

Abstract
Age-related macular degeneration (AMD) is a common cause of visual loss in the elderly, with increasing prevalence due to increasing life expectancy. While the introduction of anti-VEGF therapy has improved outcomes, there are still major unmet needs and gaps in the understanding of underlying biological processes. These include early, intermediate, and atrophic disease stages. Recent studies have assessed therapeutic approaches addressing various disease-associated pathways, including complement inhibitors. Drug-delivery aspects are also relevant, as many agents have to be administered repeatedly. Herein, relevant pathogenetic factors and underlying mechanisms as well as recent and potential therapeutic approaches are reviewed.
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible blindness in industrialized countries (1). Recent advances in retinal imaging technology, including noninvasive, high-resolution spectral-domain optical coherence tomography (SD-OCT) and confocal scanning laser ophthalmoscopy imaging, have markedly improved early diagnosis as well as disease monitoring during treatment. The development and application of anti-VEGF therapy has led to an unprecedented improvement in functional outcomes for patients affected by the “wet,” neovascular form of the disease, measurably reducing the incidence of blindness in the elderly (2–4). However, there are still many unmet needs in AMD therapy. Anti-VEGF therapy must be administered repeatedly, sometimes every month, over a long period of time. As AMD is a chronic disease, patients need to return regularly for monitoring and treatment visits. Real-life observational studies have shown that this burden, along with adherence aspects, often leads to undertreatment and subsequent visual loss (5). Recent studies indicate that, despite optimal individualized treatment, visual function may slowly deteriorate due to other AMD-related disease processes (6, 7). Additionally, there is not yet a treatment available to slow or halt progression of the nonexudative late-stage “dry” manifestation of AMD, i.e., geographic atrophy (GA).
The natural history of AMD is characterized by the progression from early to intermediate stages of the disease and subsequently the development of the two major advanced forms of AMD, i.e., GA and neovascular AMD (Figure 1). Histologically, areas of GA are characterized by loss of retinal pigment epithelium (RPE) cells and outer layers of the neurosensory retina as well as the choriocapillaris (8, 9). Neovascular or “wet” AMD is characterized by the formation of choroidal neovascularization (CNV), the ingrowth of new blood vessels from the choriocapillaris through Bruch’s membrane into the subpigmentepithelial or subretinal space. Retinal angiomatous proliferations (RAP) have recently been recognized as a variant of neovascular AMD, with neovascularizations within the retina that may secondarily invade into the subretinal space or communicate with choroidal vessels (10). Characteristic for neovascular AMD is the leakage of plasma or blood into the surrounding tissue. Ultimately, CNV may evolve into fibrovascular scar tissue. Although the advanced forms have distinct pathologic mechanisms, they converge on cellular pathways that lead to photoreceptor death, which is the ultimate cause of visual loss in AMD. The formation of focal extracellular deposition under the RPE (classical drusen) or above (reticular pseudodrusen) is predictive of severe, late-stage forms of AMD. These phenotypic hallmarks may be associated with variable degrees of degenerative changes at the photoreceptor level. Further, a thickening of Bruch’s membrane along with the accumulation of diffuse lipid aggregates precedes disabling later manifestations of the disease. Choroidal involvement is still debated, while changes of the inner layer of the choroid underneath Bruch’s membrane, namely the choriocapillaris, may be of importance, as normal perfusion in this vasculature is a prerequisite for normal function of the outer retina.
Figure 1. Schematic showing morphological changes in the macula during evolution of early/intermediate AMD, exudative/neovascular AMD, and GA, respectively, along with several known pathogenetic factors.

The course of the disease, however, can vary from case to case. High-resolution SD-OCT images show typical findings of the different AMD stages. Images reproduced with permission from Investigative Ophthalmology Visual Science (85).
Various cell populations are involved in the disease process. Each cell type possesses essential features to maintain visual function: (a) the photoreceptors, which are the light-responsive elements that initiate signaling by phototransduction; (b) the RPE cell monolayer, the functions of which include phagocytosis of constantly shed photoreceptor outer segments, participation in the visual cycle, maintenance of the outer blood-retina barrier, secretion of neurotrophic, inflammatory, and vasculotrophic growth factors, water transport out of the subretinal space, and regulation of bidirectional ion and metabolic transport between the retina and the choroid (11); (c) Bruch’s membrane, which affects the diffusion of molecules between the RPE and the choroid, provides physical support for RPE cell adhesion and migration, and restricts choroidal and retinal cellular migration (12); (d) and the choroid, which supplies oxygen and metabolites to the outer retina and the RPE.
Recent discoveries in genetic and genomic studies have contributed significantly to the understanding of AMD pathogenesis. Significant associations were found with a number of complement pathway-associated genes including CFH, CFB, C3, and CFI. These complement-related variations account for a large proportion of the genetic risk; however, many patients with AMD do not have these variations. Other variants were detected for ARMS2 and HTRA1 genes on chromosome 10, as well as genes involved in lipid metabolism, extracellular matrix remodeling, and angiogenesis (13).
One of the challenges in developing AMD therapies is the lack of good animal models. Mice and rats — among other distinguishing features — do not have a macula or a foveola. The neovascular phenotype can be simulated to some extent by laser-induced CNV animal models; however, this wound model has no aspects of aging changes in outer retinal layers, the RPE, or Bruch’s membrane. Animal models for drusen or GA are also limited. These should at least depict one single phenotypic characteristic of AMD, such as focal and diffuse Bruch’s membrane changes, RPE lipofuscin accumulation, or RPE and photoreceptor cell death. Disease modeling could be a future alternative, e.g., using differentiated autologous stem cells from affected patients.
Pathogenetic pathways and therapeutic targets in late-stage AMD
AMD is now one of the best-understood complex, multifactorial diseases. While knowledge of this multifactorial, complex disease has been expanded in recent years, there are still many open questions. Besides various nonspecific, age-associated alterations that affect the photoreceptors, the RPE, Bruch’s membrane, and the choriocapillaris layer of the choroid, various additional pathways have been implicated, including oxidative stress, impairment of lysosomal degradation with accumulation of retinal toxins, local inflammation, complement system dysfunction, and VEGF hyperexpression.
Oxidative stress
The retina provides an ideal environment for the generation of ROS due to its specific anatomical and metabolic characteristics: (a) oxygen consumption by the outer retina/RPE complex is greater than in any other human tissue; (b) the retina is subjected to high levels of cumulative irradiation; (c) the neurosensory retina and RPE contain abundant photosensitizers; and (d) photoreceptor outer segment membranes are rich in polyunsaturated fatty acids, which are readily oxidized and can initiate a cytotoxic chain reaction. Reducing oxidative stress is therefore a target in the prophylaxis and treatment of AMD.
The Age-Related Eye Disease Study 1 (AREDS1), a multicenter, prospective study including 4,757 probands, demonstrated that daily high doses of antioxidants, including β-carotene, vitamins C and E, and zinc, reduced the progression to late-stage AMD by 25% over a 5-year period (14). The recently completed AREDS2 trial (NCT00345176, ref. 15) investigated the additional intake of two long-chain omega-3 fatty acids, docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), which did not show beneficial effects, although previous studies had demonstrated a significant association between fish or fish oil intake and the delayed incidence of AMD (16, 17). The absence of a beneficial effect of DHA and EPA in the AREDS2 trial could be explained by the fact that patients in this study were well educated and many were already taking fish oil supplements and eating healthy diets, so that additional supplementation may have had no further effect. Substitution of lutein and zeaxanthin for β-carotene, which may have adverse effects on patients with a smoking history, was recommended based on the AREDS2 data. None of the formulations had an impact on the incidence or progression of atrophic AMD (15).
Accumulation of retinal toxins
With increasing age and disease, lipofuscin accumulates in the lysosomal compartment of postmitotic RPE cells due to the incomplete degradation of the photoreceptor outer segments (reviewed by Boulton, ref. 18). Lipofuscin consists of a multitude of molecules, including the dominant fluorophore N-retinyl-N-retinylidene ethanolamine (A2E), a by-product of the visual cycle (19, 20). A number of molecular mechanisms have been identified by which these nondegradable components may interfere with normal cellular function, including detergent and phototoxic effects as well as impairment of autophagy. Clinical findings using fundus autofluorescence imaging have indicated that the accumulation of fluorophores, including A2E in the RPE, precedes the development of outer retinal atrophy (reviewed by Schmitz-Valckenberg et al., ref. 21).
Local inflammation and complement system dysfunction
In exudative AMD, CNV has been shown to be associated with inflammatory cells (22). Furthermore, activation of immune cells constitutively present in the retina has been proposed to contribute to GA. These include microglia, Mueller cells, RPE cells, and macrophages as well as immune cells present in the choroid, including pericapillary macrophages, giant cells, and mast cells (23, 24). The appearance of these innate immune cells at sites of cellular deposits on Bruch’s membrane may indicate that the phagocytic capacity of the aged postmitotic RPE cells is impaired. Drusen largely consist of lipoprotein particles and RPE cell remnants, but also contain various inflammatory proteins, including apolipoprotein E, coagulation proteins, acute phase proteins, IgG, complement components, and complement activators. This is in accordance with the assumed local low-grade inflammatory process along with the accumulation of drusen material in the ECM, a process that is highly similar to atherosclerosis (25). Interestingly, low-grade chronic inflammation is also a feature in other retinal diseases, such as diabetic retinopathy, retinal vein occlusions, and proliferative vitreoretinopathy following retinal detachment.
The complement system, one of the main effectors of the innate immune system, can be activated via a classic pathway, a lectin-based pathway, and an alternative pathway. In each pathway a proteolytic cascade is amplified and effectors such as anaphylatoxins, the membrane attack complex, and opsonins may be activated. Excessive activation of the alternative complement cascade generates a variety of proinflammatory responses, and it has been suggested that aging causes increased activation of the alternative complement system in the blood (26, 27).
The role of the alternative complement pathway in AMD pathogenesis has been underscored by the discovery of an association between CFH (28–31) and other risk-conferring polymorphisms in this pathway, including CFB, C2, C3, and CFI (32–36). Subsequent studies of the functional consequences of the CFH risk variants provided the first direct insights into molecular mechanisms that may lead to AMD (37). The CFH gene encodes two functional proteins, namely CFH and the isoform FHL-1, which have been shown to act as negative regulators of complement activation (38, 39). The risk variant at codon 402 obviously alters the capacity of CFH and FHL-1 to regulate the complement system. As a consequence, hyperactivation of the complement system in patients with the risk variant may lead to a chronic attack on the aging macula (40). While complement-related variations account for a proportion of the genetic risk for AMD (e.g., the common variants in CFH, the Y402H substitution [rs1061170, c. 1204C>T] and the intronic rs1410996 SNP) explain 17% of AMD liability (41), many patients do not have such polymorphisms.
VEGF
VEGF is a potent endothelial cell mitogen and vascular permeability factor that plays a pivotal role in the pathogenesis of exudative AMD. VEGF has been detected in CNV membranes obtained from AMD patients (42). Further, there were significantly increased VEGF levels in the aqueous humor of AMD patients as compared with control eyes without ocular or systemic disease (43). Experimental in vivo models have provided evidence for a temporal relationship between VEGF expression and the development of CNV (44). Intravitreal injection of a recombinant humanized monoclonal antibody directed toward VEGF prevented the formation of CNV in response to argon laser burns in a nonhuman primate model (45).
Under physiological conditions, VEGF is a major factor in embryonic vascular development as well as blood vessel formation in the adult. Additionally, VEGF plays a role in inflammatory processes, including immunity and wound healing. It acts as a survival factor for endothelial cells and as a neuroprotectant for neurons of the CNS, including the retina (reviewed by Grisanti et al., ref. 46).
The PDGF family includes VEGF-A, -B, -C, -D, -E, and placental growth factor (PlGF), with VEGF-A being its best-examined representative. Alternative splicing of VEGFA results in the generation of four major homodimeric polypeptides: VEGF-A121, -A165, -A189, and -A206. Solubility of these VEGF-A isoforms depends on their binding domains, and they differ in their ability to interact with receptor-related structures. The VEGF family members bind with different affinities to three related receptor tyrosine kinases, and VEGF-A binds mainly to VEGFR1 and VEGFR2.
Current successful therapies for exudative AMD are based on VEGF inhibition (see below). However, potential long-term consequences of intraocular VEGF suppression on the retina due to inhibition of physiological VEGF function have to be taken into account. Data from animal and in vitro studies suggest that VEGF-A may be a survival factor for retinal neurons and that chronic inhibition of VEGF-A function in normal adult animals leads to a significant loss of retinal ganglion cells (47). Furthermore, it has been demonstrated in mice that the absence of soluble VEGF isoforms leads to the development of focal choriocapillaris atrophy with subsequent RPE and photoreceptor cell loss (48).
Current treatments, unmet needs, and future directions
Exudative/neovascular AMD.
The results of pivotal phase 3 clinical trials with anti-VEGF represented a breakthrough in treating patients with neovascular AMD. Previously, patients were treated with laser photocoagulation or verteporfin photodynamic therapy (PDT), which at best slowed the progression of the disease. Eventually, these patients became legally blind (49–51).
The first phase 3 randomized controlled trial for treating exudative AMD with pegaptanib sodium, an oligonucleotide aptamer specifically targeting VEGF-A165, resulted in a better outcome when compared with the natural course of the disease (Table 1). However, an average decline in central visual function was noted (VISION trial, ref. 52). In contrast, ranibizumab, a humanized antigen-binding antibody fragment (Fab) targeting all isoforms of VEGF-A, stabilized or improved vision in over 90% of patients and significantly improved vision in over 30% of patients (53, 54) (MARINA, NCT00056836; ANCHOR trial, NCT00061594).
Table 1.
Clinical trials for exudative AMD
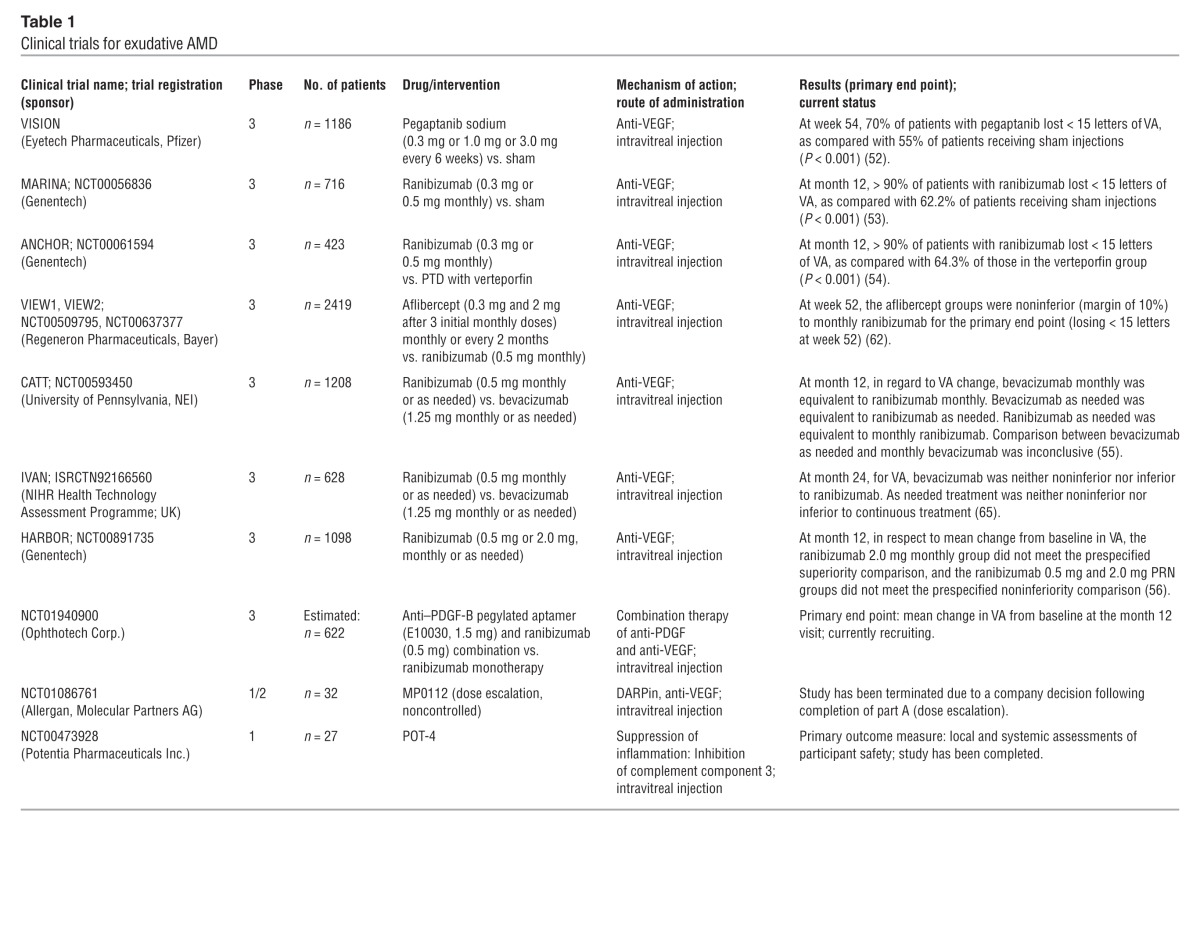
The beneficial effects of ranibizumab were achieved by monthly intravitreal injections; however, it is particularly difficult for elderly patients to adhere to this treatment strategy. Furthermore, a number of patients may be overdosed by this regimen. Clinical studies with individualized pro re nata (PRN) approaches showed similar outcomes with a lower number of injections (CATT, ref. 55, NCT00593450; HARBOR, ref. 56, NCT00891735; PrONTO, ref. 57, NCT00344227; SUSTAIN, ref. 58). The minor differences in effectiveness between a monthly or PRN regime would not warrant monthly dosing in all patients with active neovascular AMD.
The course of the disease is obviously highly variable, and an individualized approach appears sensible. As there are no predictive markers to indicate individual outcome or requirements with regard to the number of injections, patients need to be seen regularly to monitor disease activity. Retreatments are mandatory as soon as new disease activity is detected, either by means of SD-OCT or functional parameters.
Bevacizumab, initially developed for oncologic indications, is a fully humanized antibody directed against VEGF and administered to AMD patients off-label. The CATT study demonstrated that ranibizumab and bevacizumab had similar effects on visual acuity over a two-year period (55). The pharmacokinetics of bevacizumab differ from that of ranibizumab due to its molecular structure, including the Fc fragment, and it has a longer half-life in the systemic circulation, which might explain signals for possible differences in systemic safety profiles. Systemic side effects of anti-VEGF treatment overall are rare; however, it is still a matter of debate whether treatment with anti-VEGF could increase the risk of thromboembolic events (59) or cause kidney toxicity (60). While in the CATT study, there were no differences in rates of death or arteriothrombotic events, a higher rate of serious adverse events was reported for bevacizumab-treated patients (55).
Recently, aflibercept (VEGF trap-eye), a fusion protein consisting of key domains of the human VEGF1 and VEGF2 receptors coupled to the Fc part of a human IgG molecule, has been approved for neovascular AMD (46). The compound has a high affinity for all VEGF isoforms as well as PlGF (reviewed by Mitchell et al., ref. 50). Theoretical models suggested that this molecule may have a longer duration of action compared with current treatments (61). In recent phase 3 clinical trials, aflibercept, administered every second month after three monthly loading doses over 12 months, proved to be noninferior to monthly injections of ranibizumab, although with increased VEGF affinity, long-term follow-up for evaluation of neuronal degeneration is important (VIEW 1 and VIEW 2 studies, ref. 62; NCT00509795, NCT00637377).
The currently available treatment regimes for neovascular AMD leave a number of unmet needs. Observations from postapproval studies including the WAVE (5) (3,470 patients) and AURA studies (63) (2,500 patients) have shown that outcomes may be suboptimal due to underdosing. During the first year of treatment, an average of seven to eight injections are required (CATT, ref. 55, NCT00593450; IVAN, refs. 64, 65, ISRCTN92166560); however, only an average of four to five injections were actually administered. A number of factors may account for this underdosage, but a major reason seems to be the burden of monthly disease monitoring and the many individualized retreatments by intravitreal injections over a long period of time. To reduce the treatment and monitoring burden, differential routes of drug delivery (Figure 2) and the development of long-acting drug delivery systems are desirable. While this has been accomplished for the treatment of retinal diseases with corticosteroids, such devices are more challenging for less stable biological compounds such as antibodies, antibody fragments, or fusion proteins (66).
Figure 2. Schematic of various suitable routes of drug delivery for treatment of AMD.

Drugs to be delivered to the target area (macula with foveola) can be administered topically to the surface of the eye (eye drops), underneath the conjunctiva for transscleral delivery (subconjunctival injection/implant, parabulbar injection), directly into the vitreous (intravitreal injection/implant), or systemically (oral, intravenous infusion).
Designed ankyrin repeat proteins (DARPins) represent a novel class of drugs. DARPins are genetically engineered antibody mimetic proteins that exhibit highly specific, high-affinity target protein binding. The DARPin MP0112 is a potent VEGF inhibitor. Animal studies indicate that dosing frequency in patients may be reduced compared with current therapy (67). A phase I dose escalation study demonstrated the overall safety and efficacy of MP0112 (NCT01086761) (68).
Combining existing and future therapies that address different pathways may also result in less frequent administration while achieving greater efficacy. The combined inhibition of PDGF and VEGF is currently being evaluated. In a phase 2b study, the combination of ranibizumab with an anti-PDGF aptamer achieved better visual outcomes than anti-VEGF monotherapy (69). A phase 3 clinical trial of this combination has just been initiated (NCT01940900).
Dry AMD/GA.
The treatment of dry AMD/GA represents a major challenge, as there is no approved therapy available for patients. Incidence and prevalence of the disease are rising, and GA is approximately 4 times more common than neovascular AMD in patients over 85 (70). As outlined above, various pathways have been identified that appear to play a role in pathogenesis and may be suitable for pharmacological intervention.
Modulation of the visual cycle is expected to reduce the accumulation of retinal toxins, including A2E, by inhibiting a key enzyme of the visual cycle, thereby lowering the supply of A2E precursors. In an animal study of Stargardt disease, a monogenetic macular disease mimicking some features of AMD, the synthetic retinoid derivative fenretinide was shown to decrease the formation of A2E and other fluorophores (71). The first interventional phase 2 clinical trial in patients with GA did not, however, demonstrate a significant therapeutic response (72). Currently, a placebo-controlled, phase 2b/3 study with orally administered emixustat, an inhibitor of the visual cycle enzyme RPE65, is underway in over 400 patients with GA (NCT01002950) (Table 2). Emixustat exhibits effects that are quantifiable by electrophysiologic examination of the retina (electroretinogram [ERG]), proving its interaction with the visual cycle. The main outcome parameter is an objective anatomical end point, i.e., GA lesion growth over time visualized in vivo by fundus autofluorescence imaging.
Table 2.
Clinical trials for GA
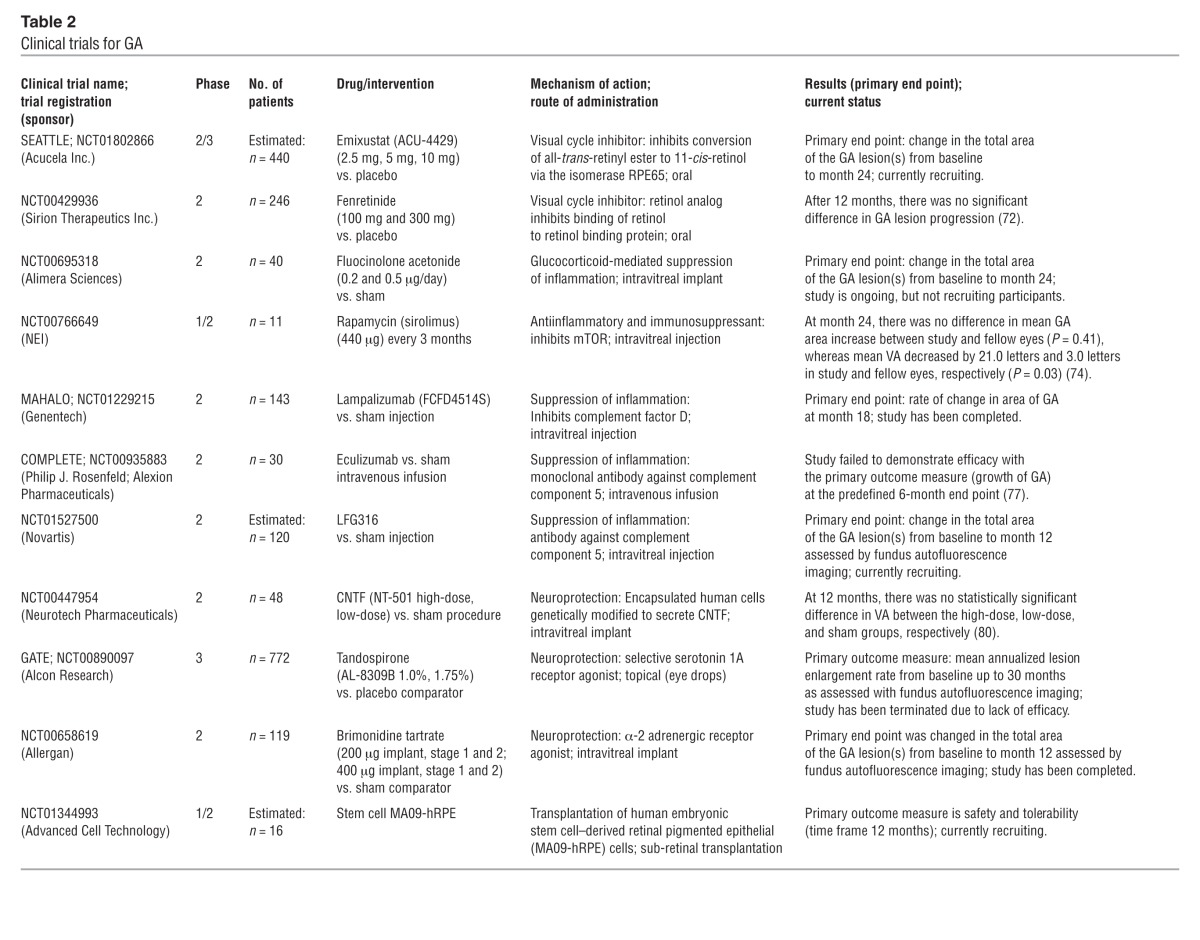
Several antiinflammatory agents are currently being tested in humans. A nonbioerodible polyimide tube containing fluocinolone has been designed for intravitreal injection. The active component is supposed to diffuse into the eye over a period of three years. A phase 2 study enrolling patients with bilateral GA is currently underway (NCT00695318).
Rapamycin (sirolimus) was originally approved as an antifungal agent and later used in transplant patients in various proliferative diseases such as polycystic kidney diseases as well as in oncology because of its ability to inhibit mTOR. Its spectrum of actions includes inhibition of inflammation, angiogenesis, fibrosis, and hyperpermeability. The rationale for mTOR pathway inhibition as a therapeutic strategy for retinal degenerative diseases was the finding that pharmacological inhibition of mTOR with rapamycin in metabolic and oxidative stress mouse models blunted key aspects of RPE dedifferentiation and preserved photoreceptor function (73). A phase 1/2 study sponsored by the National Eye Institute (NEI) assessed whether rapamycin could be safely administered to patients with GA and whether it could preserve vision (NCT00766649). Thus far, there has been no evidence of efficacy, but concerns about safety have increased (74).
The use of complement inhibitors was proposed when genetic studies elucidated the role of the complement system as an important factor in AMD. Factor D is a rate-limiting enzyme in the activation of the alternative complement pathway. Lampalizumab, a recombinant mAb fragment directed against factor D, has proved to be safe and to slow lesion growth in eyes with GA in a phase 2 study (MAHALO study; NCT01802866) (75). For the first time, efficacy in terms of slowing atrophy progression has been demonstrated when lampalizumab was administered intravitreally in monthly intervals (75).
POT-4 is a cyclic, 13-aa peptide that prevents the conversion of C3 to C3a and C3b. A phase 1 study in neovascular AMD has been completed (NCT00473928). The therapeutic effect of POT-4 may not be sufficient to prevent complement activation, as it can still be activated via the classical and the lectin pathways. A treatment trial is planned in patients with dry AMD. Another C5 inhibitor, LFG316, is an intravitrally administered antibody. It is currently being evaluated in a phase 2 study in patients with GA (NCT01527500).
Eculizumab is a humanized monoclonal antibody that inhibits the complement cascade at C5, preventing the formation and release of the downstream anaphylatoxin C5a and the formation of the cytolytic membrane attack complex. Eculizumab is FDA approved for the intravenous treatment of paroxysmal nocturnal hemoglobinuria, also a complement-mediated condition; however, its chronic systemic use is associated with increased risk of Neisseria meningitides infection (76). This highlights the dangers of immune suppression during chronic complement inhibition. A phase 2 trial (COMPLETE Study; NCT00935883) failed to demonstrate efficacy of intravenously administered eculizumab in patients with advanced dry AMD (77). A possible explanations for the lack of a treatment effect could be that the dose of eculizumab was too low or that the drug should have been delivered as a direct intravitreal injection to achieve an adequate level of drug in the retina or the RPE (77).
In advanced dry AMD, neuronal cells of the outer retina, including photoreceptors, degenerate and, ultimately, die. Therefore, neuroprotective agents have been proposed to preserve the neuronal structure and function. The ciliary neurotrophic factor (CNTF) is a neuroprotective agent that has been shown to inhibit photoreceptor cell apoptosis in animal models of retinal degeneration (78, 79). A sustained-release platform with encapsulated human RPE cells engineered to release CNTF for one year or longer has been developed and was tested in a phase 2 study in patients with GA (NCT00447954). Stabilization of visual acuity at 12 months was achieved; however, the difference was not statistically significant between the high-dose, low-dose, and sham-treated groups (80).
Serotonin-1A agonists possess neuroprotective properties in the CNS in animal models. Tandospirone is a selective serotonin-1A agonist that has been shown to protect the retina in rats from the severe photooxidative injury induced by exposure to blue light (81). The precise molecular mechanisms involved in serotonin-1A receptor–mediated neuroprotection remain to be defined, but prevention of complement deposition may play an important role (81). A phase 3 trial (GATE study; NCT00890097) assessing the effect of topical tandospirone (1% and 1.75%) on the rate of GA progression was discontinued due to lack of efficacy.
Brimonidine is a highly selective α2-adrenergic agonist that has gained attention for its role in reducing intraocular pressure in the treatment of glaucoma. More recent experimental and animal models suggest brimonidine protects retinal ganglion cells, bipolar cells, and photoreceptors from degeneration after a number of types of insults, including retinal ischemia and retinal phototoxicity (reviewed by Wheeler et al., ref. 82). The neuroprotective effects of brimonidine are likely multifactorial. Proposed mechanisms include upregulation of trophic factors such as brain-derived neurotrophic factor in retinal ganglion cells and of intrinsic cell survival signaling pathways and antiapoptotic genes (reviewed by Saylor et al., ref. 83). Rimonidine has been formulated as an intravitreal implant, which delivers the drug over a 3-month period. Clinical studies are underway to assess efficacy and safety in patients with GA (e.g., NCT00658619).
Stem cell therapy has been proposed to both restore retinal function and protect viable tissue from degeneration. Subretinal transplantation of human embryonic stem cell–derived RPE cells (MA09-hRPE) has been performed in two patients. One patient had GA due to AMD (84) (NCT01344993). Patients were systemically immunosuppressed during the trial, and no safety concerns arose during the observation period. Further clinical investigations in AMD are planned.
Outlook
While legal blindness in the elderly can now be prevented in a larger proportion of patients with neovascular AMD, there are still many unresolved issues, including serial intraocular injections as well as dry AMD manifestations. Ongoing preclinical and clinical studies are addressing various pathways thought to be operative in AMD pathogenesis. A first positive result for halting the progression of GA, the advanced form of dry AMD, has recently been demonstrated for a humanized anti–factor D antibody administered monthly into the eye. Long-acting drug delivery systems are currently being explored in order to avoid frequent intraocular injections and to improve long-term outcomes.
Acknowledgments
The authors gratefully acknowledge support and funding from the Federal Ministry of Education and Research (BMBF), Germany (grant FKZ 13N10349), the German Research Foundation (DFG) (grants Ho1926/1-3 and FL658/4-1), and the Research Commission of the Medical Faculty at Bonn University (BONFOR) (grant O-137-0012).
Footnotes
Conflict of interest: The authors receive research support from Acucela, Allergan, Bayer, Genentech, Heidelberg Engineering, Novartis, Optos, Roche, and Zeiss.
Citation for this article:J Clin Invest. 2014;124(4):1430–1438. doi:10.1172/JCI71029.
References
- 1.Lim LS, Mitchell P, Seddon JM, Holz FG, Wong TY. Age-related macular degeneration. Lancet. 2012;379(9827):1728–1738. doi: 10.1016/S0140-6736(12)60282-7. [DOI] [PubMed] [Google Scholar]
- 2.Campbell JP, Bressler SB, Bressler NM. Impact of availability of anti-vascular endothelial growth factor therapy on visual impairment and blindness due to neovascular age-related macular degeneration. Arch Ophthalmol. 2012;130(6):794–795. doi: 10.1001/archophthalmol.2011.2480. [DOI] [PubMed] [Google Scholar]
- 3.Bloch SB, Larsen M, Munch IC. Incidence of legal blindness from age-related macular degeneration in denmark: year 2000 to 2010. Am J Ophthalmol. 2012;153(2):209–213. doi: 10.1016/j.ajo.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Belkin M, Kalter-Leibovici O, Chetrit A, Skaat A. Time trends in the incidence and causes of blindness in Israel. Am J Ophthalmol. 2013;155(2):404. doi: 10.1016/j.ajo.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Finger RP, Wiedemann P, Blumhagen F, Pohl K, Holz FG. Treatment patterns, visual acuity and quality-of-life outcomes of the WAVE study — a noninterventional study of ranibizumab treatment for neovascular age-related macular degeneration in Germany. Acta Ophthalmol. 2013;91(6):540–546. doi: 10.1111/j.1755-3768.2012.02493.x. [DOI] [PubMed] [Google Scholar]
- 6.Singer MA, et al. HORIZON: an open-label extension trial of ranibizumab for choroidal neovascularization secondary to age-related macular degeneration. Ophthalmology. 2012;119(6):1175–1183. doi: 10.1016/j.ophtha.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Rofagha S, et al. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: a multicenter cohort study (SEVEN-UP). Ophthalmology. 2013;120(11):2292–2299. doi: 10.1016/j.ophtha.2013.03.046. [DOI] [PubMed] [Google Scholar]
- 8.Sarks JP, Sarks SH, Killingsworth MC. Evolution of geographic atrophy of the retinal pigment epithelium. Eye (Lond). 1988;2(pt 5):552–577. doi: 10.1038/eye.1988.106. [DOI] [PubMed] [Google Scholar]
- 9.Green WR, Key SN., 3rd Senile macular degeneration: a histopathologic study. Trans Am Ophthalmol Soc. 1977;75:180–254. [PMC free article] [PubMed] [Google Scholar]
- 10.Yannuzzi LA, et al. Retinal angiomatous proliferation in age-related macular degeneration. Retina. 2001;21(5):416–434. doi: 10.1097/00006982-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Thumann G, Dou G, Wang Y, Hinton DR. Cell biology of the retinal pigment epithelium. In: Ryan SJ, ed.Retina . London, United Kingdom: Elsevier; 2013:401–414. [Google Scholar]
- 12.Booij JC, Baas DC, Beisekeeva J, Gorgels TG, Bergen AA. The dynamic nature of Bruch’s membrane. Prog Retin Eye Res. 2010;29(1):1–18. doi: 10.1016/j.preteyeres.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 13.Fritsche LG, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013;45(4):433–439. doi: 10.1038/ng.2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Age-Related Eye Disease Study Research Group A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss: AREDS report no. 8. Arch Ophthalmol. 2001;119(10):1417–1436. doi: 10.1001/archopht.119.10.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chew EY, et al. Lutein/zeaxanthin for the treatment of age-related cataract: AREDS2 randomized trial report no. 4. JAMA Ophthalmol. 2013;131(7):843–850. doi: 10.1001/jamaophthalmol.2013.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Christen WG, Schaumberg DA, Glynn RJ, Buring JE. Dietary ω-3 fatty acid and fish intake and incident age-related macular degeneration in women. Arch Ophthalmol. 2011;129(7):921–929. doi: 10.1001/archophthalmol.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schleicher M, Weikel K, Garber C, Taylor A. Diminishing risk for age-related macular degeneration with nutrition: a current view. Nutrients. 2013;5(7):2405–2456. doi: 10.3390/nu5072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulton M. Ageing of the retina and retinal pigment epithelium. In: Holz FG, Pauleikhoff D, Spaide RF, Bird AC, eds.Age-Related Macular Degeneration . Heidelberg, Germany: Springer; 2013:45–63. [Google Scholar]
- 19.Suter M, et al. Age-related macular degeneration. The lipofusion component N-retinyl-N-retinylidene ethanolamine detaches proapoptotic proteins from mitochondria and induces apoptosis in mammalian retinal pigment epithelial cells. J Biol Chem. 2000;275(50):39625–39630. doi: 10.1074/jbc.M007049200. [DOI] [PubMed] [Google Scholar]
- 20.Palczewska G, et al. Noninvasive multiphoton fluorescence microscopy resolves retinol and retinal condensation products in mouse eyes. Nat Med. 2010;16(12):1444–1449. doi: 10.1038/nm.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitz-Valckenberg S, Fleckenstein M, Scholl HP, Holz FG. Fundus autofluorescence and progression of age-related macular degeneration. Surv Ophthalmol. 2009;54(1):96–117. doi: 10.1016/j.survophthal.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Grossniklaus HE, et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol Vis. 2002;8:119–126. [PubMed] [Google Scholar]
- 23.Cherepanoff S, McMenamin P, Gillies MC, Kettle E, Sarks SH. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol. 2010;94(7):918–925. doi: 10.1136/bjo.2009.165563. [DOI] [PubMed] [Google Scholar]
- 24.Lutty G, Bhutto I, Seddon J, McLeod D. Mast cell degranulation in AMD choroid. Invest Ophthalmol Vis Sci. 2013;54(6):3051. [Google Scholar]
- 25.Ebrahimi KB, Handa JT. Lipids, lipoproteins, and age-related macular degeneration. J Lipids. 2011;2011:802059. doi: 10.1155/2011/802059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hecker LA, et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum Mol Genet. 2010;19(1):209–215. doi: 10.1093/hmg/ddp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loyet KM, et al. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2012;53(10):6628–6637. doi: 10.1167/iovs.12-9587. [DOI] [PubMed] [Google Scholar]
- 28.Haines JL, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308(5720):419–421. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 29.Klein RJ, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hageman GS, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102(20):7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308(5720):421–424. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 32.Maller J, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38(9):1055–1059. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 33.Gold B, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38(4):458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39(10):1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- 35.Yates JR, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357(6):553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 36.Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17(1):100–104. doi: 10.1038/ejhg.2008.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fritsche L, Friedrich U, Weber BH. Genetics. In: Holz FG, Pauleikhoff D, Spaide RF, Bird AC, eds.Age-Related Macular Degeneration . Heidelberg, Germany: Springer; 2013:33–43. [Google Scholar]
- 38.Giannakis E, et al. Multiple ligand binding sites on domain seven of human complement factor H. Int Immunopharmacol. 2001;1(3):433–443. doi: 10.1016/S1567-5769(00)00040-0. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41(4):355–367. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Nitsch D, Douglas I, Smeeth L, Fletcher A. Age-related macular degeneration and complement activation-related diseases: a population-based case-control study. Ophthalmology. 2008;115(11):1904–1910. doi: 10.1016/j.ophtha.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 41.Raychaudhuri S, et al. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat Genet. 2011;43(12):1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Transdifferentiated retinal pigment epithelial cells are immunoreactive for vascular endothelial growth factor in surgically excised age-related macular degeneration-related choroidal neovascular membranes. Invest Ophthalmol Vis Sci. 1996;37(5):855–868. [PubMed] [Google Scholar]
- 43.Tong JP, et al. Aqueous humor levels of vascular endothelial growth factor and pigment epithelium-derived factor in polypoidal choroidal vasculopathy and choroidal neovascularization. Am J Ophthalmol. 2006;141(3):456–462. doi: 10.1016/j.ajo.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Wang F, Rendahl KG, Manning WC, Quiroz D, Coyne M, Miller SS. AAV-mediated expression of vascular endothelial growth factor induces choroidal neovascularization in rat. Invest Ophthalmol Vis Sci. 2003;44(2):781–790. doi: 10.1167/iovs.02-0281. [DOI] [PubMed] [Google Scholar]
- 45.Krzystolik MG, et al. Prevention of experimental choroidal neovascularization with intravitreal anti-vascular endothelial growth factor antibody fragment. Arch Ophthalmol. 2002;120(3):338–346. doi: 10.1001/archopht.120.3.338. [DOI] [PubMed] [Google Scholar]
- 46.Grisanti S, Lüke J, Peters S. Anti-VEGF therapy: basics and substances. In: Holz FG, Pauleikhoff D, Spaide RF, Bird AC, eds.Age-Related Macular Degeneration . Heidelberg, Germany: Springer; 2013:225–232. [Google Scholar]
- 47.Nishijima K, et al. Vascular endothelial growth factor-A is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am J Pathol. 2007;171(1):53–67. doi: 10.2353/ajpath.2007.061237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci U S A. 2009;106(44):18751–18756. doi: 10.1073/pnas.0905010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barakat M, Steinle N, Kaiser PK. Combination therapies for treatment of AMD. In: Holz FG, Pauleikhoff D, Spaide RF, Bird AC, eds.Age-Related Macular Degeneration . Heidelberg, Germany: Springer; 2013. [Google Scholar]
- 50.Mitchell P, Foran S. Anti-VEGF therapy for AMD: results and guidelines. In: Holz FG, Pauleikhoff D, Spaide RF, Bird AC, eds.Age-Related Macular Degeneration . Heidelberg, Germany: Springer; 2013. [Google Scholar]
- 51.Schmidt-Erfurth U, Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration. Surv Ophthalmol. 2000;45(3):195–214. doi: 10.1016/S0039-6257(00)00158-2. [DOI] [PubMed] [Google Scholar]
- 52.Gragoudas ES, Adamis AP, Cunningham ET, Jr, Feinsod M, Guyer DR. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351(27):2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 53.Rosenfeld PJ, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 54.Brown DM, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–1444. doi: 10.1056/NEJMoa062655. [DOI] [PubMed] [Google Scholar]
- 55.Martin DF, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–1398. doi: 10.1016/j.ophtha.2012.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Busbee BG, et al. Twelve-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2013;120(5):1046–1056. doi: 10.1016/j.ophtha.2012.10.014. [DOI] [PubMed] [Google Scholar]
- 57.Lalwani GA, et al. A variable-dosing regimen with intravitreal ranibizumab for neovascular age-related macular degeneration: year 2 of the PrONTO Study. Am J Ophthalmol. 2009;148(1):43–58. doi: 10.1016/j.ajo.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 58.Holz FG, et al. Safety and efficacy of a flexible dosing regimen of ranibizumab in neovascular age-related macular degeneration: the SUSTAIN study. Ophthalmology. 2011;118(4):663–671. doi: 10.1016/j.ophtha.2010.12.019. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt-Erfurth U. Clinical safety of ranibizumab in age-related macular degeneration. Expert Opin Drug Saf. 2010;9(1):149–165. doi: 10.1517/14740330903418422. [DOI] [PubMed] [Google Scholar]
- 60.Pelle G, et al. Systemic and kidney toxicity of intraocular administration of vascular endothelial growth factor inhibitors. Am J Kidney Dis. 2011;57(5):756–759. doi: 10.1053/j.ajkd.2010.11.030. [DOI] [PubMed] [Google Scholar]
- 61.Ohr M, Kaiser PK. Aflibercept in wet age-related macular degeneration: a perspective review. Ther Adv Chronic Dis. 2013;3(4):153–161. doi: 10.1177/2040622312446007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heier JS, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–2548. doi: 10.1016/j.ophtha.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 63.Sivaprasad S, et al. Retrospective analysis of the real-world utilization of ranibizumab in wAMD. Invest Ophthalmol Vis Sci. 2013;54(6):3836. [Google Scholar]
- 64.Chakravarthy U, et al. Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology. 2012;119(7):1399–1411. doi: 10.1016/j.ophtha.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 65.Chakravarthy U, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet. 2013;382(9900):1258–1267. doi: 10.1016/S0140-6736(13)61501-9. [DOI] [PubMed] [Google Scholar]
- 66.El Sanharawi M, Kowalczuk L, Touchard E, Omri S, de Kozak Y, Behar-Cohen F. Protein delivery for retinal diseases: from basic considerations to clinical applications. Prog Retin Eye Res. 2010;29(6):443–465. doi: 10.1016/j.preteyeres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 67.Stahl A, et al. Highly potent VEGF-A-antagonistic DARPins as anti-angiogenic agents for topical and intravitreal applications. Angiogenesis. 2013;16(1):101–111. doi: 10.1007/s10456-012-9302-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wolf S, et al. Phase I Mp0112 wet AMD study: results of a single escalating dose study with DARPin(R) MP0112 in wet AMD. Invest Ophthalmol Vis Sci. 2011;52(6):1655. doi: 10.1167/iovs.10-6003. [DOI] [PubMed] [Google Scholar]
- 69.Boyer D. A phase 2b study of fovista & trade; a platelet derived growth factor (PDGF) inhibitor in combination with a vascular endothelial growth factor (VEGF) inhibitor for neovascular age-related Macular Degeneration (AMD). Invest Ophthalmol Vis Sci. 2013;54(6):2175. [Google Scholar]
- 70.Klein R, Klein BE, Knudtson MD, Meuer SM, Swift M, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007;114(2):253–262. doi: 10.1016/j.ophtha.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 71.Radu RA, et al. Reductions in serum vitamin A arrest accumulation of toxic retinal fluorophores: a potential therapy for treatment of lipofuscin-based retinal diseases. Invest Ophthalmol Vis Sci. 2005;46(12):4393–4401. doi: 10.1167/iovs.05-0820. [DOI] [PubMed] [Google Scholar]
- 72.Mata NL, Lichter JB, Vogel R, Han Y, Bui TV, Singerman LJ. Investigation of oral fenretinide for treatment of geographic atrophy in age-related macular degeneration. Retina. 2013;33(3):498–507. doi: 10.1097/IAE.0b013e318265801d. [DOI] [PubMed] [Google Scholar]
- 73.Zhao C, et al. mTOR-mediated dedifferentiation of the retinal pigment epithelium initiates photoreceptor degeneration in mice. J Clin Invest. 2011;121(1):369–383. doi: 10.1172/JCI44303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong WT, et al. Treatment of geographic atrophy with subconjunctival sirolimus: results of a phase I/II clinical trial. Invest Ophthalmol Vis Sci. 2013;54(4):2941–2950. doi: 10.1167/iovs.13-11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Holz F. The MAHALO Phase II Study: Safety, Tolerability and Evidence of Activity of Lampalizumab (Anti-factor D) in Patients with Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration (AMD). Paper presented at: 13th Euretina Congress; September 27, 2013; Hamburg, Germany. [Google Scholar]
- 76.Parker C. Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet. 2009;373(9665):759–767. doi: 10.1016/S0140-6736(09)60001-5. [DOI] [PubMed] [Google Scholar]
- 77.Yehoshua Z, et al. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study of ophthalmology. Ophthalmology. 2013:S0161-6420(13)00889-0. doi: 10.1016/j.ophtha.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.LaVail MM, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39(3):592–602. [PubMed] [Google Scholar]
- 79.Cayouette M, Behn D, Sendtner M, Lachapelle P, Gravel C. Intraocular gene transfer of ciliary neurotrophic factor prevents death and increases responsiveness of rod photoreceptors in the retinal degeneration slow mouse. J Neurosci. 1998;18(22):9282–9293. doi: 10.1523/JNEUROSCI.18-22-09282.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang K, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108(15):6241–6245. doi: 10.1073/pnas.1018987108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Collier RJ, et al. Agonists at the serotonin receptor (5-HT(1A)) protect the retina from severe photo-oxidative stress. Invest Ophthalmol Vis Sci. 2011;52(5):2118–2126. doi: 10.1167/iovs.10-6304. [DOI] [PubMed] [Google Scholar]
- 82.Wheeler L, WoldeMussie E, Lai R. Role of alpha-2 agonists in neuroprotection. Surv Ophthalmol. 2003;48(suppl 1):S47–S51. doi: 10.1016/S0039-6257(03)00004-3. [DOI] [PubMed] [Google Scholar]
- 83.Saylor M, McLoon LK, Harrison AR, Lee MS. Experimental and clinical evidence for brimonidine as an optic nerve and retinal neuroprotective agent: an evidence-based review. Arch Ophthalmol. 2009;127(4):402–406. doi: 10.1001/archophthalmol.2009.9. [DOI] [PubMed] [Google Scholar]
- 84.Schwartz SD, et al. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012;379(9817):713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- 85.Smailhodzic D, et al. Central areolar choroidal dystrophy (CACD) and age-related macular degeneration (AMD): differentiating characteristics in multimodal imaging. Invest Ophthalmol Vis Sci. 2011;52(12):8908–8918. doi: 10.1167/iovs.11-7926. [DOI] [PubMed] [Google Scholar]