

Abstract
The genes encoding RAS family members are frequently mutated in juvenile myelomonocytic leukemia (JMML) and acute myeloid leukemia (AML). RAS proteins are difficult to target pharmacologically; therefore, targeting the downstream PI3K and RAF/MEK/ERK pathways represents a promising approach to treat RAS-addicted tumors. The p110α isoform of PI3K (encoded by Pik3ca) is an essential effector of oncogenic KRAS in murine lung tumors, but it is unknown whether p110α contributes to leukemia. To specifically examine the role of p110α in murine hematopoiesis and in leukemia, we conditionally deleted p110α in HSCs using the Cre-loxP system. Postnatal deletion of p110α resulted in mild anemia without affecting HSC self-renewal; however, deletion of p110α in mice with KRASG12D-associated JMML markedly delayed their death. Furthermore, the p110α-selective inhibitor BYL719 inhibited growth factor–independent KRASG12D BM colony formation and sensitized cells to a low dose of the MEK inhibitor MEK162. Furthermore, combined inhibition of p110α and MEK effectively reduced proliferation of RAS-mutated AML cell lines and disease in an AML murine xenograft model. Together, our data indicate that RAS-mutated myeloid leukemias are dependent on the PI3K isoform p110α, and combined pharmacologic inhibition of p110α and MEK could be an effective therapeutic strategy for JMML and AML.
Introduction
HSCs are defined by their ability to both self-renew and to differentiate into all of the blood lineages. Hematopoietic growth factors, such as erythropoietin, FLT3 ligand, c-kit, and thrombopoietin, control HSC and progenitor proliferation, differentiation, self-renewal, and survival. These growth factors activate a variety of signaling pathways, but the PI3K pathway is a common downstream signaling pathway for all of them (1). The PI3Ks are lipid kinases that convert phosphatidyl inositol phosphate-2 (PIP2) to PIP3. Class IA PI3Ks can be activated by tyrosine kinase receptors (RTKs), GPCRs, and oncogenes such as RAS. They exist as heterodimers, consisting of a catalytic 110-kDa subunit termed p110, which in hematopoietic cells has 3 isoforms (p110α, p110β, and p110δ), and 5 regulatory subunits (p85α, p85β, p55α, p55β, and p55γ) (2). The class IB PI3K, termed PI3Kγ, is activated by GPCRs and uses the p110γ catalytic isoform and p101 regulatory isoform (1). The serine/threonine kinase AKT, the major effector of PI3K, regulates survival, growth, and cell cycle control through its many substrates, including the mammalian target of rapamycin (MTOR) and the FOXO transcription factors (3). The phosphatase PTEN counteracts the function of PI3K by converting PIP3 to PIP2, thereby inhibiting AKT signaling (1).
Dysregulation of the PI3K pathway has been implicated in many human malignancies, and chemical inhibitors are rapidly being developed to target this pathway for cancer therapy (4–6). However, since most hematopoietic growth factors activate PI3K signaling, it can be expected that inhibition of PI3K signaling may also affect normal hematopoiesis and HSC self-renewal. In fact, studies using chemical inhibitors or retroviral expression of dominant-negative AKT in cell lines and human CD34+ cells suggest that PI3K may be important for hematopoiesis (7). Targeted deletion of both the p85α and p85β regulatory subunits of PI3K in mice reduces the number of fetal liver progenitors and impairs the repopulating ability of fetal liver HSCs (2, 8). Likewise, deletion of both AKT1 and AKT2 impairs the self-renewal of fetal liver HSCs (9). Furthermore, HSC-specific deletion of PTEN, which activates AKT signaling, causes rapid cycling of HSCs and reduced self-renewal (10, 11). Constitutive activation of AKT, or HSC-specific deletion of the AKT targets FOXO or TSC1, all lead to a similar HSC burnout phenotype (12–14). All of these mouse models suggest that PI3K signaling may play an important role in normal hematopoiesis and HSC maintenance. However, it is still not known whether PI3K is required for adult HSC self-renewal, and even less is known about the contributions of its individual catalytic isoforms to HSC homeostasis.
Of the class IA PI3K catalytic isoforms, p110α (encoded by PIK3CA) and p110β are ubiquitously expressed, whereas expression of p110δ is primarily restricted to leukocytes. While germline deletion of either p110α or p110β results in embryonic lethality, homozygous germline deletion of p110δ produces viable and fertile mice with normal blood counts (15–17). This suggests that p110δ may be dispensable in HSCs. Given the early embryonic lethality resulting from p110α or p110β deletion, the roles of these isoforms in hematopoiesis have been difficult to investigate. The only isoform that is mutated in a broad range of human malignancies is p110α (4). In mouse fibroblasts, deletion of p110α impaired AKT signaling induced by receptor tyrosine kinases, such as IGF-1R, EGFR, and PDGFR, and transformation by oncogenic signals, such as mutated EGFR or NEUT-HER2 (18). Furthermore, p110α is required for transformation by HER2-NEU in a murine model of breast cancer, by KRAS in a murine model of lung cancer, and by HRAS in a murine model of skin cancer (19, 20). Therefore, there is a keen interest in developing isoform-selective inhibitors to target p110α in several human malignancies (6). For example, the p110α-specific inhibitor BYL719 has preclinical activity in several mouse tumor models (21) and is currently being tested in a phase I clinical trial in advanced breast cancer with PIK3CA mutations (22). Data are also emerging that p110α-selective inhibitors may have some activity in AML blasts in vitro (23). However, the efficacy and safety of p110α-selective inhibitors in hematologic malignancies in vivo is unknown. Furthermore, it is not clear in which molecular contexts leukemic cells may be dependent upon p110α.
RAS family members are mutated in 25% of juvenile myelomonocytic leukemia (JMML) and in 15% of acute myeloid leukemia (AML) (24, 25). RAS proteins have been notoriously difficult to target pharmacologically in the clinic, and recent efforts have focused on synthetic lethal approaches or on targeting downstream signaling pathways (25). Among the most important downstream signaling pathways are the PI3K pathway and the RAF/MEK/ERK pathway (26). Genetic or pharmacologic targeting of MEK or ERK has resulted in moderate success in murine models of JMML driven by oncogenic KRAS or by loss of neurofibromin 1 (27–29). However, targeting of ERK may result in excessive hematologic toxicity, as ERK has an important role in normal hematopoiesis (27). Therefore, while MEK and ERK are promising therapeutic targets for RAS-mutated tumors, safer approaches are needed for targeting RAS in hematologic malignancies. Furthermore, there is a large body of evidence that oncogenic RAS proteins signal via multiple pathways, as well as mounting evidence that combination approaches are needed to treat RAS-mutated malignancies (30, 31).
We hypothesized that signaling through the p110α isoform is important for hematopoiesis and HSC self-renewal and may also be important in leukemia. To examine the role of p110α in HSCs and in leukemic cells, we generated Pik3caf/f;Mx1-Cre mice (referred to herein as FA/FA;Cre mice) to enable inducible conditional deletion of Pik3ca in HSCs. We observed that, after postnatal deletion of p110α, mice developed mild anemia, with a defect in terminal erythroblast maturation. However, p110α-deficient HSCs were still capable of long-term reconstitution in vivo. Remarkably, loss of p110α caused a significant improvement in survival in the lox-stop-lox-KrasG12D;Mx1-Cre murine model of JMML (KMX mice). Furthermore, we found that the p110α-selective inhibitor BYL719 (32) partially inhibited KMX BM colony formation in growth factor–free methylcellulose and sensitized these cells to a low dose of the MEK inhibitor MEK162 (33). In addition, we found that the human RAS-mutated cell lines THP1 and NOMO1 were sensitive to BYL719, and even more sensitive to the combination of BYL719 and MEK162. Our results suggest that targeting the p110α isoform of PI3K in combination with low-dose MEK inhibition may be a safer and more effective approach for treating RAS-mutated myeloid leukemias.
Results
Postnatal deletion of Pik3ca results in mild anemia.
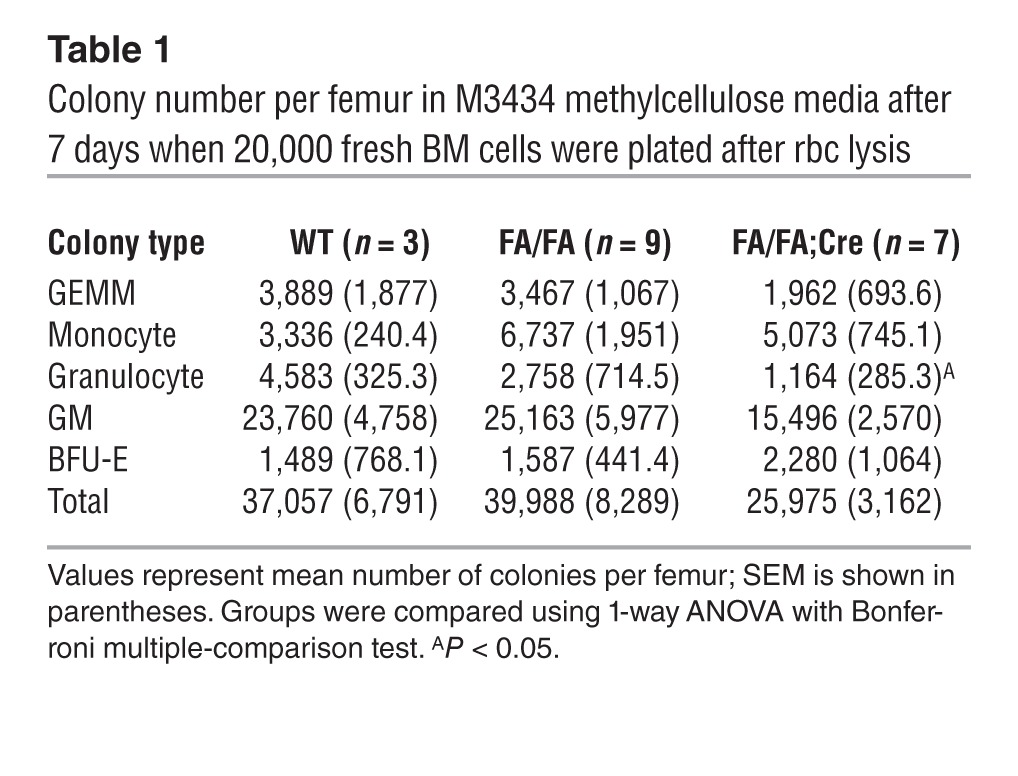
Mice with germline deletion of Pik3ca die between E9.5 and E10.5 from a general proliferative defect (15). We therefore bred Pik3caf/f (FA/FA) mice (18) to Mx1-Cre transgenic mice (34) to enable the conditional deletion of Pik3ca in HSCs, generating FA/FA;Cre mice. The Mx1 promoter, which is expressed in long-term HSCs and some other cell types, can be induced by interferon or by the synthetic ribonucleotide polyI-polyC (pIpC) (34). At 4 weeks after administration of 2 i.p. pIpC injections to 4 week-old mice, p110α protein levels were significantly decreased in the BM of Pik3caf/+;Mx1-Cre mice (FA/+;Cre mice) and undetectable in the BM of FA/FA;Cre mice compared with littermate or age-matched WT controls (Figure 1A). Peripheral blood (PB) counts after pIpC administration revealed a mild but statistically significant anemia in FA/FA;Cre mice compared with age-matched FA/FA littermate controls (Figure 1B). We did not see any significant difference in hemoglobin between WT;Mx1-Cre mice (WT;Cre mice) and FA/FA mice (data not shown). However, wbc and platelet counts of FA/FA;Cre mice were normal. FA/FA;Cre mice also had significantly decreased spleen and thymus weights, as well as mildly decreased BM cellularity (Figure 1C). Flow cytometry revealed no significant differences in the distribution of differentiated myeloid or lymphoid cells in the BM and spleen (Figure 1, D and E, and Supplemental Figure 1, A and B; supplemental material available online with this article; doi: 10.1172/JCI69927DS1). To determine whether loss of p110α affects myeloid colony formation, we plated BM cells from FA/FA;Cre mice after pIpC treatment on M3434 methylcellulose media (see Methods), which contains myeloid growth factors. We did not see any significant differences in the number of total myeloid colonies after loss of p110α, although there was a slight decrease in the number of granulocyte CFUs per femur (Table 1). We observed FA/FA;Cre mice for up to 9 months after pIpC administration; the animals appeared healthy for this time period, with no worsening of blood counts (data not shown). Therefore, p110α appeared to have a minimal role in myeloid and lymphoid development and maintenance in adult mice.
Figure 1. HSC-specific deletion of p110α has minimal effects on hematopoiesis.

(A) Western blot analysis of BM lysates from mice treated with pIpC and sacrificed after 4 weeks. Lanes were run on the same gel but were noncontiguous. (B) PB cell counts obtained from FA/FA;Cre mice and FA/FA and FA/+;Cre littermate controls treated with pIpC at 4 weeks of age and analyzed 3 weeks later. **P < 0.005, ***P < 0.001, 1-way ANOVA with Tukey post-test. (C) BM cell counts from 2 femurs and 2 tibia, spleen weights, and thymus weights of FA/FA;Cre mice, FA/FA and FA/+;Cre littermate controls, and age-matched WT;Cre controls sacrificed 4 weeks after pIpC treatment. 1-way ANOVA with Tukey post-test was used to compare groups. (D and E) Representative flow cytometry plots of BM and spleen single-cell suspensions from FA/FA;Cre mice and FA/FA littermate controls sacrificed 4 weeks after pIpC treatment. See Supplemental Figure 1 for dot plots of individual data for each cell population.
Table 1.
Colony number per femur in M3434 methylcellulose media after 7 days when 20,000 fresh BM cells were plated after rbc lysis
Erythroblast maturation and maintenance is impaired in the absence of p110α.
To understand why FA/FA;Cre mice have anemia, we examined erythroid populations in BM and spleen cell suspensions by flow cytometry with antibodies against CD71 and Ter119. We found a decrease in the frequency of total Ter119+ erythroid cells in FA/FA;Cre BM and spleen (Figure 2A). The Ter119+ population can be further subdivided into stages I–IV of erythroblast maturation (35). We uncovered a block in erythroblast development in the spleens of FA/FA;Cre mice, with an accumulation of cells at stage II of erythroblast maturation (basophilic erythroblasts; Figure 2B). There was also a decrease in the size of the stage IV population (orthochromatophilic erythroblasts; Figure 2B). To determine whether p110α is required for the earliest stages of adult erythroid specification and proliferation, we plated BM and spleen cells in M3334 methylcellulose media, which is supplemented only with erythropoietin and transferrin, to select for the outgrowth of burst-forming unit–erythroid (BFU-E) colonies. Compared with cells from littermate controls, BM and spleen cells from FA/FA;Cre mice formed fewer BFU-E colonies in response to erythropoietin (Figure 2C), which suggested that p110α is specifically required for adult BFU-E formation and for terminal erythroblast maturation.
Figure 2. Deletion of p110α impairs erythroblast differentiation.

(A) Representative flow cytometry plots of BM and spleen single-cell suspensions prepared without rbc lysis 4 weeks after pIpC treatment. Dot plots of individual data are also shown. 2 tailed Student’s t test (BM) or 1-way ANOVA with Tukey post-test (spleen) was used for analysis. (B) The Ter119+ gate was further subdivided into stages I–IV of erythroblast maturation, and the frequency of each cell population in splenocytes was quantified. Representative flow cytometry plots and dot plots of individual data are shown. 2-tailed Student’s t test was used to compare groups. (C) BFU-E colony assays in M3334 methylcellulose media of BM and spleen cells. BFU-E colonies were scored 7 days after plating. Data represent a composite of 3 separate experiments. 1-way ANOVA with Tukey post-test (BM) or 2-tailed Student’s t test (spleen) was used for analysis.
The erythroid stress response to phenylhydrazine is delayed in FA/FA;Cre mice.
To determine whether stress erythropoiesis is affected by loss of p110α, we injected FA/FA and FA/FA;Cre mice with 30 mg/kg phenylhydrazine (PHZ) on 2 consecutive days and then followed the hematocrit (HCT) and reticulocyte index. It has been previously demonstrated that PHZ induces rapid hemolysis, which leads to an erythroid stress response (35). We found that FA/FA;Cre mice were able to respond to PHZ, with a rise in the reticulocyte index and a drop in the HCT to a nadir similar to that of controls (Figure 3, A and B). However, the decrease in the reticulocyte index was delayed, resulting in delayed HCT recovery. Because the spleen is the primary site of stress erythropoiesis in mice, we next performed flow cytometry with CD71 and Ter119 antibodies on mouse spleens 9 days after PHZ treatment to examine erythroblast maturation. This demonstrated an accentuated block in erythroblast maturation at stages I and II and a corresponding decrease at stage IV in FA/FA;Cre spleens (Figure 3C). In controls, the proportion of erythroblasts at stage II increased to 25% after PHZ treatment from a steady-state baseline of 5%, whereas in FA/FA;Cre spleens, this proportion rose to 65% from a baseline of 10% (Figure 2B and Figure 3C). This could explain the sluggish reticulocyte response in the absence of p110α. These data suggested that p110α is particularly important during stress erythropoiesis.
Figure 3. FA/FA;Cre mice have a sluggish response to erythroid stress after PHZ administration.

(A) 6 FA/FA mice and 4 FA/FA;Cre mice were injected with PHZ (30 mg/kg s.c.) on 2 consecutive days. PB was obtained before and 3, 6, and 9 days after the first injection. The experiment was performed twice. (B) Reticulocyte count was determined using methylene blue staining, and the reticulocyte index was calculated (assuming a normal HCT of 45%) as reticulocyte count (%) × (HCT/45) and expressed as a percentage. (C) Amplified block in erythroblast maturation in the spleen after PHZ injection. Spleens from PHZ-treated mice were harvested on day 9 after the first injection, subjected to staining with Ter119 and CD71, and analyzed using FACSCalibur. The Ter119+ gate was further subdivided into stages I–IV of erythroblast maturation, and the frequency of each cell population in splenocytes was quantified. Representative flow cytometry plots and bar graphs of the individual data are shown. **P < 0.005, ***P < 0.001, 2-tailed Student’s t test.
HSC and progenitor numbers are unchanged in FA/FA;Cre mice.
To determine whether postnatal deletion of p110α affects the HSC and progenitor compartments in the BM, we performed multiparameter flow cytometry on the BM of FA/FA;Cre mice 3 weeks after pIpC treatment. We found no significant differences in the total number or frequency of Lin–Sca1+c-Kit+ (LSK) cells — which contain HSCs and multipotential progenitors (MPPs) — or Lin–Sca1–c-Kit+ myeloid progenitors in FA/FA;Cre versus littermate FA/FA or age-matched WT;Cre mice (Figure 4A and data not shown). Furthermore, we did not see any differences in the number or frequency of LSK CD150+CD48– cells, which contain the long-term HSCs, or the LSK CD150–CD48– MPPs (Figure 4B). There were also no differences in the number or distribution of the myeloid progenitor categories: common myeloid progenitors (CMPs), granulocyte-macrophage progenitors (GMPs), or megakaryocyte-erythroid progenitors (MEPs) (Figure 4C). To determine whether loss of p110α affects the cycling properties of HSCs and progenitors, we performed Hoechst and pyronin Y staining on BM from pIpC-treated FA/FA;Cre mice and FA/FA controls. LSK cells from p110α-deleted BM had a normal cell cycle profile (Supplemental Figure 2A). Overall, loss of p110α had no significant effect on stem or progenitor cell populations in the BM.
Figure 4. Deletion of p110α does not affect the number of HSCs or myeloid progenitors in the BM.

BM was stained with a lineage cocktail of antibodies and with antibodies recognizing Sca1, c-Kit, CD150, CD48, and Flk2. (A) Representative flow plots gated on the live lineage-low gate, with LSK and Lin–Sca1–c-Kit+ myeloid progenitor (M Prog) populations indicated. Dot plot showing absolute number of LSK cells per femur is also shown. (B) Representative flow plots gated on LSK cells, with CD150+CD48– HSC and CD150–CD48– MPP populations indicated. Dot plots showing absolute number of HSCs and MPPs per femur are also shown. (C) Representative flow plots, gated on the myeloid progenitor gate, with CMP, GMP, and MEP subpopulations indicated. Dot plots showing absolute numbers of cells per femur are also shown. All statistical analyses were performed using 1-way ANOVA with Tukey post-test.
p110α-deficient HSCs are capable of in vivo long-term multilineage reconstitution.
Although deletion of p110α had no effect on the number of HSCs or progenitors in adult BM, it was still possible that loss of p110α could affect HSC self-renewal. To determine the effect of p110α on HSC fitness in vivo, we performed noncompetitive repopulation experiments (36). BM was harvested from FA/FA;Cre and FA/FA littermate donors 4 weeks after pIpC treatment, and 1 × 106 cells were injected into the tail veins of lethally irradiated B6.SJL recipients. PB was collected from the recipients 4, 8, and 16 weeks after transplant, and donor chimerism was determined by flow cytometry with an anti-CD45.2 antibody (since CD45.2 is expressed in the donor C57BL/6 strain, but not in the recipient B6.SJL strain). Recipients in both groups survived for at least 16 weeks, and we could not detect any significant difference in donor chimerism in the PB over the time course of the experiment (Figure 5A). To test for more subtle defects in HSC self-renewal in direct comparison with WT controls, we performed competitive repopulation experiments (36). BM cells from FA/FA;Cre and FA/FA littermate donors were harvested 4 weeks after pIpC treatment and mixed in a 3:1 ratio with BM cells from WT F1 C57BL/6×B6.SJL mice (whose cells express both CD45.1 and CD45.2), and this mixture was injected into the tail veins of lethally irradiated B6.SJL recipients. Throughout the time course of these experiments, we did not detect any significant difference in donor contribution to the total PB by p110α-deleted BM cells compared with controls (Figure 5B). Furthermore, donor contribution to the myeloid (Mac1+Gr1+) or B cell (B220+) lineages was not significantly different at 16 weeks after transplantation (Figure 5C). We confirmed by PCR that excision of exon 1 of Pik3ca was maintained up to 16 weeks after transplantation, both in the whole BM and in the long-term HSCs of FA/FA;Cre transplant recipients (Supplemental Figure 2, B and C, and ref. 18). Therefore, HSCs maintained full long-term multilineage repopulating ability in the absence of p110α and were as fit as their WT counterparts.
Figure 5. p110α is not essential for HSC self-renewal or for stress hematopoiesis.

(A) Noncompetitive repopulation of BM from pIpC-treated FA/FA;Cre mice or FA/FA littermate controls. PB was collected from transplant recipients at 4, 8, and 16 weeks after transplantation, and donor chimerism was determined by flow cytometry as the frequency of CD45.2+ donor-derived cells. (B) Competitive repopulation using BM from pIpC-treated FA/FA;Cre mice or FA/FA littermate controls, mixed in a 3:1 ratio with competitor BM from WT F1 C57BL/6×B6.SJL mice and injected into lethally irradiated B6.SJL recipients. Donor chimerism was determined as in A. (C) Donor contribution to the Mac1+Gr1+ myeloid and B220+ B cell populations in the PB in competitive repopulation experiments. (D) 5-fluorouracil administration to pIpC-treated FA/FA;Cre mice and FA/FA littermate controls. Statistical analyses were performed using 2-tailed Student’s t test. Experiments were performed twice.
Preserved stress hematopoiesis in FA/FA;Cre mice.
During times of hematopoietic stress, extra demands are placed on HSCs to proliferate and differentiate, in order to enable the rapid regeneration of the hematopoietic system. This becomes especially important during infection or after chemotherapy treatment. To determine whether p110α is required during stress hematopoiesis, we treated FA/FA;Cre and FA/FA mice with a single dose of 5-fluorouracil (200 mg/kg) 4 weeks after pIpC treatment, and then followed the serial wbc counts (37). While the proliferative burst observed at day 15 was slightly dampened in FA/FA;Cre mice, the time course of hematopoietic recovery was unaffected, and all of the animals survived for at least 36 days after treatment (Figure 5D). This suggests that even under times of hematopoietic stress, p110α is not essential for HSC function. The minimal overall importance of p110α in hematopoiesis was surprising, given its important role in growth factor signaling observed in other tissues (18). However, this result is encouraging, as it suggests that p110α could be a relatively safe drug target for the treatment of human malignancies.
Deletion of p110α prolongs the latency of the myeloproliferative neoplasm driven by oncogenic KRAS.
Given the known essential role for p110α in transducing oncogenic RAS signals in solid tumors (20), we asked whether p110α might also play a role in leukemias induced by oncogenic RAS mutations. It has been previously demonstrated that induction of oncogenic KRAS in HSCs using the KMX system results in a rapidly lethal myeloproliferative neoplasm (MPN) in mice, resembling human JMML (38, 39). To determine whether oncogenic KRAS is dependent on p110α in this disease model, we generated mice with the genotype Pik3caf/f;lox-stop-lox-KrasG12D;Mx1-Cre (FAKMX mice) and treated these animals and KMX controls with pIpC at 4 weeks of age. While KMX controls became moribund with weight loss, splenomegaly, hepatomegaly, and elevated wbc counts, with a median survival of 18 days after pIpC treatment, FAKMX animals had a significantly prolonged median survival of 55 days (P = 0.0005; Figure 6A). This suggests that p110α is important in mediating signaling downstream of oncogenic KRAS in this murine model of JMML. However, FAKMX mice eventually developed symptoms similar to those of controls and died from a similar MPN, characterized by infiltration of the PB, spleen, and liver with myeloid cells and expansion of Mac1+Gr1+ myeloid cells in the BM and spleen (Figure 6, B and C, and data not shown). To examine how resistance may progress to p110α loss in the MPN induced by oncogenic KRAS, we performed Western blot analysis on BM lysates from FAKMX and KMX mice 9 days after pIpC and at the time of death (Figure 6, D and E, and Supplemental Figure 3, A and B). First, we confirmed that p110α protein could not be detected after pIpC treatment in FAKMX mice (Figure 6, D and E), to exclude the possibility that incomplete excision was responsible for disease development in these mice. Interestingly, we found that phosphorylation of AKT and the ribosomal protein S6 (a target of MTOR) were detectable in all tested BM samples from KMX and FAKMX mice at the early time point and were variably detectable in both groups at the time of death (Figure 6, D and E). To determine whether expression of other PI3K isoforms may be upregulated in the absence of p110α in FAKMX BM, we also examined the protein expression of p110δ, p110β, and p110γ in BM lysates of moribund mice. The expression levels of p110β and p110γ were undetectable by Western blotting in the BM of both KMX and FAKMX mice (data not shown). While p110δ expression was variably detected in both KMX and FAKMX BM, there was no significant difference in the mean levels of p110δ protein expression between groups (Supplemental Figure 4). However, there appeared to be a correlation between levels of p110δ expression and AKT phosphorylation in both groups. We did not see any differences in the levels of STAT5 phosphorylation between KMX and FAKMX samples (Supplemental Figure 4), which suggests that this is not a likely mechanism to explain resistance to p110α loss. Overall, these data suggest the existence of multiple compensatory mechanisms in JMML, at least one of which involves the activation of AKT and MTOR downstream of oncogenic KRAS in the absence of p110α, through an alternative mechanism.
Figure 6. KRASG12D in a murine model of JMML is dependent upon p110α.

(A) Kaplan-Meier survival curves of FAKMX mice and KMX controls after 1 dose of pIpC (100 μg) at 4 weeks of age. MS, median survival. The log-rank test was used to compare survival between groups. (B) Representative blood smears and liver histology sections from moribund FAKMX mice and KMX controls at the time of sacrifice. Original magnification, ×60. (C) Representative flow cytometry plots of BM and spleen cells from moribund FAKMX mice and KMX controls stained with Mac1 and Gr1 antibodies. (D) Western blot analysis of BM lysates from FAKMX mice and KMX controls sacrificed 9 days after pIpC treatment. The same membrane was stripped and incubated with anti-AKT and anti-ERK antibodies. (E) Western blot analysis of BM lysates from moribund FAKMX mice and KMX controls. The membrane was stripped and incubated with anti-AKT antibody. Lanes were run on the same gel but were noncontiguous. See Supplemental Figure 3, A and B, for quantification of signal intensities.
Other PI3K isoforms can partially compensate for p110α downstream of KRASG12D in JMML.
One possibility to explain this phenomenon is that another p110 catalytic isoform or isoforms of PI3K can compensate for p110α downstream of KRASG12D. To test this, we plated BM cells from pIpC-treated KMX mice in growth factor–free M3231 methylcellulose media and treated the plates with isoform-selective pharmacologic PI3K inhibitors (each at its maximal isoform-selective dose) or with the pan-PI3K inhibitor BKM120 (40). It was previously demonstrated that BM cells from KMX mice are able to form myeloid colonies in the absence of growth factors (38, 39). We found that KMX BM colony formation was only modestly decreased after treatment with the p110α-selective inhibitor BYL719, the p110δ-selective inhibitor GS1101 (also known as CAL101) (41), or the p110β-selective inhibitor TGX221 (42); however, the combination of BYL719 with either GS1101 or TGX221 inhibited colony formation by about 2-fold, as effectively as the pan-PI3K inhibitor BKM120 (Figure 7A). Notably, BYL719 would be expected to lose stability after 7 days in aqueous culture (C. Fritsch, unpublished observations), so these data may underestimate the effects of BYL719 due to constraints of the experimental design. The p110γ-selective inhibitor NVS-PI3-5 (43) did not inhibit colony formation at this dose (Figure 7A). After short-term inhibitor treatment of KMX BM in vitro, BYL719, GS1101, and BKM120 all reduced phosphorylation of AKT at Ser473 (Supplemental Figure 5A).
Figure 7. A combination of PI3K and MEK inhibitors reduces myeloid colony formation by KMX BM.

(A–C and E) Number of total colonies observed 7 days after plating 1 × 105 BM cells from pIpC-treated KMX (A and C) or FAKMX mice (B and E) in M3231 growth factor–free methylcellulose media. The final concentration of each inhibitor in methylcellulose is as follows: BYL719, 1 μM; GS1101, 1 μM; TGX221, 1 μM; BKM120, 1 μM; NVS-PI3-5, 100 nM; MEK162, 100 nM. PI3K inhibitors were used at the highest dose at which isoform selectivity could be expected for each. Each experiment was performed 3 times. *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA with Bonferroni multiple-comparison post-test. (D) Freshly harvested KMX BM cells were cultured for 1 hour in the presence of the same inhibitors, stimulated with 1 ng/ml GM-CSF for 5 minutes, lysed, and subjected to Western blotting. Lanes were run on the same gel but were noncontiguous. The experiment was performed 3 times. The membrane was stripped and incubated with anti-AKT and anti-ERK antibodies. See Supplemental Figure 5B for quantification of signal intensities.
In BM from pIpC-treated FAKMX mice, GS1101 reduced colony number about 2-fold as a single agent, nearly as effectively as BKM120 (Figure 7B). Inhibition of p110β, p110γ, or p110α was not effective in blocking colony formation of p110α-deleted cells. Therefore, other p110 isoforms of PI3K were able to compensate for p110α downstream of KRASG12D in BM cells. Remarkably, inhibition of only 1 additional PI3K isoform together with p110α was sufficient to reduce colony formation to the same extent as pan-PI3K inhibition (Figure 7, A and B). Of the other p110 isoforms, p110δ appeared to be the most important for transducing oncogenic KRAS signals in BM the absence of p110α (Figure 7B). However, even the pan-PI3K inhibitor BKM120 failed to completely inhibit colony formation, highlighting the potential importance of other parallel signaling pathways downstream of oncogenic KRAS.
RAS proteins also signal via the RAF/MEK/ERK pathway, and MEK inhibitors have shown preclinical efficacy in RAS-mutated malignancies, including in this murine model of JMML (25, 28, 29). Therefore, we tested the MEK inhibitor MEK162 in the KMX BM growth factor–free methylcellulose colony assay (Figure 7C). We hypothesized that in RAS-mutated leukemia cells, a minimally effective low dose of a MEK inhibitor would be more effective combined with a PI3K inhibitor. Myeloid cells in JMML are characterized by hypersensitivity to GM-CSF (38). Therefore, to test this hypothesis, we titrated down the dose of MEK162 to 100 nM and tested this inhibitor alone and in combination with PI3K inhibitors in the presence of increasing concentrations of GM-CSF. The doses of GM-CSF used in this experiment were lower than the minimal dose required for myeloid colony formation by WT BM (38). At this dose, MEK162 caused only moderate inhibition of colony formation by KMX BM (Figure 7C). However, addition of BYL719 or BKM120 sensitized KMX cells to MEK162, resulting in a more significant decrease in colony formation, especially with increasing doses of GM-CSF (Figure 7C). We then treated cultured BM cells from KMX mice with the same inhibitors at the same doses for 1 hour, followed by stimulation with 1 ng/ml GM-CSF for 5 minutes. Western blotting revealed that MEK162 reduced levels of phospho-ERK in KMX BM at this dose, without affecting levels of phospho-AKT (Figure 7D). In contrast, MEK162 combined with either BYL719 or BKM120 reduced both phospho-AKT and phospho-ERK in response to GM-CSF (Figure 7D and Supplemental Figure 5B).
As expected, MEK162 at 100 nM was also effective in reducing colony formation in BM from FAKMX mice in the absence of growth factors, and was more effective in combination with BKM120 (Figure 7E). Furthermore, we found that GS1101 could be substituted for BKM120 in combination with MEK162 in p110α-deleted cells with similar results. In short-term drug treatment assays of FAKMX BM, MEK162 combined with either GS1101 or BKM120 reduced both phospho-AKT and phospho-ERK in response to GM-CSF (Supplemental Figure 6). Therefore, the combined inhibition of MEK and PI3K effectively blocked myeloid colony formation and AKT and ERK phosphorylation in the KMX JMML murine model, both in the absence of growth factors and in the presence of GM-CSF. Furthermore, a p110α-selective inhibitor could be substituted for a pan-PI3K inhibitor in combination with a MEK inhibitor in these assays with similar results. These data suggest that compensatory signaling through the RAF/MEK/ERK pathway is at least partially responsible for the emergence of resistant JMML after deletion of p110α.
The combination of a PI3K inhibitor and a low-dose MEK inhibitor reduces proliferation in RAS-mutated AML cell lines.
We were encouraged by the effectiveness of the combined MEK and PI3K inhibition in the KMX murine model of JMML. To determine whether a similar approach may be effective in RAS-mutated AML cells, we tested 2 human AML cell lines: THP1 cells, which have an NRAS mutation, and NOMO1 cells, which have a KRAS mutation (44, 45). We treated these cell lines in 72-hour MTS proliferation assays with PI3K inhibitors, MEK162, and combinations thereof. First, we found that both THP1 and NOMO1 cells were only mildly sensitive to PI3K isoform–selective inhibitors, but were more sensitive to the combination of BYL719 and GS1101 and to the pan-PI3K inhibitor BKM120 (Figure 8, A and B). Based on 72-hour MTS proliferation assays in THP1 cells, the 50% growth inhibition dose (GI50) was 1.5 μM for BYL719, 750 nM for BKM120, and 15 nM for MEK162. To determine whether the combination of PI3K inhibitors and low-dose MEK inhibitors could be more effective than the individual inhibitors, we treated THP1 and NOMO1 cells with 25 nM MEK162 combined with 1 μM BYL719 or 1 μM BKM120. These combinations reduced proliferation to a greater extent than MEK162 alone (Figure 8, A and B, and Supplemental Figure 7A).
Figure 8. A combination of PI3K and MEK inhibitors blocks proliferation and AKT and MAPK signaling in RAS-mutated AML cell lines.

(A–D) 72-hour MTS proliferation assays. To calculate proliferation, each absorbance value was divided by the mean absorbance of DMSO-treated cells. The concentration of each inhibitor used is as follows: BYL719, 1 μM; GS1101, 1 μM; TGX221, 1 μM; BKM120, 1 μM; NVS-PI3-5, 100 nM; MEK162, 25 nM. Each experiment was performed 3 times. *P < 0.05, **P < 0.01, ***P < 0.001, 1-way ANOVA with Dunnett multiple-comparison test (vs. DMSO control) or with Tukey post-test (as denoted by brackets). (E) Western blot analysis of AKT and MAPK signaling in THP1 cells after drug treatment for the indicated times, at the concentrations listed above. The same membrane was stripped and incubated with anti-AKT, anti-ERK, and anti-S6 antibodies. Lanes were run on the same gel but were noncontiguous. Each experiment was performed 3 times. See Supplemental Figure 10 for quantification of signal intensities.
However, these AML cell lines have multiple different genetic alterations, so sensitivity to these inhibitors cannot be attributed only to mutated RAS. To examine the effects of oncogenic KRAS in isolation, we also tested murine BaF3 cells transduced with a retrovirus expressing KRASG12V (another oncogenic KRAS mutation), which are transformed to growth factor independence. These cells exhibited a pattern of inhibitor sensitivity similar to that seen in the human RAS-mutated AML cell lines (Figure 8C). In KRASG12V cells, the GI50 for MEK162 in the 72-hour MTS proliferation assay was 25 nM. Addition of either BYL719 or BKM120 sensitized BaF3 KRASG12V cells to this low dose of MEK162, resulting in more significant inhibition of proliferation than with MEK162 alone (Figure 8C and Supplemental Figure 7B). Interestingly, these combinations had no effect on BaF3 cells transduced to growth factor independence with a retrovirus expressing FLT3-ITD (Figure 8D). This suggested that sensitivity to these drug combinations cannot be generalized to all molecular subtypes of leukemia and may be specific for leukemias harboring RAS mutations. Furthermore, these drug combinations at these doses did not have a generalized cytotoxic effect.
One rationale for combining MEK inhibitors and PI3K inhibitors in the treatment of RAS-mutated tumors is that both downstream pathways converge on the tuberous sclerosis complex, thereby activating MTOR, which ultimately leads to phosphorylation of the ribosomal protein S6 (46). To determine whether this could be a possible mechanism for the combined effects of MEK and PI3K inhibitors that we observed in AML cell lines, we performed Western blot analysis on lysates from THP1 cells treated with each inhibitor or combinations thereof (Supplemental Figure 7C). Of the PI3K inhibitors tested, only BYL719, GS1101, and BKM120 could decrease both phospho-AKT and phospho-S6 in these cells. We obtained similar results in BaF3 KRASG12V and NOMO1 cells (Supplemental Figure 7C, Supplemental Figure 8, and Supplemental Figure 9).
To examine the dynamic effects of combining MEK162 with either BYL719 or BKM120 in AML cells, we performed time course assays in which cultured THP1 cells were treated for 15 minutes or for 1, 6, or 24 hours. At all time points tested, the PI3K inhibitors BYL719 and BKM120 both reduced phospho-AKT at Ser473 and phospho-S6 at Ser235/236, while not affecting phospho-ERK (Figure 8E and Supplemental Figure 10). After at least 1 hour of treatment, MEK162 reduced both phospho-ERK and phospho-S6, without affecting phospho-AKT. However, MEK162 in combination with either BYL719 or BKM120 led to a reduction in phospho-AKT, phospho-ERK, and particularly phospho-S6, an effect that was still detectable after 24 hours of treatment. We observed similar results in time course assays on NOMO1 cells (Supplemental Figure 11). These results suggested that the convergent effects of combined MEK and PI3K inhibition on MTOR may explain, at least in part, their cooperative effect on RAS-mutated AML cell lines.
BYL719 and MEK162 have in vivo efficacy in a murine xenograft model of AML.
Although BYL719 and MEK162 are currently in clinical trials for solid tumors, both individually and in combination (ClinicalTrials.gov ID NCT01449058), these drugs have not been tested in AML in vivo. To test the in vivo efficacy of BYL719 and MEK162 in AML, we performed xenograft studies in mice. 4.5 × 106 THP1 cells transduced with a luciferase-expressing lentiviral vector were injected into the tail veins of NOG mice, and drug administration was initiated 5 days later by daily oral gavage. Bioluminescence images obtained after 7 days of drug treatment revealed a decrease in disease burden in the MEK162 and combination treatment groups (Figure 9A). All animals were sacrificed after 11 days of drug treatment for disease burden analysis, which included assessment of liver weight and of human CD45+ leukemic cell frequency in BM, spleen, and PB by flow cytometry. Animals treated with 40 mg/kg/d BYL719 had significantly lower liver weights and a trend toward a lower percentage of human leukemic cells in all tissues tested (Figure 9B and Supplemental Figure 12A). Animals treated with MEK162 at 10 mg/kg/d had a significantly decreased disease burden by all parameters tested. These doses were close to the doses currently used in patients in clinical trials (C. Fritsch, unpublished observations). At this dose of MEK162, the addition of BYL719 did not cause any further decrease in disease burden.
Figure 9. BYL719 and MEK162 reduce disease burden in a THP1 murine xenograft AML model.

(A) Bioluminescence imaging of mice 12 days after injection with 4.5 × 106 THP1-luciferase cells, 7 days after the initiation of drug treatment. The intensity scale of the bioluminescence signal applies to all images. (B) Liver weights of THP1 xenografted mice 11 days after initiation of treatment with 40 mg/kg/d BYL719, 10 mg/kg/d MEK162, or the combination. (C) Liver weights of mice xenografted with 4.5 × 106 THP1 cells, then treated for 11 days with vehicle or low-dose MEK162 (2 mg/kg/d), alone or in combination with 40 mg/kg/d BYL719. *P < 0.05, ***P < 0.001, 1-way ANOVA with Bonferroni multiple-comparison post-test.
There are concerns with potential toxicity in studies using combined MEK and PI3K inhibitors, and MEK inhibitors in particular have been associated with frequent adverse events (33). To determine whether MEK162 could be effective at a lower, presumably less toxic, dose when combined with BYL719, we tested 2 mg/kg/d MEK162 alone and in combination with 40 mg/kg BYL719 in a second cohort of mice xenografted with THP1 cells. At this dose, MEK162 caused a mild, nonsignificant decrease in liver weight compared with the vehicle control at 11 days of treatment (Figure 9C). However, addition of BYL719 to MEK162 led to a significant decrease in liver weight, suggesting a further decrease in disease burden. Furthermore, at these doses, there was little evidence of toxicity in the mice, with minimal weight loss throughout the course of treatment (Supplemental Figure 12B). These data suggested that both BYL719 and MEK162 could be effective in treating AML in vivo and that it may be possible to use a lower dose of MEK162 when it is combined with BYL719.
Discussion
The PI3K/AKT pathway is frequently mutated or upregulated in human malignancies, and many efforts have been focused on targeting this pathway safely and effectively in cancer. In preclinical studies, certain RAS-mutated solid tumors are dependent specifically on the p110α isoform of PI3K (20, 31). However, it is not known whether RAS-mutated leukemias are also dependent on p110α, or what role p110α has in normal hematopoiesis. Here, we present evidence that the p110α isoform of PI3K is not essential for HSC self-renewal, but does have a role in terminal erythroblast maturation. Furthermore, our data demonstrated that combined pharmacologic inhibition of p110α together with MEK may be an effective therapeutic approach for RAS-mutated JMML and AML.
Given the many roles that the PI3K/AKT pathway plays in regulating apoptosis, the cell cycle, and protein synthesis, inhibition of the entire PI3K enzyme family at sufficiently high doses to achieve a therapeutic effect could lead to untoward adverse effects. To minimize toxicity from PI3K inhibition, isoform-selective approaches for targeting PI3K are currently under investigation in the clinic for some types of cancer as well as for other clinical indications (22). However, the specific in vivo roles of each of the catalytic isoforms of PI3K in hematopoiesis are unknown. To determine whether the p110α isoform of PI3K is essential for hematopoiesis and leukemia, we generated FA/FA;Cre mice (genotype Pik3caf/f;Mx1-Cre) to allow for inducible deletion of p110α in HSCs. Surprisingly, deletion of p110α in the hematopoietic system did not affect the number of HSCs or myeloid progenitors, and p110α-deleted BM cells were capable of long-term multilineage reconstitution in lethally irradiated recipients, even when competing against WT cells. Furthermore, deletion of p110α also had no effect on hematopoietic recovery after chemotherapy. This was unexpected, given the crucial role of p110α previously demonstrated in growth factor–induced AKT signaling in fibroblasts (18). However, this result may be explained in part by the increased redundancy of PI3K catalytic isoforms in hematopoietic cells, which have 4 class I p110 isoforms (α, β, γ, and δ), unlike most other cell types, which have only 2 (α and β). This heightened redundancy may speak to the importance of tight regulation of PI3K/AKT in HSCs. To further address this question, compound deletion of several or all class I PI3K isoforms will need to be performed in HSCs in order to determine whether PI3K is truly essential for HSC self-renewal.
After postnatal deletion of p110α in HSCs, FA/FA;Cre mice developed a mild but reproducible anemia, characterized by a decrease in total Ter119+ cells in the BM and spleen. BM from p110α-deleted mice demonstrated defective BFU-E colony formation in response to erythropoietin, suggestive of a defect in erythroid progenitor maturation. This is consistent with the previously described essential role of PI3K in mediating erythroblast progenitor proliferation in response to erythropoietin (47–49) and with the binding of erythropoietin receptor to the p85α regulatory subunit of PI3K (48). We did not observe any changes in MEP numbers or in platelet counts in FA/FA;Cre mice, which suggests that the defect is unlikely to be at the level of MEP specification. We also observed a block in erythroblast maturation at the basophilic erythroblast stage, which led to a decrease in mature erythroblasts. This block may also be due to decreased responsiveness to erythropoietin, which has been widely implicated in promoting erythroblast survival and is required for terminal erythroblast maturation (50). Our results underscored the importance of PI3K in erythropoietin signaling and provided the first evidence for a specific role for PI3K p110α in erythroblast proliferation and maturation. However, the mild degree of anemia observed in FA/FA;Cre mice suggests that pharmacologic treatment with a p110α-selective inhibitor would be unlikely to cause life-threatening anemia in patients.
Overall, the mild effects of p110α deletion on hematopoiesis suggest that p110α could be a safe pharmacologic target for cancer therapy. Because of the design of the present study, we cannot comment on nonhematologic toxicities of the pharmacologic targeting of p110α in humans. However, limited manageable toxicities have been reported in early clinical trials with the p110α-specific inhibitor BYL719, mainly related to the role of p110α in insulin signaling (22). While there is preclinical evidence for the dependence of certain solid tumors on p110α, no such evidence exists in hematologic malignancies. Surprisingly, we found that postnatal deletion of p110α significantly prolonged survival in the murine model of JMML induced by oncogenic KRAS. Furthermore, treatment with the p110α-selective inhibitor BYL719 reduced colony formation in the absence of growth factors by KMX BM cells and reduced phosphorylation of AKT in KMX BM cells and in RAS-mutated AML cell lines. Despite the redundancy between PI3K isoforms in hematopoietic cells, oncogenic RAS still maintained some dependence on the p110α isoform in myeloid leukemic cells.
Although myeloid leukemic cells in both JMML and AML appeared to have some dependence on p110α, this effect appeared to be short-lived, as FAKMX mice did eventually develop JMML-like disease. Our analysis of colony formation by BM cells from these mice and from KMX cells cultured with isoform-selective PI3K inhibitors suggests that compensation by other p110 isoforms of PI3K can only partially explain this resistance to p110α loss. Here we demonstrated that it is crucial to simultaneously inhibit both PI3K and MAPK signaling downstream of RAS mutations in myeloid malignancies. Furthermore, we showed that the combination of a PI3K inhibitor and a MEK inhibitor could function, at least in part, through a combinatorial effect on MTOR signaling, as has been demonstrated in other cell types (46). Other potential mechanisms could also explain the effectiveness of this powerful combination, including overlapping effects on the apoptosis machinery (51). The limitation for this combination approach in the clinic has been increased toxicity (52, 53). One way to overcome this toxicity is to use a lower dose of the MEK inhibitor, which may be easier when it is combined with a PI3K inhibitor. Using an isoform-selective PI3K inhibitor (such as BYL719) instead of a pan-PI3K inhibitor could further minimize potential toxicity. Our data suggested that a p110α-specific PI3K inhibitor can exhibit efficacy similar to that of a pan-PI3K inhibitor in both JMML and AML with RAS mutations when combined with a low-dose MEK inhibitor.
Several pharmaceutical companies are currently developing isoform-selective PI3K inhibitors, and several of these are in clinical trials (6). Therefore, it is essential to understand the therapeutic applications of these inhibitors in hematologic malignancies as well as their potential toxicity to the hematopoietic system. Here we presented the first in vivo evidence that inactivation of p110α is not essential for HSC self-renewal in mice. Furthermore, we demonstrated that p110α inhibitors could be used effectively in combination with MEK inhibitors to treat myeloid malignancies with RAS mutations, both in vitro and in vivo. It will be interesting to see whether this combination could also be applied to safely and effectively treat RAS-mutated solid tumors.
Methods
Mice.
FA/FA;Cre (Pik3caf/f;Mx1-Cre) mice were backcrossed against C57BL/6 inbred mice for 9 generations (18). KrasG12D;Mx1-Cre mice were obtained by breeding KrasG12D mice (obtained from T. Jacks, Massachusetts Institute of Technology, Cambridge, Massachusetts, USA) to Mx1-Cre mice (from G. Gilliland, Brigham and Women’s Hospital, Boston, Massachusetts, USA) and were backcrossed against C57BL/6 inbred mice for 9 generations. CIAE NOG mice were purchased from Taconic. All mice were housed in a pathogen-free animal facility at Children’s Hospital Boston or at Dana-Farber Cancer Institute in microisolator cages. pIpC (GE Healthcare) was dissolved in PBS and injected (250 μg/mouse i.p.) twice on alternating days at 4 weeks postnatally, except where otherwise indicated. Genotyping and excision analysis for Pik3ca by PCR was performed as previously described (18). Littermate controls were used for all experiments, unless stated otherwise in the figure legends. PB was obtained by retro-orbital venous blood sampling. PB counts were analyzed on a Hemavet 950FS blood analyzer (Drew Scientific).
Bioluminescence imaging.
Mice were anesthetized with ketamine and xylazine, and d-luciferin (Promega) was administered i.p. to monitor luciferase gene expression in vivo. Images were obtained with Kodak Image Station 4000MM and analyzed with KODAK Molecular Imaging Software (version 4.5.0b6 SE).
Histology.
Recipient mice were sacrificed by CO2 asphyxiation when they began to show signs of disease (weight loss, splenomegaly, or difficulty breathing). Organs were fixed in formalin with 5% paraformaldehyde, and histology slides were prepared and stained by the Brigham and Women’s core Rodent Histology Facility. Digital images were acquired using a Nikon Eclipse E400 microscope equipped with a digital camera and analyzed using Spot Advanced software.
Flow cytometry.
BM cells, splenocytes, and thymocytes were harvested and subjected to rbc lysis using rbc lysis solution (Qiagen), except in Figure 2, A and B. Fresh or frozen cells were stained with the antibodies PE-conjugated Mac1, APC-conjugated Gr1, APC-conjugated c-Kit, PE-conjugated CD71, APC-conjugated Ter119, PE-conjugated B220, and APC-conjugated Thy1.2 (Becton-Dickinson), then analyzed on BD FACSCalibur or BD FACSCanto II instruments. Staining for multiparameter flow cytometry was performed as previously described (12), and cells were analyzed on BD FACSAria or BD FACSCanto II instruments. Cell cycle analysis using Hoechst 33342 and pyronin Y was performed as previously described (12).
Colony-forming assays.
BM and spleen cells were harvested, subjected to rbc lysis, and resuspended in IMDM with 10% FBS and 5% penicillin-streptomycin. Cells were plated in duplicate in M3434 methylcellulose media (Stem Cell Technologies) at 1 × 104 cells/dish for BM and 5 × 104 cells/dish for spleen cells. For plating in M3334 methylcellulose media (Stem Cell Technologies), 5 × 105 spleen cells were plated per dish. For plating in M3231 methylcellulose media (Stem Cell Technologies), 1 × 105 BM cells were plated per dish. Colonies were scored after 7 days. BYL719, BKM120, NVS-PI3-5, and MEK162 were obtained from Novartis Institutes of Biomedical Research through a Material Transfer Agreement with Dana-Farber Cancer Institute. GS1101 was purchased from Selleck Chemicals. TGX221 was purchased from Chemdea. All inhibitors were maintained as stocks of 10–200 μM in DMSO at –20°C.
Western blotting.
Whole-cell protein lysates were prepared from single-cell suspensions of BM cells or cell lines. Western blotting was performed as previously described (13), with the following modifications. Nitrocellulose 0.45-μM membranes (Millipore) were used for transfer. Blocking was performed in 1× TBS, 5% nonfat milk. Primary antibodies used were as follows: p110α (catalog no. 4249; Cell Signaling), AKT (catalog no. 9272; Cell Signaling), phospho-AKT (Ser473) (catalog no. 44-621G; BioSource), S6 (catalog no. 2217; Cell Signaling), phospho-S6 (catalog no. 2211; Cell Signaling), ERK (catalog no. 9102; Cell Signaling), phospho-ERK (catalog no. 9101; Cell Signaling), α-tubulin (catalog no. T9026; Sigma-Aldrich). The directly fluorophore-conjugated secondary antibodies anti-mouse 680 and anti-rabbit 800 (LICOR) were used at dilutions of 1:5,000 or 1:10,000, respectively. The final wash was performed in 1× TBS. Membranes were developed on the LICOR Odyssey instrument. Signal intensity with background correction was quantified using LICOR Image Studio software. Stripping was performed using Re-blot Plus Strong solution (Millipore) for 15 minutes at room temperature.
PHZ stress test.
PHZ (Sigma-Aldrich) was diluted in PBS and injected on 2 consecutive days (30 mg/kg s.c.). Blood was collected prior to injection and at days 3, 6, and 9 for HCT and reticulocyte count measurements. Reticulocyte count was measured by counting the number of reticulocytes and rbcs on blood smears stained with new methylene blue “n” (Ricca Chemical) (35). At least 500 cells were counted per time point. Corrected reticulocyte count was calculated as described in the Figure 3 legend.
Repopulation and transplantation assays.
Donors were backcrossed against C57BL/6 inbred mice for 9 generations. For competitive repopulation experiments, WT competitor BM was obtained from age-matched F1 offspring from mating between C57BL/6 and B6.SJL mice. Transplant recipients were 6- to 8-week-old B6.SJL mice. For lethal irradiation, mice were given 2 doses of 550 cGy at least 3 hours apart on the day of transplant. Donor BM was injected into the tail vein or into the retro-orbital plexus. Recipient animals were maintained on acidified water or Baytril water for 2 weeks after transplant.
5-fluorouracil administration.
After pIpC treatment, 5-fluorouracil (Sigma-Aldrich) was injected once (200 mg/kg i.p.). PB was collected before and 3, 7, 14, 21, 30, and 36 days after injection.
Cell culture.
BaF3, NOMO1, and THP1 cells were obtained from G. Gilliland (Brigham and Women’s Hospital, Boston, Massachusetts, USA) and maintained in RPMI with 10% FCS. For generation of luciferase-expressing THP1 cells, 6 μg pLENTI-Luciferase-Blasticidin vector (gift from H. Chang, Dana-Farber Cancer Institute, Boston, Massachusetts, USA), 2 μg of the packaging plasmid VSVG and 1 μg of D-8-9 helper plasmid were cotransfected into HEK293T packaging cells with Fugene 6 (Roche). After 72 hours, the culture supernatants containing lentivirus were collected, filtered (0.45 μm), and stored at 4°C until use. THP1 cells were transduced twice with lentivirus at 37°C overnight. 2 days after transduction, cells were selected in 35 μg/ml blasticidin for 10 days. Polyclonal cells were analyzed and confirmed for luciferase expression using the Promega Luciferase assay system (E1500), then expanded for transplantation. Cells were confirmed to be mycoplasma- and pathogen-free prior to injection into mice.
MTS proliferation assays.
Cell lines were plated at 2 × 105 cells/well in 24-well plates and incubated in drug solution for 72 hours. An aliquot was removed every 24 hours and mixed with Aqueous One reagent (Promega), then incubated for 1 hour at 37°C, after which absorbance readings at 490 nM were taken.
Statistics.
Graphpad Prism 4.0 was used for all statistical analyses. The specific statistical test used is indicated in each figure legend. In all graphs, error bars reflect mean ± SEM. A P value less than 0.05 was considered significant.
Study approval.
All use of animals described herein was approved by the IACUC of Dana-Farber Cancer Institute.
Supplementary Material
Acknowledgments
We thank James Griffin for critical review of the manuscript; David Tuveson and Tyler Jacks for KrasLSL-G12D mice; Gary Gilliland for Mx1-Cre mice, cell lines, and valuable advice; Benjamin Ebert for use of the BD FACSCalibur and BD FACSCanto II in the Brigham and Women’s Hospital Hematology Division; and Michael Kharas, Steven Lane, Demetrios Kalaitzidis, Lukas Baitsch, and all members of the Roberts and Zhao labs for helpful discussions. This work was supported by an American Society of Hematology Fellow Scholar Award (to K. Gritsman), by the Friends of Dana-Farber Fund (to K. Gritsman), by NCI grant K08 CA149208 (to K. Gritsman), by NIH grant R01 CA172461 (to J.J. Zhao), and by Dana-Farber Fund grant 8508054 (to J.J. Zhao).
Footnotes
Conflict of interest: Christine Fritsch, Sauveur-Michel Maira, and Gregory Hollingworth are employees of Novartis Institutes of Biomedical Research. Thomas M. Roberts is a consultant for Novartis Pharmaceuticals Corp.
Citation for this article:J Clin Invest. 2014;124(4):1794–1809. doi:10.1172/JCI69927.
References
- 1.Polak R, Buitenhuis M. The PI3K/PKB signaling module as key regulator of hematopoiesis: implications for therapeutic strategies in leukemia. Blood. 2012;119(4):911–923. doi: 10.1182/blood-2011-07-366203. [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci. 2005;30(4):194–204. doi: 10.1016/j.tibs.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 5.Jia S, Roberts TM, Zhao JJ. Should individual PI3 kinase isoforms be targeted in cancer? Curr Opin Cell Biol. 2009;21(2):199–208. doi: 10.1016/j.ceb.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brana I, Siu LL. Clinical development of phosphatidylinositol 3-kinase inhibitors for cancer treatment. BMC Med. 2012;10(1):161. doi: 10.1186/1741-7015-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buitenhuis M, et al. Protein kinase B (c-akt) regulates hematopoietic lineage choice decisions during myelopoiesis. Blood. 2008;111(1):112–121. doi: 10.1182/blood-2006-07-037572. [DOI] [PubMed] [Google Scholar]
- 8.Haneline LS, et al. Genetic reduction of class IA PI-3 kinase activity alters fetal hematopoiesis and competitive repopulating ability of hematopoietic stem cells in vivo. Blood. 2006;107(4):1375–1382. doi: 10.1182/blood-2005-05-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Juntilla MM, Patil VD, Calamito M, Joshi RP, Birnbaum MJ, Koretzky GA. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–4038. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441(7092):475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441(7092):518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 12.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Kharas MG, et al. Constitutively active AKT depletes hematopoietic stem cells induces leukemia in mice. Blood. 2010;115(7):1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205(10):2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110α subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274(16):10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 16.Jou ST, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110δ in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22(24):8580–8591. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110β catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13(3):169–172. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 18.Zhao JJ, et al. The p110α isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103(44):16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Utermark T, et al. The p110α and p110β isoforms of PI3K play divergent roles in mammary gland development and tumorigenesis. Genes Dev. 2012;26(14):1573–1586. doi: 10.1101/gad.191973.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110α is required for ras-driven tumorigenesis in mice. Cell. 2007;129(5):957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 21. Fritsch C, et al. Abstract 3748: NVP-BYL719, a novel PI3Kalpha selective inhibitor with all the characteristics required for clinical development as an anti-cancer agent. Poster presented at: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; March 31, 2012–April 4, 2012; Chicago, Illinois, USA. Abstract nr 3748. [Google Scholar]
- 22. Juric D, et al. Abstract CT-01: BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first-in-human study. Paper presented at: Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research; March 31, 2012–April 4, 2012; Chicago, Illinois, USA. Abstract nr CT-01. [Google Scholar]
- 23.Xing Y, Gerhard B, Hogge DE. Selective small molecule inhibitors of p110α and δ isoforms of phosphoinosityl-3-kinase are cytotoxic to human acute myeloid leukemia progenitors. Exp Hematol. 2012;40(11):922–933. doi: 10.1016/j.exphem.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 24.Patel JP, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120(17):3397–3406. doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellano E, Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol. 2010;346:143–169. doi: 10.1007/82_2010_56. [DOI] [PubMed] [Google Scholar]
- 27.Staser K, et al. Normal hematopoiesis neurofibromin-deficient myeloproliferative disease require Erk. J Clin Invest. 2013;123(1):329–334. doi: 10.1172/JCI66167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang T, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–339. doi: 10.1172/JCI63193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyubynska N, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3(76):76ra27. doi: 10.1126/scitranslmed.3001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sos ML, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106(43):18351–18356. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14(12):1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Furet P, et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase α inhibitor selected for clinical evaluation. Bioorg Med Chem Lett. 2013;23(13):3741–3748. doi: 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 33.Ascierto PA, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14(3):249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 34.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 35.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(–/–)5b(–/–) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–3273. doi: 10.1182/blood.V98.12.3261. [DOI] [PubMed] [Google Scholar]
- 36.Growney JD, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106(2):494–504. doi: 10.1182/blood-2004-08-3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng T, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 38.Braun BS, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci U S A. 2004;101(2):597–602. doi: 10.1073/pnas.0307203101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan IT, et al. Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease. J Clin Invest. 2004;113(4):528–538. doi: 10.1172/JCI20476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koul D, et al. Antitumor activity of NVP-BKM120 — a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clin Cancer Res. 2012;18(1):184–195. doi: 10.1158/1078-0432.CCR-11-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herman SE, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. 2010;116(12):2078–2088. doi: 10.1182/blood-2010-02-271171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaussade C, et al. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. Biochem J. 2007;404(3):449–458. doi: 10.1042/BJ20070003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bruce I, et al. Development of isoform selective PI3-kinase inhibitors as pharmacological tools for elucidating the PI3K pathway. Bioorg Med Chem Lett. 2012;22(17):5445–5450. doi: 10.1016/j.bmcl.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 44.Morgan MA, Dolp O, Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood. 2001;97(6):1823–1834. doi: 10.1182/blood.V97.6.1823. [DOI] [PubMed] [Google Scholar]
- 45.Scholl C, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 47.Ghaffari S, et al. AKT induces erythroid-cell maturation of JAK2-deficient fetal liver progenitor cells and is required for Epo regulation of erythroid-cell differentiation. Blood. 2006;107(5):1888–1891. doi: 10.1182/blood-2005-06-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bouscary D, et al. Critical role for PI 3-kinase in the control of erythropoietin-induced erythroid progenitor proliferation. Blood. 2003;101(9):3436–3443. doi: 10.1182/blood-2002-07-2332. [DOI] [PubMed] [Google Scholar]
- 49.Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood. 2006;107(3):907–915. doi: 10.1182/blood-2005-06-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. doi: 10.1016/0092-8674(95)90234-1. [DOI] [PubMed] [Google Scholar]
- 51.Rahmani M, et al. The BH3-only protein Bim plays a critical role in leukemia cell death triggered by concomitant inhibition of the PI3K/Akt and MEK/ERK1/2 pathways. Blood. 2009;114(20):4507–4516. doi: 10.1182/blood-2008-09-177881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimizu T, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18(8):2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 53.LoRusso P, et al. A first-in-human phase Ib study to evaluate the MEK inhibitor GDC-0973, combined with the pan-PI3K inhibitor GDC-941, in patients with advanced solid tumors. J Clin Oncol (Meeting Abstracts). 2012;30(15 Suppl.):2566. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.