48 hours after injection, suggesting a normal response to peritoneal inflammation in MARCO−/− mice (Fig. 4, F and G). However, IMP treatment abrogated the influx of Ly6Chi/CD11b+ ΦIM into the peritoneum of wild-type, but not MARCO−/−, animals (Fig. 4, F and G). Furthermore, IMP uptake by ΦIM was significantly reduced in MARCO−/− mice (Fig. 4H; P < 0.05), as determined by the reduced number of IMP+ ΦIM isolated from the spleens of MARCO−/− mice compared to wild-type controls (Fig. 4H). As expected, no IMP uptake was observed in vehicle control wild-type or MARCO−/− animals (Fig. 4H).
The data implicate MARCO in the uptake and efficacy of IMPs. However, it is important to determine whether ΦIM migrate in response to MARCO+ marginal zone macrophages that have taken up IMPs or because inflammatory monocytes have taken up IMPs in the circulation. To directly address the role of marginal zone macrophages, these cells were deleted using clodronate liposomes (36). With this method, it has been described that marginal zone macrophages and marginal zone metallophilic macrophages can be deleted for up to 6 to 8 days without affecting other splenic macrophage and DC populations (36). We observed rapid deletion within 14 hours that lasted beyond 3 days (fig. S3B). Deletion of marginal zone macrophages using this method did not reduce the capacity of PS-IMPs to inhibit ΦIM migration into the TG-inflamed peritoneum (Fig. 4, I to K). Furthermore, this reduction was observed to correspond with accumulation of ΦIM in the spleen (Fig. 4K). This was further confirmed in the WNV encephalitis model, with IMP treatment in clodronate-mediated marginal zone macrophage–depleted animals resulting in reduced ΦIM in the brain and increased ΦIM in the spleen (Fig. 4, L and M). Together, the data suggest that IMP-induced monocyte accumulation is not a secondary response to IMP uptake by marginal zone macrophages, and are consistent with the possibility that ΦIM take up IMPs in a MARCO-dependent fashion in the vasculature and subsequently traffic to the spleen. The rapid reduction of circulating ΦIM after IMP treatment potentially supports this hypothesis (Fig. 2G and fig. S2A). Further support is provided by the fact that within the peripheral blood compartment, MARCO expression is limited to Ly6Chi/CD11b+/CD11c− ΦIM Ly6Chi ΦIM in WNV-infected mice (Fig. 5, A and B) and the fact that this expression was not perturbed by clodronate liposome treatment (fig. S3C).
Fig. 5. Uptake of PS-IMPs by MARCO+ inflammatory monocytes induces apoptosis.
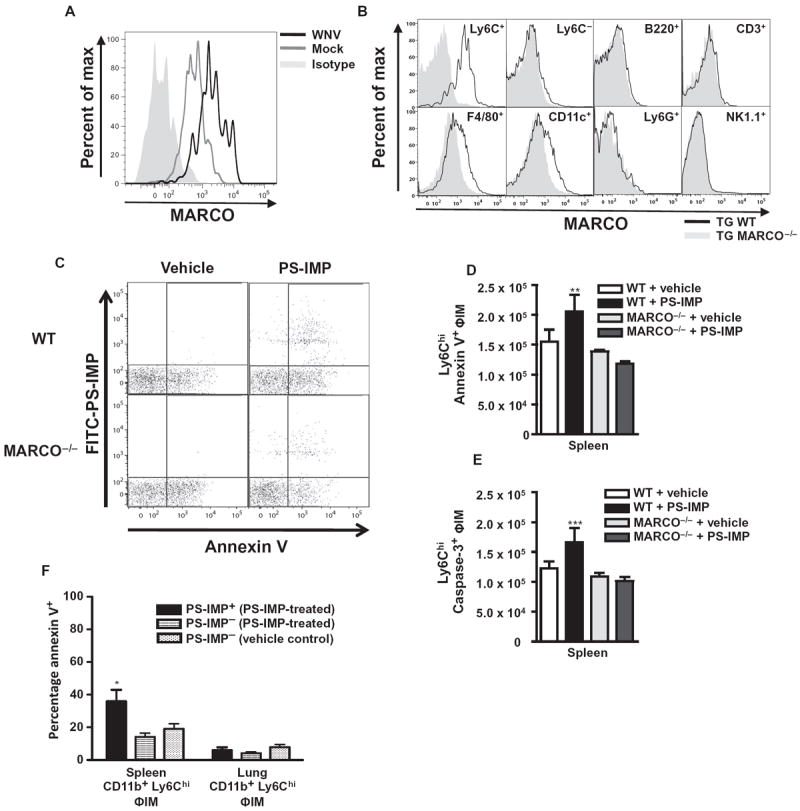
(A and B) MARCO expression on peripheral blood inflammatory monocytes (ΦIM) from WNV-infected animals at day 7 after infection (A), as well as on peripheral blood leukocyte populations in WT or MARCO−/− mice 24 hours after TG injection (B). (C to E) With the TG model of colitis, the numbers of splenic annexin V+ CD11b+ Ly6Chi Ly6G− ΦIM (C and D) and caspase-3+ CD11b+ Ly6Chi Ly6G− ΦIM (E) were examined. (F) In the spleen and lung, the percentage of annexin V+ CD11b+ Ly6Chi Ly6G− ΦIM that were PS-IMP+ or PS-IMP− was compared. Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Statistical analysis was conducted with one-way ANOVA and Tukey-Kramer post-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing the WT PS-IMP groups to all other groups.
ΦIM undergo apoptosis in the spleen
We next investigated the fate of the ΦIM accumulating in the spleen. Scavenger receptor activation can trigger numerous intracellular signaling pathways, including those involved in macrophage cholesterol transport, immune tolerance induction, cellular differentiation, cytokine production, and apoptosis (33, 34, 37, 38). MARCO, for example, plays a direct role in the apoptosis of macrophages after silica particle uptake (28). In wild-type mice, IMP infusion resulted in significantly increased numbers of annexin V+ ΦIM (Fig. 5, C and D; P < 0.005). Annexin V binds to cell surface phosphatidylserine and is a marker of early apoptosis and also serves as a target for receptor-mediated removal of cellular debris from the circulation (39-41). Furthermore, IMP treatment also resulted in increased caspase-3+ ΦIM (Fig. 5E; P < 0.001). Both annexin V+ (P < 0.005) and caspase-3+ (P < 0.001) ΦIM were significantly reduced in IMP-treated MARCO-deficient mice (Fig. 5, D and E). This occurred with IMPs derived from nonbiodegradable polystyrene (Fig. 5, C to E). At the time point examined, apoptosis was predominantly found to be occurring in splenic IMP+ ΦIM (Fig. 5, C to E), with the percentage of IMP− ΦIM from the spleen or control IMP+/− lung macrophages observed to be much lower (Fig. 5F). Together, these results show that IMPs induce apoptosis in ΦIM, potentially implicating a phosphatidylserine-mediated clearance pathway for circulating IMP+ ΦIM (39-41).
IMP therapy inhibits ΦIM in experimental autoimmune encephalomyelitis
To address the potential for IMPs as a treatment in other disorders and to garner further mechanistic understanding, we also examined a number of unrelated inflammatory disease models. IMPs were tested in the noninfectious CNS inflammatory model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE). In EAE mice, circulating ΦIM differentiate predominantly into CD11c+ DCs that promote T cell activation and epitope spreading (2, 3, 11, 42). Daily intravenous infusion of biodegradable PLGA-IMPs over 10 days from the time of disease onset both ameliorated disease during treatment and was associated with a posttreatment period of 14 days devoid of symptoms (Fig. 6A). For clinical translation, IMP treatment given at the initiation of relapse terminated symptoms of disease relapse, with treated mice showing reduced EAE scores (Fig. 6B). Reduced disease scores in animals treated at onset correlated with reduced inflammation in the spinal cord, as determined by flow cytometry (Fig. 6, C, D, and F), with the most marked reduction observed within the ΦIM-derived DC compartment (Fig. 6, D and F). Similar to observations in animals suffering WNV encephalitis or peritonitis, the reduction in ΦIM in the CNS of EAE mice correlated with a significant accumulation of ΦIM in the spleen (Fig. 6, E and F; P < 0.001).
Fig. 6. PLGA-IMPs ameliorate disease in EAE and cardiac infarction.
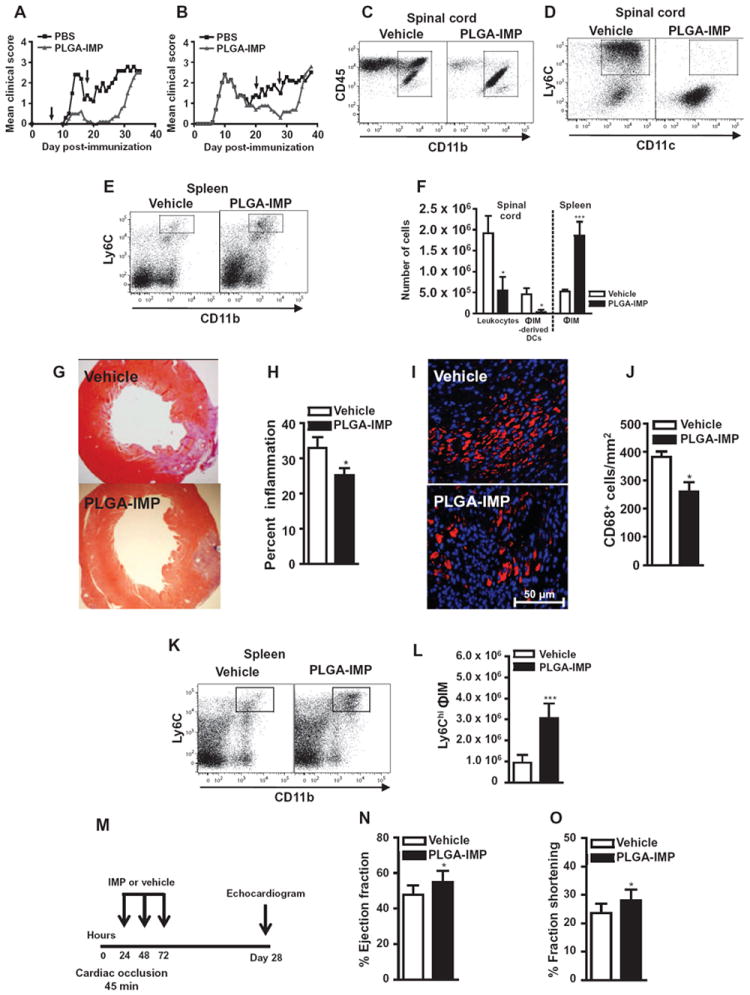
(A and B) EAE disease scores in mice treated for 10 days with PLGA-IMPs or vehicle at onset (A) or relapse (B). (C to E) CD45+/CD11b+/Ly6Chi/CD11c+ ΦIM-derived DCs in spinal cords (C and D) and CD45+/CD11b+/Ly6Chi ΦIM in spleens (E) of mice treated for 7 days at disease onset. (F) Cell numbers in spinal cord and spleen. (G and H) Cardiac histology (G) and infarction size quantification (H) were performed 7 days after permanent left anterior descending artery occlusion in mice treated with three daily infusions of PLGA-IMPs or vehicle beginning 24 hours after occlusion. (I and J) CD68+ macrophages were enumerated by immunohistochemistry. (K and L) Spleens were processed for flow cytometry 7 days after occlusion, and CD45+/CD11b+/Ly6C+/ΦIM were gated and enumerated. (M to O) Function after temporary cardiac arterial occlusion was determined by echocardiography 28 days after occlusion (M), the percentage cardiac ejection fraction (N), and fraction shortening (O) 28 days after reperfusion. EAE mean clinical score data are representative of three separate experiments with 10 to 20 mice per group. Flow cytometry data are means ± SD and represent three separate experiments with four to five mice per group. Cardiac infarction data are representative of two experiments with at least three mice per group. Image analysis was performed as described in Materials and Methods. Statistical analysis was conducted with unpaired, two-tailed Student’s t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing PLGA-IMP and vehicle control group.
IMP therapy reduces cardiac and kidney reperfusion injury
The pathologic effect of ΦIM is also found in other organs. Reducing immigration of ΦIM into the heart and kidney after myocardial infarction and transplantation is associated with reduced tissue pathology (10, 42-44). Using a permanent left anterior descending artery occlusion model (45), we determined the impact of 3 days of PLGA-IMP treatment, initiated 12 hours after occlusion, by histopathology and immunohistochemistry 1 week after occlusion. One week after occlusion, vehicle-treated mice had a strong infiltration of monocytes into the myocardium, with up to 40% of the left ventricular wall involved (Fig. 6G). IMP treatment significantly reduced the size of the inflammatory focus, reducing overall cardiac inflammation by 20 to 25% (Fig. 6H; P < 0.05). Furthermore, a significant reduction in CD68+ macrophage numbers within the infarct area was observed, with IMP treatment resulting in a 30% reduction in the number of CD68+ cells/mm2, relative to the vehicle-treated control (Fig. 6, I and J; P < 0.05). This reduction again correlated with a significant increase in the number of ΦIM in the spleen (Fig. 6, K and L; P < 0.001). Functional analysis in the complete occlusion model is not considered to be reflective of natural physiology. To determine whether IMP-mediated inhibition of ΦIM was associated with enhanced cardiac function after myocardial infarction, we performed echocardiography 30 days after mice had undergone temporary occlusion (45 min) of the left anterior descending artery followed by 3 days of IMP infusion initiated 12 hours after reperfusion (Fig. 6, M to O). Strikingly, animals treated with IMPs showed enhanced cardiac function, as determined by increased ejection fractions (Fig. 6N) and fraction shortening (Fig. 6O) in comparison to control vehicle-treated mice. In parallel with these studies, we also examined the ability of IMP infusion to reduce ischemia-reperfusion injury in the kidney. Similar to the temporary cardiac occlusion, the renal artery was temporarily ligated for 45 min. IMP treatment was initiated immediately and continued for three consecutive days (Fig. 7A). Relative to vehicle-treated mice, IMP treatment significantly enhanced serum creatinine clearance at days 1 (P < 0.001) and 5 (P < 0.005) after reperfusion (Fig. 7B). This increase in kidney function in IMP-treated mice also correlated with reduced tubular atrophy (Fig. 7C) at these time points. Together, these data suggest that IMP-mediated ΦIM immigration inhibition reduced ischemia-reperfusion injury of the heart and kidney and may support organ recovery.
Fig. 7. PS-IMP ameliorates disease in kidney injury and colitis.
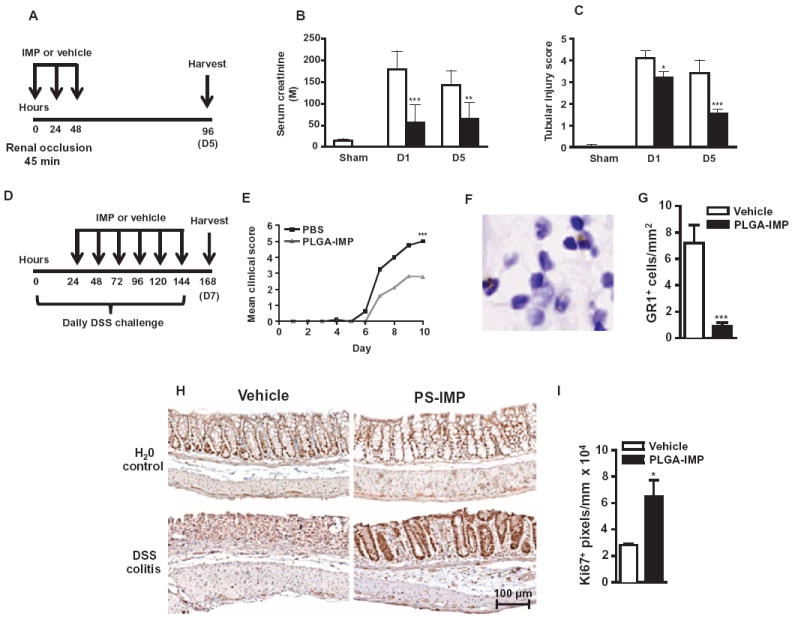
(A to C) PLGA-IMP treatment was tested after temporary renal artery occlusion (A) using serum creatinine (B) and tubular injury score (C) as functional measures at days 1 and 5 after reperfusion. (D) PS-IMP or vehicle control was also injected daily intravenously into mice with DSS-induced colitis or control (water vehicle) from day 1 to day 6 after disease induction. (E to G) Daily mean clinical scores (E) and GR1+ monocytes were enumerated at day 7 (F and G). (H) Mouse colon tissue was processed for immunohistology on day 9 of DSS challenge, and Ki67+ staining (brown) was performed on transverse colon sections. (I) Image analysis was conducted comparing DSS-induced colitis vehicle to PS-IMP. Renal artery occlusion and DSS-induced colitis data are representative of at least three experiments with at least 5 to 20 mice per group. Image analysis was performed as described in Materials and Methods. Statistical analysis was conducted with unpaired, two-tailed Student’s t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, comparing PLGA-IMP and vehicle control group.
IMP therapy alleviates inflammatory bowel disease symptoms
ΦIM have also been implicated in the pathology observed in inflammatory bowel disease (IBD) (46). We therefore examined the ability of IMP therapy to inhibit ΦIM-mediated pathology in the dextran sodium sulfate (DSS)–induced colitis model of IBD (Fig. 7D) (46). Relative to vehicle-treated controls, daily PS-IMP treatment resulted in significantly diminished IBD symptoms (Fig. 7E; P < 0.001), which correlated with reduced inflammatory monocyte migration into the colon. This was demonstrated by the reduced number of GR1+ cells with monocytic morphology in the mucosa and submucosa of PLGA-IMP–treated, but not vehicle-treated, animals (Fig. 7, F and G; P < 0.001). IMP-induced reduction in ΦIM immigration into the gut was also associated with increased epithelial tissue repair and proliferation as determined by general histology and labeling with the proliferation marker Ki67 in PS-IMP–treated animals (Fig. 7, H and I; P < 0.05).
DISCUSSION
This work demonstrates the ability of carboxylated microparticles to trigger alterations in the migratory behavior and function of circulating Ly6Chi ΦIM. We show that this interaction can be harnessed therapeutically to reduce ΦIM-mediated pathology and promote repair mechanisms in a number of unrelated disease models, including WNV encephalitis, multiple sclerosis, ulcerative colitis, kidney reperfusion injury, myocardial infarction, and peritonitis.
Important advances have been made in micromedicine research that have focused on the use of microparticles as drug carriers or as contrast agents for imaging purposes. Here, we extend their potential utility by showing that microparticles with negative surface charge characteristics are taken up by Ly6Chi blood ΦIM, which are consequently diverted to the spleen where they undergo apoptosis, thereby abrogating macrophage-mediated inflammatory activity in target tissues. Whereas opsonin-mediated clearance is one mechanism for microparticle removal, IMPs and other particles, such as silica and dextran–superparamagnetic ion oxide particles and some bacterial components (29, 47, 48), are efficiently taken up through the scavenger receptor MARCO. Whereas the precise molecular interactions resulting in binding of IMPs to MARCO remain to be elucidated, MARCO contains a collagen-like domain characterized by a lysine positively charged at the ε amino acid. Electrostatic interactions between these domains are important for particle uptake, with mutations in the collagen-like or cysteine-rich regions of MARCO impeding particle uptake (29).
The precise role of apoptosis after IMP uptake remains ambiguous and requires further examination. However, IMP treatment results in a MARCO-dependent increase in annexin V+ ΦIM in the spleen, indicative of the presence of phosphatidylserine flipped onto the external leaflet of the plasma membrane in these cells, which is an early marker of apoptosis. Indeed, phosphatidylserine mediates the splenic removal of senescing red blood cells and circulating cellular debris (39-41). It is thus plausible that a similar phosphatidylserine-related mechanism mediates the sequestration of ΦIM in the spleen as a prelude to apoptosis of these cells. Phosphatidylserine flipping and other early apoptosis events, mediated in part by p53, are reversible if the apoptotic stimulus is removed within a reasonable time frame (49). Hence, in the absence of a spleen, IMPs may still trigger ΦIM phosphatidylserine flipping. However, in the absence of the spleen, reversal of these p53- mediated apoptosis pathways may occur once IMPs have been processed by circulating monocytes. This hypothesis requires further investigation, but it may also help to address the differential apoptosis observed between IMP+ macrophage populations in the spleen and the lung. Lung macrophages in IMP-treated mice did not show significant amounts of apoptosis relative to spleen ΦIM. Physiologically, lung macrophages are exposed to numerous nonpathological environmental particulates recognized by MARCO without undergoing apoptosis. This may be the result of differential macrophage subpopulation responses to IMP or the absence of particular apoptosis signals delivered within the spleen microenvironment that are not readily available in peripheral tissues like the lung. Furthermore, although it is known that the liver can also remove circulating phosphatidylserine-positive red blood cells (41), in our studies the liver seems to be unable to compensate for the absence of the spleen. Thus, given the currently defined apoptotic cell clearance mechanisms (34, 38) and considering that ΦIM are evidently recruited to areas of inflammation as dispensable “foot soldiers” of the immune system that are quickly lost once reaching their tissue target, we propose that IMP internalization by ΦIM triggers the cascade of cellular and splenic mechanisms important not only for clearance of senescent blood cells but also for containment of potentially infectious or noxious substances.
Our data extend earlier findings highlighting a role for MARCO in apoptosis induction and cellular trafficking and strongly suggest that MARCO-mediated uptake and associated downstream signaling is a primary mechanism for IMP-induced ΦIM inhibition. The biochemical characteristics of the MARCO receptor likely explain the key difference observed between the outcome of positively charged and negatively charged microparticle infusion in vivo. Nevertheless, such multifarious roles, combined with the inherent functional redundancy in the scavenger receptor family (33, 37, 38, 48, 50-52), justify a more detailed investigation of MARCO signaling pathways, as well as the potential contribution to the efficacy of IMP treatment of other scavenger receptors in this family.
IMPs are cleared rapidly from the circulation and spleen in treated rodents. However, the therapeutic effects of IMPs were seen to extend well beyond the final dose in animals suffering EAE. Furthermore, IMP treatment resulted in not only an immediate reduction in immune pathology in WNV-infected animals but also the development of long-term WNV-specific immunity. Together, it appears that acute modulation of ΦIM using IMPs may augment, or allow for the development of, other immune regulatory mechanisms that may otherwise be inhibited by the proinflammatory environment generated by ΦIM. This, as well as findings suggesting that IMPs may promote tissue regeneration in heart, kidney, and colon mouse models of disease, requires further examination. Note that the role of ΦIM is not defined for all disease conditions, and during translation, care should be used to avoid using IMPs in disorders where ΦIM are important for disease control or resolution.
In conclusion, safe and specific therapies inhibiting ΦIM migration and function have remained elusive, with clinical utilization of CCR2/CCL2 antagonists and anti–VLA-4 antibodies failing to provide specific, safe, and efficacious ΦIM inhibition (11, 18). Chemokine redundancy and the ubiquitous expression of CCR2/CCL2 are barriers hampering the clinical application of CCR2/CCL2 antagonists; expression of VLA-4 on activated T cells and JC virus infection has limited potential clinical uses of anti–VLA-4 antibodies (53). Our results not only highlight the importance of microparticle-scavenger receptor interactions for ΦIM function but also indicate that IMP therapy may provide a specific, safe, and cost-effective tool for inhibiting ΦIM-mediated pathology in a number of inflammatory conditions. However, there still remain important unresolved questions regarding the role of other scavenger receptors and the molecular processes through which IMPs interact with circulating ΦIM under laminar flow conditions. The availability of IMPs derived from the U.S. Food and Drug Administration–approved polymer, PLGA, which breaks down into acetic and glycolic acid within hours of infusion, and the recent discovery of human monocyte subsets expressing CCR2+ should enable accelerated clinical translation of these findings.
MATERIALS AND METHODS
Study design
This study was designed to test the hypothesis that negatively charged microparticles, defined as IMPs, bind to circulating inflammatory monocytes, subsequently directing them to the spleen where they undergo programmed cell death. Furthermore, the safety and mechanism of action of IMPs were tested in numerous inflammatory disease models where inflammatory monocytes have been implicated in pathology. Flow cytometry (both intracellular and cell surface labeling), high-speed cell sorting, histology, immunohistochemistry, liquid chromatography–mass spectrometry (LC-MS), and disease models affecting numerous organ systems were used. More specifically, with a mouse model of WNV encephalitis, initial experiments examined important particle characteristics, including charge and dose volume. IMP-induced changes in monocyte trafficking were examined with adoptive transfer techniques. The induction of apoptosis and the role of the scavenger receptor MARCO were examined with flow cytometry and mice deficient in MARCO. The disease models examined included mouse models of WNV encephalitis, EAE, TG peritonitis, DSS-induced colitis, and cardiac and kidney ischemia-reperfusion models. Sample sizes were predetermined on the basis of previous experience using at minimum three groups of mice, and all experiments were replicated at least twice to confirm findings. Statistical analyses were conducted with a two-tailed t test or one-way ANOVA as described below. Mice were randomly assigned to treatment groups, and where possible, treatment groups were blinded until statistical analysis. No animals or potential outliers were excluded from the data sets presented in this study.
Mice
For WNV and IBD studies, 8-week-old female C57BL/6 mice were obtained from the Animal Resources Centre (ARC) (Western Australia, Australia). All procedures were performed with permission of the University of Sydney Animal Ethics Committee. For EAE studies, 8-week-old female SJL/J mice were obtained from Harlan Laboratories. For TG studies, 8-week-old female BALB/c mice were obtained from the National Cancer Institute (Bethesda, MD), or 8-week-old female C57BL/6 mice were obtained from ARC. MARCO−/− animals on the BALB/c background were provided by L. Kobzik (Harvard University, Boston, MA). For cardiac inflammation studies, 12-week-old male C57BL/6 mice were purchased from The Jackson Laboratory. All procedures were performed with permission from the Northwestern University Institutional Animal Care and Use Committee. All animals were housed under specific pathogen–free conditions with food and water provided ad libitum.
WNV infection
WNV (Sarafend) was derived from the brains of neonatal mice and propagated in Vero cell cultures as described previously (2, 24). WNV infection was conducted in C57BL/6 animals as previously described (2, 24). Mice were intranasally infected with 6 × 104 or 6 × 103 PFU of WNV in 10 μl of sterile PBS (Invitrogen). Mock infections were conducted with sterile PBS only. After infection, mice were weighed daily.
TG-induced peritonitis
Peritonitis was induced by intraperitoneal injection of 1 ml of a 4% (w/v) TG broth prepared in sterile water (Sigma-Aldrich). Leukocytes were isolated at defined time points by intraperitoneal lavage (wash) with ice-cold 0.05 mM EDTA-PBS solution.
EAE induction
PLP139–151 (HSLGKWLGHPDKF) peptide–induced EAE was induced in SJL/J mice as previously reported (34). Individual animals were observed daily, and clinical scores were assessed in a blinded fashion on a 0 to 5 scale as follows: 0, no abnormality; 1, limp tail or hindlimb weakness; 2, limp tail and hindlimb weakness; 3, hindlimb paralysis; 4, hind limp paralysis and forelimb weakness; and 5, moribund. These data are reported as mean clinical scores. Paralyzed animals were afforded easier access to food and water.
Cardiac infarction and ischemia-reperfusion injury
Myocardial infarction was conducted in C57BL/6 mice. The left anterior descending artery was permanently or temporarily (45 min) occluded surgically, as previously described (45).
Inflammatory bowel disease induction
The DSS-induced colitis model of IBD was induced as previously described (54). DSS (molecular weight, 36,000 to 44,000 daltons; ICN Biomedicals) [2.5% (w/v)] dissolved in tap water was administered ad libitum for nine consecutive days (46). Control groups received tap water only during this time. Body weight and clinical assessments were measured daily. Percentage body weight loss was calculated as follows: (mean weight at day 9/weight on day 0) × 100. After body weight measurements, mice were examined for clinical parameters, including mobility and gait, vocalizations, group interactions, and grooming. Each parameter was scored a value between 0 and 2, where a total value of 0 to 1 represents normal, 2 to 4 mild, 5 to 7 moderate, and 8 to 10 severe (46). Fecal assessment was measured daily where possible. Scores were given for fecal consistency, hematochezia, and rectal bleeding. A total value between 0 and 6 reflected the fecal score, where a value of 0 represents feces of normal appearance and a value of 6 represents severe diarrhea, rectal bleeding, and presence of blood in stools.
Kidney ischemia-reperfusion model
Induction of kidney ischemia-reperfusion injury was as previously described (55-57). Briefly, with a midline abdominal incision, both renal pedicles were clamped for 45 min with microaneurysm clamps. During the period of ischemia, body temperature was maintained by placing the mice on a 32°C heat pad. After removal of the clamps, the kidneys were inspected for 1 min for restoration of blood flow, returning to their original color. The abdomen was closed. Sham-operated mice received identical surgical procedures except that microaneurysm clamps were not applied. To maintain fluid balance, we supplemented all mice with 1 ml of saline administered subcutaneously. Mice were sacrificed 1 and 5 days after reperfusion (n = 5 to 7 per group). Blood was collected, and kidney tissues were fixed in 10% neutral buffered formalin. Serum creatinine was used as a measure of kidney function with the modified Jaffe rate reaction by the Biochemistry Department of The Royal Prince Alfred Hospital, Sydney, Australia.
Intravenous delivery of microparticles
FITC or bright blue Fluoresbrite neutral and carboxylated PS-NPs (500 nm in diameter) were obtained from Polysciences. FITC PLGA and FITC polystyrene neutral, carboxylated, and aminated microparticles (500 nm in diameter) were made to specification by Phosphorex or Polysciences. Carboxylated 500-nm microdiamonds were generated at Macquarie University or Sydney University (New South Wales, Australia). Microparticles were diluted to the indicated concentration in sterile PBS, and 200 μl was injected intravenously as indicated. Specific particle properties are defined in table S1. For opsonization experiments, particles were incubated at 37°C with plasma isolated from mock- or WNV-infected mice.
Flow cytometry
Mice were anesthetized, blood was collected, and animals were perfused with 50 ml of sterile PBS. Spleen, brain, bone marrow, and peritoneal fluid were isolated and processed into single-cell suspensions as previously described (2). Cells were incubated with anti-CD16/32, and live cells were counted with trypan blue exclusion, which routinely showed >95% viability. Multiparametric flow cytometry was used. Cells were then incubated with fluorescently labeled antibodies against CD45. The most common combination of antibody cocktail included FITCCD45 (BioLegend) or allophycocyanin (APC)–Cy7 CD45 (BioLegend) or V500 CD45 (Becton Dickinson), APC-Cy7 CD11b (BioLegend) or Alexa Fluor 700–CD11b (BioLegend), APC-Ly6C (BioLegend) or Brilliant Violet 570–Ly6C (BioLegend), peridinin chlorophyll protein (PerCP)–F4/80 (BioLegend), phycoerythrin (PE)–Ly6G (BioLegend) or Brilliant Violet 421–Ly6G (BioLegend), PE-CD11c (BioLegend) or PE-Cy7-CD11c (BioLegend), APC-NK1.1 (BioLegend) or Alexa Fluor 700–NK1.1 (BioLegend), PerCP-B220 (BioLegend) or FITC-B220 (BioLegend), PerCP-CD3 (BioLegend) or APC-CD3 (BioLegend), PE-MARCO (BioLegend) or biotinylated MARCO (BioLegend), followed by streptavidin-PE (BioLegend) and GR1-APC (BioLegend). Cell surface molecule expression was measured on a fluorescence-activated cell sorting (FACS) LSR II with FACSDiva program (Becton Dickinson). Viable populations were gated by forward and side scatter, and identified fluorescent populations were determined by forward gating thereafter. Acquired FACS data files were analyzed with FlowJo (Tree Star Inc.). Quantification of cell populations of interest was calculated on the basis of flow cytometry percentages at analysis and absolute cell counts from each organ.
Adoptive transfer
As previously described (2), the bone marrow of WNV-infected animals was processed into single-cell suspensions on day 6 after infection and incubated with fluorescently labeled antibodies against CD11b, Ly6C, and Ly6G. CD11b+, Ly6Chi, Ly6G− ΦIM were sorted on a FACSAria with the FACSDiva program (Becton Dickinson), with stringencies set to achieve >98% purity. Cells were then labeled with the fluorescent membrane dye PKH26 (Invitrogen) according to the manufacturer’s instructions. Matched mock- and WNV-infected recipients were intravenously injected with 2.0 × 106 sorted Ly6Chi ΦIM on day 6 after infection, delivered in 200 μl of PBS. Brain and spleens were isolated from recipients on day 7 after infection (24 hours after transfer) and processed for flow cytometry, as described above.
Histology and immunohistochemistry
Mice were anesthetized and perfused with 50 ml of sterile PBS. With the exception of the heart and intestines, which were processed into paraffin blocks, all organs were isolated and snap-frozen in optimum cutting temperature (OCT) compound (Tissue-Tek). Tissue sections (8 mm) were cut on a cryostat microtome, air-dried overnight, and then stored at −80°C until required. Frozen sections were thawed, and histology (standard hematoxylin and eosin staining) or immunohistochemistry was performed as previously described (2). Before staining heart and intestine sections, proteinase K treatment or citrate buffer microwave antigen retrieval was conducted, as previously described (2). Spleen, brain, and heart sections were incubated with biotinylated antibodies against CD11c (eBioscience), CD11b (eBioscience), F4/80 (R&D Systems), MARCO (R&D Systems), SIGN-R (R&D Systems), SIGLEC-1 (R&D Systems), CD68 (Abcam), or biotinylated Griffonia simplicifolia Lectin (Sigma-Aldrich), followed by secondary staining with streptavidin Alexa Fluor 594 or Alexa Fluor 488 (Life Technologies). The nuclear counterstain DAPI was then applied (Vector Laboratories). Sections of intestine were stained with biotinylated GR1 (BioLegend) or Ki67 (Abcam), followed by amplification with a tyramide signal amplification kit (Perkin Elmer) and staining with the chromogen diaminobenzidine (Vector Laboratories). Slides were then counterstained with hematoxylin. Images were acquired on an Olympus BX-51 microscope with a DP-70 camera and DP manager 2.2.1 software (Olympus).
Marginal zone macrophage deletion
Clodronate-loaded liposomes (provided by N. Van Roijen) were used as previously described (2, 35) to deplete marginal zone macrophages. PBS-loaded liposomes were used as a nondepleting control.
Statistics
Graphs were made and statistical analyses were performed in GraphPad Prism (GraphPad Software). To compare two samples, we conducted an unpaired, two-tailed Student’s t test. To compare three or more samples, we performed a one-way ANOVA with a Tukey-Kramer post-test. For survival data, the Mantel-Haenszel log-rank test was conducted. For these tests, P ≤ 0.05 (*) was deemed significant, P ≤ 0.01 (**) very significant, and P ≤ 0.001 (***) extremely significant.
Supplementary Material
Acknowledgments
We acknowledge B. Wu at Phosphorex for the production of IMPs made to authors’ specifications. Clodronate liposomes were prepared and supplied by N. Van Roijen.
Funding: This work was funded by National Health and Medical Research Council grants 1030897 (to N.J.C.K. and D.R.G.) and 512413 (to N.J.C.K. and I.L.C.) and NIH grants NS-026543, EB-013198 (to S.D.M.), HL-097021 (to E.B.T.), and 1R21HL092357 (to G.S.K.). D.R.G. and N.J.C.K. also received grant support from the Ryan Marrion Foundation.
Footnotes
Author contributions: All authors contributed to paper production, including intellectual input and result analysis. Specifically, D.R.G. and N.J.C.K. uncovered the IMP therapeutic potential, designed the experiments, and, with S.D.M. and R.L.T., wrote the paper. N.J.C.K., D.R.G., J.K., and W.H. designed the therapeutic approach associated with weight loss and seizure symptom constellation in the WNV model. N.J.C.K., D.R.G., and I.L.C. provided intellectual, design, resource, and experimental input into early CNS studies defining responses in the WNV model. D.R.G., M.M., and M.T.G. performed the initial studies defining therapeutic potential of IMP in WNV and carried out pilot EAE experiments. R.L.T. defined the negative charge potentials and tested the IMP generated from other materials. T.M.A., C.D., and C.v.V. performed immunohistology, clodronate depletion, and adoptive transfer experiments. D.R.G., S.D.M., and R.L.T. designed the EAE and MARCO-deficient mouse experiments. R.L.T., D.M., and A.M. performed the majority of EAE experiments. G.S.K., D.R.G., and R.L.T. designed peritonitis model experiments, with R.L.T. performing the experiments. N.J.C.K. and S.B. designed and C.v.V., H.W., and J.M. performed kidney reperfusion injury experiments. L.D.S., D.R., J.S., and L.B. provided nanoparticle design and manufacturing advice. J.S. and L.B. designed and chemically modified nanodiamonds and undertook analytical testing (size and z potential determination) for quality control of product. S. J. Cordwell and M.Y.W. designed and performed LC-MS experiments. D.R.G., E.B.T., S.D.M., and N.J.C.K. designed and R.L.T. performed cardiac infarction experiments. S.B., D.R.G., and N.J.C.K. designed and B.C. performed colitis model experiments.
Competing interests: D.R.G., N.J.C.K., and R.L.T. are co-authors on a patent application for modified immune-modulating nanoparticles (#WO2012065153A2). D.R.G., N.J.C.K., S.D.M., and L.D.S. consult for Cour Pharmaceutical Development Company, the license holder of this patent application. These authors are also stakeholders in Cour Pharmaceutical Development Company.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/6/219/219ra7/DC1
Materials and Methods
Fig. S1. PS-IMPs reduce cytokine and chemokine production in the WNV-infected brain.
Fig. S2. IMP clearance kinetics, lack of IMP localization to the brain, and PLGA-IMP treatment in TG peritonitis.
Fig. S3. IMP localization and marginal zone macrophage depletion.
Table S1. Particle physical properties.
Table S2. Opsonized proteins found on IMP.
REFERENCES AND NOTES
- 1.Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, Mowat AM. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. doi: 10.1038/mi.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113:3190–3197. doi: 10.1182/blood-2008-07-168575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol. 2011;186:508–515. doi: 10.4049/jimmunol.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiopu A, Nadig SN, Cotoi OS, Hester J, van Rooijen N, Wood KJ. Inflammatory Ly-6Chi monocytes play an important role in the development of severe transplant arteriosclerosis in hyperlipidemic recipients. Atherosclerosis. 2012;223:291–298. doi: 10.1016/j.atherosclerosis.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swirski FK, Wildgruber M, Ueno T, Figueiredo JL, Panizzi P, Iwamoto Y, Zhang E, Stone JR, Rodriguez E, Chen JW, Pittet MJ, Weissleder R, Nahrendorf M. Myeloperoxidase-rich Ly-6C+ myeloid cells infiltrate allografts and contribute to an imaging signature of organ rejection in mice. J Clin Invest. 2010;120:2627–2634. doi: 10.1172/JCI42304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A, Hogan SP. Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6Chigh CCR2+ inflammatory monocyte/macrophage-derived CCL11. J Immunol. 2011;186:5993–6003. doi: 10.4049/jimmunol.1003844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg KE, Ljungcrantz I, Andersson L, Bryngelsson C, Hedblad B, Fredrikson GN, Nilsson J, Björkbacka H. Elevated CD14++CD16− monocytes predict cardiovascular events. Circ Cardiovasc Genet. 2012;5:122–131. doi: 10.1161/CIRCGENETICS.111.960385. [DOI] [PubMed] [Google Scholar]
- 9.Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Deffrasnes C, Ashhurst TM, Radford J, Hofer M, Thomas S, Campbell IL, King NJ. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflamm. 2012;9:246. doi: 10.1186/1742-2094-9-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buckner CM, Calderon TM, Willams DW, Belbin TJ, Berman JW. Characterization of monocyte maturation/differentiation that facilitates their transmigration across the blood-brain barrier and infection by HIV: Implications for NeuroAIDS. Cell Immunol. 2011;267:109–123. doi: 10.1016/j.cellimm.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, Lang R, Haniffa M, Collin M, Tacke F, Habenicht AJ, Ziegler-Heitbrock L, Randolph GJ. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;115:e10–e19. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zimmermann HW, Seidler S, Nattermann J, Gassler N, Hellerbrand C, Zernecke A, Tischendorf JJ, Luedde T, Weiskirchen R, Trautwein C, Tacke F. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLOS One. 2010;5:e11049. doi: 10.1371/journal.pone.0011049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katschke KJ, Jr, Rottman JB, Ruth JH, Qin S, Wu L, LaRosa G, Ponath P, Park CC, Pope RM, Koch AE. Differential expression of chemokine receptors on peripheral blood, synovial fluid, and synovial tissue monocytes/macrophages in rheumatoid arthritis. Arthritis Rheum. 2001;44:1022–1032. doi: 10.1002/1529-0131(200105)44:5<1022::AID-ANR181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 16.Butovsky O, Siddiqui S, Gabriely G, Lanser AJ, Dake B, Murugaiyan G, Doykan CE, Wu PM, Gali RR, Iyer LK, Lawson R, Berry J, Krichevsky AM, Cudkowicz ME, Weiner HL. Modulating inflammatory monocytes with a unique microRNA gene signature ameliorates murine ALS. J Clin Invest. 2012;122:3063–3087. doi: 10.1172/JCI62636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: Protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Couzin-Frankel J. Drug research. Roche exits RNAi field, cuts 4800 jobs. Science. 2010;330:1163. doi: 10.1126/science.330.6008.1163. [DOI] [PubMed] [Google Scholar]
- 20.Kang YS, Cha JJ, Hyun YY, Cha DR. Novel C-C chemokine receptor 2 antagonists in metabolic disease: A review of recent developments. Expert Opin Investig Drugs. 2011;20:745–756. doi: 10.1517/13543784.2011.575359. [DOI] [PubMed] [Google Scholar]
- 21.Boros P, Ochando JC, Chen SH, Bromberg JS. Myeloid-derived suppressor cells: Natural regulators for transplant tolerance. Hum Immunol. 2010;71:1061–1066. doi: 10.1016/j.humimm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shantsila E, Wrigley B, Tapp L, Apostolakis S, Montoro-Garcia S, Drayson MT, Lip GY. Immunophenotypic characterization of human monocyte subsets: Possible implications for cardiovascular disease pathophysiology. J Thromb Haemost. 2011;9:1056–1066. doi: 10.1111/j.1538-7836.2011.04244.x. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Getts DR, Matsumoto I, Müller M, Getts MT, Radford J, Shrestha B, Campbell IL, King NJ. Role of IFN-γ in an experimental murine model of West Nile virus-induced seizures. J Neurochem. 2007;103:1019–1030. doi: 10.1111/j.1471-4159.2007.04798.x. [DOI] [PubMed] [Google Scholar]
- 25.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmström J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10:M110.006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamps JA, Scherphof GL. Receptor versus non-receptor mediated clearance of liposomes. Adv Drug Deliv Rev. 1998;32:81–97. doi: 10.1016/s0169-409x(97)00133-6. [DOI] [PubMed] [Google Scholar]
- 27.Andersen AJ, Hashemi SH, Andresen TL, Hunter AC, Moghimi SM. Complement: Alive and kicking nanomedicines. J Biomed Nanotechnol. 2009;5:364–372. doi: 10.1166/jbn.2009.1045. [DOI] [PubMed] [Google Scholar]
- 28.Chao SK, Hamilton RF, Pfau JC, Holian A. Cell surface regulation of silica-induced apoptosis by the SR-A scavenger receptor in a murine lung macrophage cell line (MH-S) Toxicol Appl Pharmacol. 2001;174:10–16. doi: 10.1006/taap.2001.9190. [DOI] [PubMed] [Google Scholar]
- 29.Chao Y, Makale M, Karmali PP, Sharikov Y, Tsigelny I, Merkulov S, Kesari S, Wrasidlo W, Ruoslahti E, Simberg D. Recognition of dextran-superparamagnetic iron oxide nanoparticle conjugates (Feridex) via macrophage scavenger receptor charged domains. Bioconjug Chem. 2012;23:1003–1009. doi: 10.1021/bc200685a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 31.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, Mack M, Charo IF. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. 2007;117:902–909. doi: 10.1172/JCI29919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chao Y, Karmali PP, Mukthavaram R, Kesari S, Kouznetsova VL, Tsigelny IF, Simberg D. Direct recognition of superparamagnetic nanocrystals by macrophage scavenger receptor SR-AI. ACS Nano. 2013;7:4289–4298. doi: 10.1021/nn400769e. [DOI] [PubMed] [Google Scholar]
- 33.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. 2012;30:1217–1224. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL, King NJ, Miller SD. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10–producing splenic macrophages and maintained by T regulatory cells. J Immunol. 2011;187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanno S, Furuyama A, Hirano S. A murine scavenger receptor MARCO recognizes polystyrene nanoparticles. Toxicol Sci. 2007;97:398–406. doi: 10.1093/toxsci/kfm050. [DOI] [PubMed] [Google Scholar]
- 36.Aichele P, Zinke J, Grode L, Schwendener RA, Kaufmann SH, Seiler P. Macrophages of the splenic marginal zone are essential for trapping of blood-borne particulate antigen but dispensable for induction of specific T cell responses. J Immunol. 2003;171:1148–1155. doi: 10.4049/jimmunol.171.3.1148. [DOI] [PubMed] [Google Scholar]
- 37.Dorrington MG, Roche AM, Chauvin SE, Tu Z, Mossman KL, Weiser JN, Bowdish DM. MARCO is required for TLR2- and Nod2-mediated responses to Streptococcus pneumoniae and clearance of pneumococcal colonization in the murine nasopharynx. J Immunol. 2013;190:250–258. doi: 10.4049/jimmunol.1202113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min KJ, Um HJ, Cho KH, Kwon TK. Curcumin inhibits oxLDL-induced CD36 expression and foam cell formation through the inhibition of p38 MAPK phosphorylation. Food Chem Toxicol. 2013;58:77–85. doi: 10.1016/j.fct.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Schwartz RS, Tanaka Y, Fidler IJ, Chiu DT, Lubin B, Schroit AJ. Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J Clin Invest. 1985;75:1965–1972. doi: 10.1172/JCI111913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J Biol Chem. 1985;260:5131–5138. [PubMed] [Google Scholar]
- 41.Lee SJ, Park SY, Jung MY, Bae SM, Kim IS. Mechanism for phosphatidylserine-dependent erythrophagocytosis in mouse liver. Blood. 2011;117:5215–5223. doi: 10.1182/blood-2010-10-313239. [DOI] [PubMed] [Google Scholar]
- 42.Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ TH-17 cells in relapsing EAE. Nat Immunol. 2007;8:172–180. doi: 10.1038/ni1430. [DOI] [PubMed] [Google Scholar]
- 43.Leuschner F, Rauch PJ, Ueno T, Gorbatov R, Marinelli B, Lee WW, Dutta P, Wei Y, Robbins C, Iwamoto Y, Sena B, Chudnovskiy A, Panizzi P, Keliher E, Higgins JM, Libby P, Moskowitz MA, Pittet MJ, Swirski FK, Weissleder R, Nahrendorf M. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J Exp Med. 2012;209:123–137. doi: 10.1084/jem.20111009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, Mulligan-Kehoe MJ, Libby P, Nahrendorf M, Pittet MJ, Weissleder R, Swirski FK. Extramedullary hematopoiesis generates Ly-6Chigh monocytes that infiltrate atherosclerotic lesions. Circulation. 2012;125:364–374. doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yeap XY, Dehn S, Adelman J, Lipsitz J, Thorp EB. Quantitation of acute necrosis after experimental myocardial infarction. Methods Mol Biol. 2013;1004:115–133. doi: 10.1007/978-1-62703-383-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Y, Hunt NH, Bao S. The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res. 2008;18:1220–1229. doi: 10.1038/cr.2008.310. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton RF, Jr, Thakur SA, Mayfair JK, Holian A. MARCO mediates silica uptake and toxicity in alveolar macrophages from C57BL/6 mice. J Biol Chem. 2006;281:34218–34226. doi: 10.1074/jbc.M605229200. [DOI] [PubMed] [Google Scholar]
- 48.Arredouani MS, Palecanda A, Koziel H, Huang YC, Imrich A, Sulahian TH, Ning YY, Yang Z, Pikkarainen T, Sankala M, Vargas SO, Takeya M, Tryggvason K, Kobzik L. MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J Immunol. 2005;175:6058–6064. doi: 10.4049/jimmunol.175.9.6058. [DOI] [PubMed] [Google Scholar]
- 49.Geske FJ, Lieberman R, Strange R, Gerschenson LE. Early stages of p53-induced apoptosis are reversible. Cell Death Differ. 2001;8:182–191. doi: 10.1038/sj.cdd.4400786. [DOI] [PubMed] [Google Scholar]
- 50.Arredouani MS, Franco F, Imrich A, Fedulov A, Lu X, Perkins D, Soininen R, Tryggvason K, Shapiro SD, Kobzik L. Scavenger receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J Immunol. 2007;178:5912–5920. doi: 10.4049/jimmunol.178.9.5912. [DOI] [PubMed] [Google Scholar]
- 51.Dahl M, Bauer AK, Arredouani M, Soininen R, Tryggvason K, Kleeberger SR, Kobzik L. Protection against inhaled oxidants through scavenging of oxidized lipids by macrophage receptors MARCO and SR-AI/II. J Clin Invest. 2007;117:757–764. doi: 10.1172/JCI29968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsushita N, Komine H, Grolleau-Julius A, Pilon-Thomas S, Mulé JJ. Targeting MARCO can lead to enhanced dendritic cell motility and anti-melanoma activity. Cancer Immunol Immunother. 2010;59:875–884. doi: 10.1007/s00262-009-0813-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tyler KL, Khalili K. Natalizumab and progressive multifocal leukoencephalopathy: Highlights of the International Workshop on JC Virus/PML and Multiple Sclerosis, June 3-4, 2005, Philadelphia PA. Rev Neurol Dis. 2005;2:144–149. [PubMed] [Google Scholar]
- 54.Bao S, Carr ED, Xu YH, Hunt NH. Gp91phox contributes to the development of experimental inflammatory bowel disease. Immunol Cell Biol. 2011;89:853–860. doi: 10.1038/icb.2011.4. [DOI] [PubMed] [Google Scholar]
- 55.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu H, Craft ML, Wang P, Wyburn KR, Chen G, Ma J, Hambly B, Chadban SJ. IL-18 contributes to renal damage after ischemia-reperfusion. J Am Soc Nephrol. 2008;19:2331–2341. doi: 10.1681/ASN.2008020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu H, Ma J, Wang P, Corpuz TM, Panchapakesan U, Wyburn KR, Chadban SJ. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21:1878–1890. doi: 10.1681/ASN.2009101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cordwell SJ, Edwards AV, Liddy KA, Moshkanbaryans L, Solis N, Parker BL, Yong AS, Wong C, Kritharides L, Hambly BD, White MY. Release of tissue-specific proteins into coronary perfusate as a model for biomarker discovery in myocardial ischemia/reperfusion injury. J Proteome Res. 2012;11:2114–2126. doi: 10.1021/pr2006928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.