

Abstract
It is increasingly evident that cancer results from altered organ homeostasis rather than from deregulated control of single cells or groups of cells. This applies especially to epithelial cancer, the most common form of human solid tumors and a major cause of cancer lethality. In the vast majority of cases, in situ epithelial cancer lesions do not progress into malignancy, even if they harbor many of the genetic changes found in invasive and metastatic tumors. While changes in tumor stroma are frequently viewed as secondary to changes in the epithelium, recent evidence indicates that they can play a primary role in both cancer progression and initiation. These processes may explain the phenomenon of field cancerization, i.e., the occurrence of multifocal and recurrent epithelial tumors that are preceded by and associated with widespread changes of surrounding tissue or organ “fields.”
Introduction
The vast majority of epithelial cancers are limited to in situ lesions that, for internal organs like breast, prostate, or lung, can remain undetected for the whole life of an individual (1, 2). The reason why only a minor fraction of these lesions progress to malignancy is not understood. In fact, many if not most of the genetic changes found in invasive and metastatic tumors are already present in premalignant lesions, raising the question of whether such changes are a primary cause or merely permissive for later cancer-spreading events. A related issue raised by deep sequencing analysis of tumors is the question of whether any of the identified driver mutations actually initiate the carcinogenic process (3). An extreme view is that none of these mutations are by themselves a driver of cancer development and that it is the ecological cellular environment that restrains or unleashes tumor growth (2, 4). Changes in tumor stroma are most frequently viewed as secondary to changes in the epithelium; however, recent evidence indicates that they may play a primary role. Such a possibility would help explain, not only dormancy of most epithelial cancers, but also field cancerization, a condition of major clinical significance defined as broader tissue and organ changes beyond localized areas of tumor development that result in multifocal and recurrent tumors (refs. 5, 6, and Figure 1).
Figure 1. Potential determinants of multifocal and recurrent epithelial cancer and field cancerization.
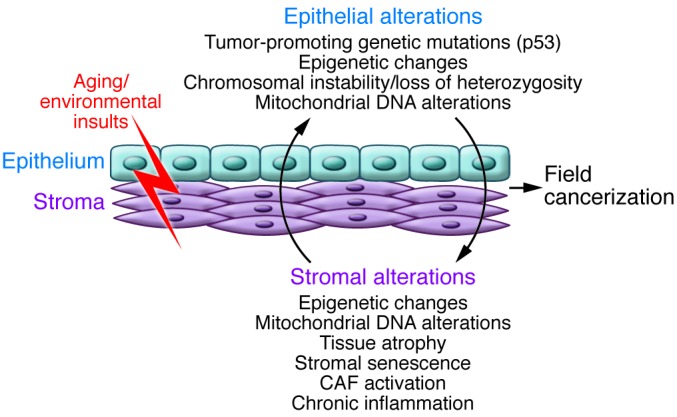
Aging and environmental insults, such as UV irradiation or smoke, can target both epithelial and stromal compartments of organs, leading to stable genetic and epigenetic changes. Cross-talk between these two compartments can induce further pro-oncogenic alterations, such as secretion of growth factors and proteases, alterations in the extracellular matrix, and recruitment of inflammatory cells. These spreading alterations in both the epithelium and stroma are a phenomenon known as field cancerization.
In this Review, I will start with an overview of the clinical problem, followed by a discussion of underlying changes in epithelial and stromal tissues. I will focus on new insights into early stromal events that precede and determine the development of epithelial cancer. A defining primary role of the stroma may be of substantial conceptual and practical value for the development of new approaches to treat and prevent epithelial cancer.
The clinical problem
An important but overlooked fact is the multifocality of cancer, with a surprisingly high frequency of multiple lesions of primary origin (with estimates ranging between 3% and 25%) of same or different histological types, with concomitant or subsequent occurrence (synchronous versus metachronous lesions), and with occurrences at proximal versus distant organ sites (7–9). An obvious difficulty is distinguishing between truly independent primary lesions and separate lesions that are the result of distant spread with single initiating events. As a result, published frequencies of multiple primary (MP) cancers depend on operational definitions adopted by various cancer registries, like those of the Surveillance, Epidemiology and End Results Program (SEER) ( http://seer.cancer.gov/) and the International Association of Cancer Registries (IACR) ( http://www.iacr.com.fr/). Typically, multifocal malignant lesions originating at same body sites, including the entire lung, are considered for epidemiological purposes as single primary cancers. However, significant differences exist in the adopted criteria, including the counting of contralateral malignant breast lesions as MP cancers according to SEER, but not IACR, rules. A number of epidemiological studies have also reported on the incidence of multiple primary tumors within the same or neighboring organs, with potentially important insights (10–17). Notably, premalignant lesions are usually excluded from cancer statistics so that real frequencies of MP lesions are likely to be significantly underestimated, a conclusion supported by the staggering numbers of premalignant and malignant lesions that are discovered by autopsy studies of individuals with other causes of death (30%–40% of cases) (18–21).
Squamous cell carcinoma of the head and neck (HNSCC) is the sixth major cause of cancer death and a problem of major clinical significance (22). The concept of field cancerization was first developed in a landmark study of these tumors, in which a link was established between the common multifocality and recurrences of HNSCC and histological abnormalities, not visible to the naked eye, in surrounding epithelial and stromal tissues (6). There have been substantial advances in surgical treatment of HNSCC, in combination with radiotherapy and targeted approaches, such as EGFR inhibitors. However, these improvements have not led to any significant decrease in locoregional recurrences, secondary tumors, or early distant metastases, and the overall five-year survival rate has improved only marginally (22). Cutaneous field cancerization with multifocal and recurrent skin squamous cell carcinomas (SCCs) is also a common occurrence in organ transplant recipient patients treated with calcineurin inhibitors, representing a major cause of death (23).
Adenocarcinomas and SCCs of the lung are also frequently multifocal (24). While adenocarcinomas tend to be peripherally located and difficult to find at early stages, lung SCCs preferentially develop in the central and pericentral airways, allowing for the observation of early tissue alterations. The use of autofluorescence bronchoscopy imaging coupled with targeted biopsies and clinical follow-up studies has failed to provide convincing evidence of a sequential progression from mild/severe dysplastic lesions to carcinoma in situ (CIS) and invasive SCC of the lung (25, 26). Importantly, while malignant progression of dysplastic lesions is an infrequent event, they are often associated with development of lung SCC at other sites. In fact, high-grade dysplasia and CIS are an indication of elevated risk of cancer in the entire lung epithelium (24, 25).
Multifocal development of breast cancer is also frequent, and it is not clear whether these lesions are monoclonally derived (13, 27). Recurrences can also appear around the site of surgical resection and at more distal sites, including the contralateral breast (27). A major disease management decision facing patients with early-stage breast cancer (stage 0–II) is between conservative limited excisions (lumpectomy) and drastic mastectomy. The risk of local or relatively distant recurrences after lumpectomy is significant (5%–22%; ref. 28), and minimal residual disease is a generally accepted explanation. Alternatively, recent experimental evidence discussed below suggests that de novo cancer cell development triggered by primary stromal alterations may take place.
In prostate cancer, multifocal lesions occur frequently, with >90% of patients diagnosed with the disease carrying two or more cancerous foci (29). There is great heterogeneity of these lesions and different rates of progression, each evolving independently of the other. This poses major clinical problems in terms of diagnosis, prognosis, and treatment (30). Systematic multiple biopsies of the prostate (“saturation biopsies”) are necessary to minimize chances that microscopic but highly aggressive lesions remain undetected (false-negative diagnosis). On the other hand, accurate prognostic and treatment decisions are hampered by the inability to predict behavior of apparently indolent lesions, with the resulting problem of overtreatment of patients (31).
In addition to the cancer types mentioned above, field cancerization plays an important role in the development of esophageal (32), gastric (33), colon (34), bladder (35), and cervical cancer (36). The association of this process with pancreatic and ovarian cancer is less appreciated but probably equally important (5). Field effects have also been implicated in nonepithelial cancer types, such as melanoma (37) and brain tumors (38). Further, bone marrow field effects have been invoked to explain the development of secondary myelodysplastic syndrome and/or acute leukemia in patients initially diagnosed with severe aplastic anemia (39, 40).
Epithelial precursor changes
Substantial attention has been devoted to genetic and/or epigenetic changes in epithelial cells as primary culprits in field cancerization leading to epithelial cancer. Such changes can occur in the absence of any histological abnormalities and at a significant distance (hundreds/thousands of cells) from tumors. They have been interpreted in the context of the multistep model of carcinogenesis, as established by classical experimental studies and clinical analysis of specific cancer types with well-defined sequential steps, like carcinoma of the colon (41).
According to this view, cancer fields result from the clonal expansion and spreading of epithelial cells with genetic alterations that have a role in cancer initiation and evolution (41). Brash and colleagues originally found that normal human skin, especially sun-exposed areas in aging individuals, contain a significant number of epidermal cells with pro-oncogenic p53 mutations (42). These cells are present as clusters that can increase in size over time. Cell populations with p53 mutations in apparently normal tissues have also been found in a number of other organs, including oral (43, 44), bronchial (45, 46), bladder (35), and esophageal (32, 47) epithelium. In concert with p53, its cousin p63 is also involved in epithelial stem cell potential and/or cell-cell adhesion (48–50). The functional implications of loss of normal p53 and/or altered p53/p63 balance seem obvious; however, the possibility that p53 mutations can be a marker of expanding clones of cells, rather than an initial or obligate cancer-triggering event, should also be considered. Consistent with this possibility is the finding that, in patients with primary oral SCCs, the presence of cells with p53 mutations in the normal epithelium is not associated with increased risk of secondary tumors (51). Additionally, discordant p53 mutations can be found in multiple cancer lesions in the same patients (52). Besides p53 mutations, other genetic events, specifically loss of heterozygosity (LOH) of common chromosomal regions, have been demonstrated in multiple lesions of the same patients and intervening normal epithelium in a number of organs, including breast (27), bronchial (53), and oral mucosa (54). LOH may contribute to clonal expansion and/or subsequent cancer development but also reflects genetic alterations that can randomly occur in a significant fraction of somatic cells, as recently reported for copy number variations in dermal fibroblasts from adults (55).
Another of the distinguishing features of cancer fields are the frequent epigenetic alterations that occur in the apparently normal cancer-surrounding epithelium (56–58). Increased DNA methylation at the promoter region of known or putative tumor-suppressing genes can lead to downregulation of their expression and function (59, 60), which may contribute to subsequent tumor development. A similar mechanism of epigenetic silencing has been found for microRNAs with putative tumor-suppressive function in gastric cancer fields (61, 62). A link has been established between Helicobacter pylori infection and epigenetic silencing of tumor-suppressing genes and miRNAs in apparently normal gastric mucosa (58). A possibly primary role of chronic inflammation and associated production of highly reactive oxygen species (ROS) in field cancerization will be considered further below. In responding epithelia, increased DNA methylation at critical transcription regulatory regions can occur as a consequence of induced DNMT1 expression (63) through a variety of mechanisms, including p53- or AP1-dependent gene transcription (64). An early consequence of increased ROS levels is also recruitment of the DNMT1 enzyme to key growth-regulatory genes like p16, thereby silencing their transcription (65). Besides DNA methylation, increased methylation of lysine 27 in the tail of histone 3 (H3K27me3) at a specific set of cell regulatory genes has also been recently reported to be a consequence of inflammation in a mouse model of colon carcinogenesis (66).
Genes involved in DNA damage repair can also be epigenetically silenced in epithelial cancer fields (67). DNA instability, with increased microsatellite variability and telomere shortening, is not limited to tumors but occurs in the surrounding epithelial tissue (68–73). Mitochondrial DNA mutations are also a hallmark of cancer found in neighboring epithelia and, to an even greater extent, underlying stroma (74, 75). As both epigenetic gene silencing and mitochondrial dysfunction are trademarks of aging (76), they likely provide an important link between the age-related increase in cancer risk and field cancerization events.
Inflammation: a primary or secondary determinant?
The determining role of inflammation in cancer development has been postulated since the 1850s (77) and is a subject of intense investigation. Many premalignant and malignant lesions are associated with an inflammatory reaction, which can have both cancer-promoting and -suppressing effects. This is clinically illustrated in the skin, in which development of actinic keratosis lesions, very common precursors of SCC and/or in situ SCC, is intimately connected with chronic inflammation. On the other hand, these lesions can be effectively reversed by treatment with TLR agonists that trigger a potent acute inflammatory reaction (78). Many studies have been dedicated to understanding how a cancer-promoting inflammatory environment can be “reeducated” to become cancer suppressing (79).
Macrophages and T cells are the primary determinants of inflammatory processes. The behavior of macrophages is very plastic and fulfills substantially different functions in acute versus chronic inflammation. These cells can be polarized into a “killing” M1 phenotype for microbe and cancer cell elimination and a permissive M2 phenotype aimed at resolving or containing acute toxic inflammation (80). It is tempting to equate M1 macrophages with an acute inflammatory reaction that can eradicate incipient tumor formation, while M2 cells can be linked with “smoldering” chronic inflammation, which promotes the carcinogenic process. This categorization is most likely an oversimplification, as M1 cytokines can have tumor-promoting effects, while M2 cytokines, such as IL-10, can be tumor suppressive (79).
Strong epidemiological and experimental evidence points to an important role for inflammation in the initial stages of cancer development. However, an important distinction needs to be made between a permissive/promoting function in expansion of “initiated” (i.e., mutated) cancer cells and a primary cancer trigger. In human skin, risk of SCC is substantially increased in clinical conditions associated with chronic inflammation (81). In mouse skin, susceptibility to chemically induced carcinogenesis is greatly influenced by transgenic expression or deletion of proinflammatory cytokines, like IL-1α (82), or enzymes, like COX-2 (83–85). However, few or no tumors developed in all these cases without prior treatment with a mutagenic carcinogen. Similarly, in classical chemical skin carcinogenesis studies, repeated treatments with proinflammatory but nonmutagenic agents, such as the tumor-promoting phorbol esters, are not sufficient for tumor initiation (81).
In contrast to the studies described above, studies in the gastrointestinal system suggest that inflammation alone can trigger cancer development. Patients with inflammatory bowel disease have a highly increased risk of colon cancer and colitis-associated cancer; a similar condition can be experimentally induced in mice by oral administration of proinflammatory tissue-damaging agents, like dextran sodium sulfate (86–89). A strong causative link exists also between H. pylori infection and stomach cancer, in which inflammation has been implicated as an underlying cause (90). Direct evidence in support of this possibility was provided by the finding that transgenic overexpression of a proinflammatory cytokine, IL-1β, in the gastric mucosa was sufficient to elicit cancer development through a cascade of NF-κB–activating cytokines and associated recruitment of immune modulatory cells (91). Interestingly, increased expression of IL-1β resulted, not only in increased proliferation and transformation of the gastric epithelium, but also in atrophy of the underlying stroma. As discussed below, stromal atrophy and associated fibroblast senescence can contribute significantly to the field cancerization process and can even have a primary determining function.
Mesenchymal stromal alterations
Epithelial cells covering the surface of organs are primary sensors of exogenous insults that trigger an inflammatory reaction. Resident cells of the stromal compartment are usually assumed to play a more secondary reactive role; however, a number of stimuli can directly affect the stroma, inducing changes that promote or even initiate the carcinogenic process. Long-wave UV (UVA) is thought to be a major cause of UV-induced skin cancer. It accounts for about 95% of total UV light exposure and, because of its greater penetration power, can directly affect the dermal compartment (92). Chronic UVA exposure leads to solar elastosis, a condition characterized by dermal atrophy and cellular and extracellular matrix alterations that, in the clinic (93) as well as a mouse model of skin field cancerization (94), precede keratinocyte tumor development. Smoking is another major cause of cancer, leading to a substantially increased risk, not only in lung, but in other organs, such as oral mucosa (22), bladder (95), and breast (96). A recent report has raised the possibility that chemicals in smoke can diffuse through the surface lung epithelium, directly targeting stromal cells of various organs (97). Interestingly, metabolites produced by the obesity-associated microbiome were recently implicated in liver cancer (98). In each of these cases, stromal cell senescence in the target organ is a putative tumor-promoting or -triggering mechanism.
Fibroblast senescence induces a program of gene expression, overlapping with that of cancer-associated fibroblasts (CAFs), including production of several diffusible growth factors and cytokines, like IL-6 (99, 100), whose increased expression can promote inflammation and proliferation of neighboring epithelial cancer cells (101). Stromal cell senescence may be more important in the initial stages of epithelial cancer than at later times, as increased rather than decreased fibroblast density, so called “tumor-associated desmoplasia,” is frequently seen around tumors, like pancreatic cancer (102). Senescent cells can be removed in vivo through a number of mechanisms, including macrophage activation (103–105), and there can be in vivo selective pressure for stromal cells with CAF properties that have escaped senescence. In fact, stromal changes coevolve with cancer development and result from a variety of epigenetic events (106–110). Chromosomal and/or genetic alterations, including loss of P53 (111–117), have also been reported to occur in the tumor stroma, although the significance of these findings has been questioned (107, 110).
To understand the complex role of stromal fibroblasts in the initiation of epithelial cancer, it is important to consider their intrinsic heterogeneity (118). Fibroblast populations from various body parts and within individual organs can have significantly different properties, including susceptibility to CAF phenotype acquisition and interactions with neighboring epithelial cells and cells of the immune/inflammatory system (119–123). These differences, and/or underlying gene expression signature, are rather stable and maintained with cultivation (124–127).
The role of mesenchymal stroma alterations in cancer initiation was proposed several years ago in the context of colon (128) and prostate (129) cancers. An important distinction needs to be made between mesenchymal outgrowths as a primary consequence of genetic alterations and secondarily increased risk of epithelial cancer and less evident stromal changes, resulting from aging and/or exogenous insults, whose main consequences are epithelial dysplastic and neoplastic lesions. Examples of genetically determined mesenchymal outgrowths with consequently increased risk of epithelial cancer are intestinal polyposis syndromes, which are characterized by multiple hamartomas and associated inflammation (130). For example, Peutz-Jeghers syndrome (PJS) results from inactivating mutations of the serine/threonine kinase 11 (LKB1) gene (131). Available evidence suggests that LKB1 plays an important function in both stromal mesenchyme and the overlying epithelium (132–134). In fact, while loss of homozygosity occurs in the adenocarcinomas that develop in patients with PJS (130), LKB1 mutations are haplosufficient for polyp formation and are found equally in mice with global myofibroblast-specific haploinsufficiency (134). While LKB1 is best known for control of AMPK/mTOR signaling (132), its role in the colonic mesenchyme is connected to decreased TGF-β expression and function (134, 135).
TGF-β signaling is a key regulator of fibroblast behavior and, depending on conditions, can induce fibrosis and/or CAF activation (136). To genetically probe the role of this pathway in the stromal mesenchyme, TGF-β type II receptor (Tgfr2) was ablated in this compartment (137). The resulting phenotype is noteworthy for both observed alterations and those that were not found. Mice developed epithelial tumors in stomach and prostate that were associated with increased proliferation and density of surrounding fibroblasts (137). Loss of Tgfr2 in gastric and/or prostate fibroblasts resulted in increased expression of hepatocyte growth factor, Wnts, and a number of proinflammatory genes that are associated with paracrine mechanisms for cancer development (137–140). In contrast to stomach and prostate, all other examined organs were normal in this mouse model, including the skin, in which effective and dermal fibroblast-specific deletion of Tgfr2 was also demonstrated (137). Given the important role of TGF-β in mesenchymal compartments of skin and other organs, this lack of effect is very surprising and may reflect the already mentioned differences of stromal fibroblasts at various body sites.
Developmental fields play a key role in tissue and organ morphogenesis. Their establishment depends on various forms of direct and indirect cell-cell communication and gradients of diffusible morphogens that instruct initially equivalent cells to assume different cell fates (141, 142). Selective adhesion of cells and physical force can also be a driving force in the morphogenetic process (143). Besides organ morphogenesis, these mechanisms may be implicated in maintenance and repair of already formed organs, for what has been termed “organ morphostats” (144). Notch/CSL signaling is an important developmental pathway and form of direct cell-cell communication (145). In the skin, a substantial body of evidence has shown that the Notch pathway promotes keratinocyte differentiation and suppresses tumor formation (146, 147). In contrast to that in the epidermis, the role of this pathway in the mesenchymal compartment of the skin has not been investigated until recently. Mice with mesenchymal deletion of CSL, the key effector of canonical Notch signaling, exhibited hair follicle abnormalities (148) and a skin phenotype with features of field cancerization, including early and widespread dermal atrophy, followed by expanding areas of inflammation and, by 2 to 4 months, multifocal keratinocyte tumors with features of actinic keratosis or in situ SCCs that eventually progressed into invasive cancer (94). Development of neoplastic lesions was significantly delayed by inhibition of inflammation, indicating that this process is an important mediator of the mesenchyme-induced epithelial lesions (94).
Further studies showed that compromised Notch/CSL signaling is likely to play a central role in intrinsic control of CAF activation, as deletion or silencing of CSL in dermal fibroblasts of either murine or human origin was sufficient to induce expression of many CAF effector proteins and transcription factors of the AP1 family (94), major determinants of skin photo-aging and cancer (149–151). The CSL protein exerts an intrinsic transcription-repressing function and binds to specific target genes in a dynamic manner (152, 153). As a result, expression of genes to which CSL binds with high affinity can be induced by either of two possible mechanisms: (a) transcriptional or posttranscriptional downregulation of CSL expression and function; (b) conversion of CSL from a transcriptional repressor into a transcriptional activator by activated Notch receptors. Consistent with this mode of action, similarly to loss of CSL function, Notch activation has also been implicated in CAF activation as well as fibrosis, in concert with other pathways and signals from the external cellular environment (refs. 102, 136, 154, 155, and Figure 2).
Figure 2. Converging control of CAF activation by multiple signaling pathways.
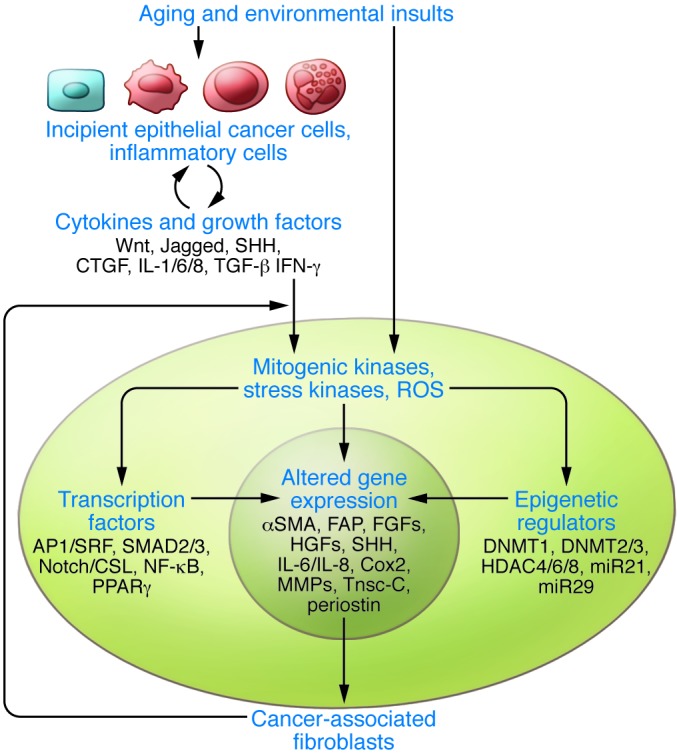
Activation of a CAF phenotype results in the increased stromal expression of a battery of molecules (representative ones are indicated in the center circle) that enhance epithelial cancer development and progression. These changes can be the combined result of several extracellular signals triggered by direct exogenous insults (smoke products, UV) or indirectly by a battery of growth factors and cytokines produced by neighboring cells (incipient epithelial cancer cells, immune/inflammatory cells). Signal amplification can then take place through a number of intermediate cytoplasmic events, like activation of kinase cascade pathways and production of ROS. Finally, establishment of a CAF phenotype is critically dependent on control of gene expression through a battery of transcription factors and epigenetic changes functioning in either a positive or negative manner. As discussed in the text, two of these pathways, linking extracellular signals to control of gene transcription, have been implicated so far in primary stromal alterations leading to epithelial cancer development. Mesenchymal loss or alterations of TGF-β/Smad signaling lead to gastric and prostate cancerous lesions (137), while compromised Notch/CSL signaling results in multiple keratinocyte tumors and skin field cancerization (94). The surprising organ-selective effects remain to be understood but could be due to heterogeneity of CAF cells of origin.
Conclusions and future directions
While metastatic spread is the principal cause of cancer-related deaths, field cancerization, with multifocal and recurrent tumors, is another clinical condition of major morbidity and lethality. As discussed above, multiplicity of lesions not amenable to surgical treatment and recurrent cancer after excision of the primary tumor are problems of foremost significance. Using a metaphor from the botanical garden, the situation can be analogous to that of a bad plant that is difficult to eradicate, because of the many roots deeply embedded in the terrain or the spreading of multiple bad seeds (Figure 3A). There is, however, another possibility — that of a bad soil that could corrupt properties of otherwise perfectly good plants (Figure 3B). In this case, unless the soil is corrected, various forms of intervention are of little or no use. Two main challenges need to be addressed. There is an urgent need to identify markers of stromal as well as epithelial alterations to guide the surgeon in tissue excision procedures and the clinician in decisions of therapeutic intervention. In this context, new in vivo imaging approaches for detection of stromal tissue alterations, including inflammation, abnormal matrix composition, proteolytic activity, fibroblast senescence, and/or altered density, could lead to important breakthroughs.
Figure 3. The seed and soil hypothesis in multifocal and recurrent epithelial cancer.

Multifocal and recurrent epithelial cancer may be analogous to a difficult to eradicate weed. (A) The theory of seed implantation suggests that multifocal recurrent tumors may be due to the ability of monoclonal cancer cells (seeds) to root deeply into the terrain and spread locally as well as disseminate to distant sites. Much like weeds, these tumor cells can grow under many conditions. (B) In contrast, the bad soil hypothesis suggests that insults and alterations in the stroma generate highly permissible soil that allows for the growth of multiple tumors of monoclonal or polyclonal origin (field cancerization). According to this latter view, unless the soil is corrected, various forms of prevention or intervention would be of little use in treating such cancers.
A second important goal is the elucidation of signaling pathways more closely connected with field cancerization as a necessary step to devise novel preventive and therapeutic treatments. In this respect, use of antiinflammatory agents is a promising venue to retard the process, even if it is probably not sufficient to totally prevent it. Targeting specific developmental signaling pathways provides another possible approach to counteract the spread of cancer fields. Equally attractive is the possibility of interfering with CAF activation (102) and, at an earlier stage, tissue aging and associated stromal cell senescence (156). In this respect, we note the complex relation between metabolism and aging (76) and the significantly increased cancer risk associated with obesity and diabetes (157, 158). One possibility is that a number of metabolism-modulatory drugs used to ameliorate these conditions could also be of substantial benefit for prevention and even reversal of field cancerization. Finally, a number of treatment regimens are already used for cancer field therapy in skin (159), and a better understanding of their mechanism of action may be instrumental in extending such treatments to other organs in which field cancerization takes place.
Acknowledgments
I thank Patrice Jichlinski, Günther Hofbauer, Maryse Fiche, and Fabio Levi for helpful feedback and discussions. This work was supported by grants from the Swiss National Foundation (grants CRSI33-130576/1; 310030B_138653/1), Oncosuisse (grant OCS-2922-02-2012), and the NIH (grant AR39190). Given the broad and complex topic, in many cases only representative papers were referenced, and I apologize to the many colleagues whose work was not included.
Footnotes
Conflict of interest: The author has declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(4):1446–1453. doi:10.1172/JCI72589.
References
- 1.Cardiff RD, Borowsky AD. Precancer: sequentially acquired or predetermined? Toxicol Pathol. 2010;38(1):171–179. doi: 10.1177/0192623309356447. [DOI] [PubMed] [Google Scholar]
- 2.Gatenby R. Perspective: Finding cancer’s first principles. Nature. 2012;491(7425):S55. doi: 10.1038/491S55a. [DOI] [PubMed] [Google Scholar]
- 3.Eifert C, Powers RS. From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat Rev Cancer. 2012;12(8):572–578. doi: 10.1038/nrc3299. [DOI] [PubMed] [Google Scholar]
- 4.Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 5.Dakubo GD, Jakupciak JP, Birch-Machin MA, Parr RL. Clinical implications and utility of field cancerization. Cancer Cell Int. 2007;7:2. doi: 10.1186/1475-2867-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953;6(5):963–968. doi: 10.1002/1097-0142(195309)6:5<963::AID-CNCR2820060515>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 7.Rosso S, et al. Multiple tumours in survival estimates. Eur J Cancer. 2009;45(6):1080–1094. doi: 10.1016/j.ejca.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 8.Schottenfeld D. The epidemiology of multiple primary cancers. CA Cancer J Clin. 1977;27(4):233–240. doi: 10.3322/canjclin.27.4.233. [DOI] [PubMed] [Google Scholar]
- 9.Weir HK, Johnson CJ, Thompson TD. The effect of multiple primary rules on population-based cancer survival. Cancer Causes Control. 2013;24(6):1231–1242. doi: 10.1007/s10552-013-0203-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Braisch U, Meyer M, Radespiel-Troger M. Risk of tobacco-related multiple primary cancers in Bavaria, Germany. BMC Cancer. 2012;12:250. doi: 10.1186/1471-2407-12-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bratslavsky G, Linehan WM. Long-term management of bilateral, multifocal, recurrent renal carcinoma. Nat Rev Urol. 2010;7(5):267–275. doi: 10.1038/nrurol.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jain KS, Sikora AG, Baxi SS, Morris LG. Synchronous cancers in patients with head and neck cancer: risks in the era of human papillomavirus-associated oropharyngeal cancer. Cancer. 2013;119(10):1832–1837. doi: 10.1002/cncr.27988. [DOI] [PubMed] [Google Scholar]
- 13.Jain S, Rezo A, Shadbolt B, Dahlstrom JE. Synchronous multiple ipsilateral breast cancers: implications for patient management. Pathology. 2009;41(1):57–67. doi: 10.1080/00313020802563502. [DOI] [PubMed] [Google Scholar]
- 14.Kuligina E, Reiner A, Imyanitov EN, Begg CB. Evaluating cancer epidemiologic risk factors using multiple primary malignancies. Epidemiology. 2010;21(3):366–372. doi: 10.1097/EDE.0b013e3181cc8871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levi F, et al. High constant incidence of second primary colorectal cancer. Int J Cancer. 2013;132(7):1679–1682. doi: 10.1002/ijc.27780. [DOI] [PubMed] [Google Scholar]
- 16.Levi F, Randimbison L, Maspoli M, Te VC, La Vecchia C. High incidence of second basal cell skin cancers. Int J Cancer. 2006;119(6):1505–1507. doi: 10.1002/ijc.22000. [DOI] [PubMed] [Google Scholar]
- 17.Levi F, Te VC, Randimbison L, Maspoli M, La Vecchia C. Second primary oral and pharyngeal cancers in subjects diagnosed with oral and pharyngeal cancer. Int J Cancer. 2006;119(11):2702–2704. doi: 10.1002/ijc.22183. [DOI] [PubMed] [Google Scholar]
- 18.Albertsen PC. Commentary: occult prostate cancer—imposter or the real deal? Int J Epidemiol. 2007;36(2):281–282. doi: 10.1093/ije/dym051. [DOI] [PubMed] [Google Scholar]
- 19.Khurana KK, Loosmann A, Numann PJ, Khan SA. Prophylactic mastectomy: pathologic findings in high-risk patients. Arch Pathol Lab Med. 2000;124(3):378–381. doi: 10.5858/2000-124-0378-PMPFIH. [DOI] [PubMed] [Google Scholar]
- 20.Welch HG, Black WC. Using autopsy series to estimate the disease “reservoir” for ductal carcinoma in situ of the breast: how much more breast cancer can we find? Ann Intern Med. 1997;127(11):1023–1028. doi: 10.7326/0003-4819-127-11-199712010-00014. [DOI] [PubMed] [Google Scholar]
- 21.Zlotta AR, et al. Prevalence of prostate cancer on autopsy: cross-sectional study on unscreened Caucasian and Asian men. J Natl Cancer Inst. 2013;105(14):1050–1058. doi: 10.1093/jnci/djt151. [DOI] [PubMed] [Google Scholar]
- 22.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 23.Hofbauer GF, Bavinck JN, Euvrard S. Organ transplantation and skin cancer: basic problems and new perspectives. Exp Dermatol. 2010;19(6):473–482. doi: 10.1111/j.1600-0625.2010.01086.x. [DOI] [PubMed] [Google Scholar]
- 24.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishizumi T, McWilliams A, MacAulay C, Gazdar A, Lam S. Natural history of bronchial preinvasive lesions. Cancer Metastasis Rev. 2010;29(1):5–14. doi: 10.1007/s10555-010-9214-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee P, et al. Color fluorescence ratio for detection of bronchial dysplasia and carcinoma in situ. Clin Cancer Res. 2009;15(14):4700–4705. doi: 10.1158/1078-0432.CCR-08-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heaphy CM, Griffith JK, Bisoffi M. Mammary field cancerization: molecular evidence and clinical importance. Breast Cancer Res Treat. 2009;118(2):229–239. doi: 10.1007/s10549-009-0504-0. [DOI] [PubMed] [Google Scholar]
- 28.Huston TL, Simmons RM. Locally recurrent breast cancer after conservation therapy. Am J Surg. 2005;189(2):229–235. doi: 10.1016/j.amjsurg.2004.07.039. [DOI] [PubMed] [Google Scholar]
- 29.Andreoiu M, Cheng L. Multifocal prostate cancer: biologic, prognostic, and therapeutic implications. Hum Pathol. 2010;41(6):781–793. doi: 10.1016/j.humpath.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Trujillo KA, Jones AC, Griffith JK, Bisoffi M. Markers of field cancerization: proposed clinical applications in prostate biopsies. Prostate Cancer. 2012;2012:302894. doi: 10.1155/2012/302894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman RM. Clinical practice. Screening for prostate cancer. N Engl J Med. 2011;365(21):2013–2019. doi: 10.1056/NEJMcp1103642. [DOI] [PubMed] [Google Scholar]
- 32.Prevo LJ, Sanchez CA, Galipeau PC, Reid BJ. p53-mutant clones and field effects in Barrett’s esophagus. Cancer Res. 1999;59(19):4784–4787. [PubMed] [Google Scholar]
- 33.Hyatt BJ, Paull PE, Wassef W. Gastric oncology: an update. Curr Opin Gastroenterol. 2009;25(6):570–578. doi: 10.1097/MOG.0b013e328331b5c9. [DOI] [PubMed] [Google Scholar]
- 34.Damania D, et al. Nanocytology of rectal colonocytes to assess risk of colon cancer based on field cancerization. Cancer Res. 2012;72(11):2720–2727. doi: 10.1158/0008-5472.CAN-11-3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hoglund M. Bladder cancer, a two phased disease? Semin Cancer Biol. 2007;17(3):225–232. doi: 10.1016/j.semcancer.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Rosenthal AN, Ryan A, Hopster D, Jacobs IJ. Molecular evidence of a common clonal origin and subsequent divergent clonal evolution in vulval intraepithelial neoplasia, vulval squamous cell carcinoma and lymph node metastases. Int J Cancer. 2002;99(4):549–554. doi: 10.1002/ijc.10362. [DOI] [PubMed] [Google Scholar]
- 37.Bastian BC. Understanding the progression of melanocytic neoplasia using genomic analysis: from fields to cancer. Oncogene. 2003;22(20):3081–3086. doi: 10.1038/sj.onc.1206463. [DOI] [PubMed] [Google Scholar]
- 38.Giannopoulos S, Kyritsis AP. Diagnosis and management of multifocal gliomas. Oncology. 2010;79(3–4):306–312. doi: 10.1159/000323492. [DOI] [PubMed] [Google Scholar]
- 39.Brodsky RA, Jones RJ. Riddle: what do aplastic anemia, acute promyelocytic leukemia, and chronic myeloid leukemia have in common? Leukemia. 2004;18(10):1740–1742. doi: 10.1038/sj.leu.2403487. [DOI] [PubMed] [Google Scholar]
- 40.Scadden DT. Rethinking stroma: lessons from the blood. Cell Stem Cell. 2012;10(6):648–649. doi: 10.1016/j.stem.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Braakhuis BJ, Tabor MP, Kummer JA, Leemans CR, Brakenhoff RH. A genetic explanation of Slaughter’s concept of field cancerization: evidence and clinical implications. Cancer Res. 2003;63(8):1727–1730. [PubMed] [Google Scholar]
- 42.Jonason AS, et al. Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A. 1996;93(24):14025–14029. doi: 10.1073/pnas.93.24.14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Houten VM, et al. Mutated p53 as a molecular marker for the diagnosis of head and neck cancer. J Pathol. 2002;198(4):476–486. doi: 10.1002/path.1242. [DOI] [PubMed] [Google Scholar]
- 44.Waridel F, et al. Field cancerisation and polyclonal p53 mutation in the upper aero-digestive tract. Oncogene. 1997;14(2):163–169. doi: 10.1038/sj.onc.1200812. [DOI] [PubMed] [Google Scholar]
- 45.Chung GT, Sundaresan V, Hasleton P, Rudd R, Taylor R, Rabbitts PH. Clonal evolution of lung tumors. Cancer Res. 1996;56(7):1609–1614. [PubMed] [Google Scholar]
- 46.Sundaresan V, et al. p53 and chromosome 3 abnormalities, characteristic of malignant lung tumours, are detectable in preinvasive lesions of the bronchus. Oncogene. 1992;7(10):1989–1997. [PubMed] [Google Scholar]
- 47.Barrett MT, et al. Evolution of neoplastic cell lineages in Barrett oesophagus. Nat Genet. 1999;22(1):106–109. doi: 10.1038/8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crum CP, McKeon FD. p63 in epithelial survival, germ cell surveillance, and neoplasia. Annu Rev Pathol. 2010;5:349–371. doi: 10.1146/annurev-pathol-121808-102117. [DOI] [PubMed] [Google Scholar]
- 49.Ferone G, et al. p63 control of desmosome gene expression and adhesion is compromised in AEC syndrome. Hum Mol Genet. 2013;22(3):531–543. doi: 10.1093/hmg/dds464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koster MI, Roop DR. Sorting out the p63 signaling network. J Invest Dermatol. 2008;128(7):1617–1619. doi: 10.1038/jid.2008.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Escher A, Piotet E, Waridel F, Iggo R, Monnier P. p53 Mutation in histologically normal mucosa of the aero-digestive tract is not a marker of increased risk for second primary carcinoma in head and neck cancer patients. Eur Arch Otorhinolaryngol. 2009;266(4):547–551. doi: 10.1007/s00405-008-0780-z. [DOI] [PubMed] [Google Scholar]
- 52.Chung KY, et al. Discordant p53 gene mutations in primary head and neck cancers and corresponding second primary cancers of the upper aerodigestive tract. Cancer Res. 1993;53(7):1676–1683. [PubMed] [Google Scholar]
- 53.Grepmeier U, et al. Deletions at chromosome 2q and 12p are early and frequent molecular alterations in bronchial epithelium and NSCLC of long-term smokers. Int J Oncol. 2005;27(2):481–488. [PubMed] [Google Scholar]
- 54.Gonzalez-Moles MA, Scully C, Ruiz-Avila I. Molecular findings in oral premalignant fields: update on their diagnostic and clinical implications. Oral Dis. 2012;18(1):40–47. doi: 10.1111/j.1601-0825.2011.01845.x. [DOI] [PubMed] [Google Scholar]
- 55.Abyzov A, et al. Somatic copy number mosaicism in human skin revealed by induced pluripotent stem cells. Nature. 2012;492(7429):438–442. doi: 10.1038/nature11629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dworkin AM, Huang TH, Toland AE. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin Cancer Biol. 2009;19(3):165–171. doi: 10.1016/j.semcancer.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo M, et al. Promoter hypermethylation of resected bronchial margins: a field defect of changes? Clin Cancer Res. 2004;10(15):5131–5136. doi: 10.1158/1078-0432.CCR-03-0763. [DOI] [PubMed] [Google Scholar]
- 58.Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40(2):142–150. doi: 10.5483/BMBRep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 59.Ramachandran K, Singal R. DNA methylation and field cancerization. Epigenomics. 2012;4(3):243–245. doi: 10.2217/epi.12.12. [DOI] [PubMed] [Google Scholar]
- 60.Asada K, et al. FHL1 on chromosome X is a single-hit gastrointestinal tumor-suppressor gene and contributes to the formation of an epigenetic field defect. Oncogene. 2013;32(17):2140–2149. doi: 10.1038/onc.2012.228. [DOI] [PubMed] [Google Scholar]
- 61.Ando T, et al. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124(10):2367–2374. doi: 10.1002/ijc.24219. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki H, et al. Methylation-associated silencing of microRNA-34b/c in gastric cancer and its involvement in an epigenetic field defect. Carcinogenesis. 2010;31(12):2066–2073. doi: 10.1093/carcin/bgq203. [DOI] [PubMed] [Google Scholar]
- 63.Arai E, Kanai Y, Ushijima S, Fujimoto H, Mukai K, Hirohashi S. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer. 2006;119(2):288–296. doi: 10.1002/ijc.21807. [DOI] [PubMed] [Google Scholar]
- 64.Kar S, et al. An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function. Epigenetics. 2012;7(9):994–1007. doi: 10.4161/epi.21568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Hagan HM, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell. 2011;20(5):606–619. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takeshima H, Ikegami D, Wakabayashi M, Niwa T, Kim YJ, Ushijima T. Induction of aberrant trimethylation of histone H3 lysine 27 by inflammation in mouse colonic epithelial cells. Carcinogenesis. 2012;33(12):2384–2390. doi: 10.1093/carcin/bgs294. [DOI] [PubMed] [Google Scholar]
- 67.Chai H, Brown RE. Field effect in cancer-an update. Ann Clin Lab Sci. 2009;39(4):331–337. [PubMed] [Google Scholar]
- 68.Heaphy CM, et al. Telomere DNA content and allelic imbalance demonstrate field cancerization in histologically normal tissue adjacent to breast tumors. Int J Cancer. 2006;119(1):108–116. doi: 10.1002/ijc.21815. [DOI] [PubMed] [Google Scholar]
- 69.Hittelman WN. Genetic instability in epithelial tissues at risk for cancer. Ann N Y Acad Sci. 2001;952:1–12. doi: 10.1111/j.1749-6632.2001.tb02723.x. [DOI] [PubMed] [Google Scholar]
- 70.Shin DM, et al. p53 protein accumulation and genomic instability in head and neck multistep tumorigenesis. Cancer Epidemiol Biomarkers Prev. 2001;10(6):603–609. [PubMed] [Google Scholar]
- 71.Stoler DL, et al. Genomic instability in invasive breast carcinoma measured by inter-simple sequence repeat PCR. Breast Cancer Res Treat. 2006;97(1):107–110. doi: 10.1007/s10549-005-9092-9. [DOI] [PubMed] [Google Scholar]
- 72.Stoler DL, et al. Comparative genomic instabilities of thyroid and colon cancers. Arch Otolaryngol Head Neck Surg. 2007;133(5):457–463. doi: 10.1001/archotol.133.5.457. [DOI] [PubMed] [Google Scholar]
- 73.Wiseman SM, Stoler DL, Anderson GR. The role of genomic instability in the pathogenesis of squamous cell carcinoma of the head and neck. Surg Oncol Clin N Am. 2004;13(1):1–11. doi: 10.1016/S1055-3207(03)00118-2. [DOI] [PubMed] [Google Scholar]
- 74.Ha PK, et al. Mitochondrial C-tract alteration in premalignant lesions of the head and neck: a marker for progression and clonal proliferation. Clin Cancer Res. 2002;8(7):2260–2265. [PubMed] [Google Scholar]
- 75.Tulah AS, Birch-Machin MA. Stressed out mitochondria: The role of mitochondria in ageing and cancer focussing on strategies and opportunities in human skin. Mitochondrion. 2013;13(5):444–453. doi: 10.1016/j.mito.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 76.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 78.Cantisani C, et al. Actinic keratosis: review of the literature and new patents. Recent Pat Inflamm Allergy Drug Discov. 2013;7(2):168–175. doi: 10.2174/1872213X11307020008. [DOI] [PubMed] [Google Scholar]
- 79.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 81.Mueller MM. Inflammation in epithelial skin tumours: old stories and new ideas. Eur J Cancer. 2006;42(6):735–744. doi: 10.1016/j.ejca.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 82.Murphy JE, Morales RE, Scott J, Kupper TS. IL-1α, innate immunity, and skin carcinogenesis: the effect of constitutive expression of IL-1α in epidermis on chemical carcinogenesis. J Immunol. 2003;170(11):5697–5703. doi: 10.4049/jimmunol.170.11.5697. [DOI] [PubMed] [Google Scholar]
- 83.Lao HC, Akunda JK, Chun KS, Flake GP, Yuspa SH, Langenbach R. Genetic ablation of cyclooxygenase-2 in keratinocytes produces a cell-autonomous defect in tumor formation. Carcinogenesis. 2012;33(11):2293–2300. doi: 10.1093/carcin/bgs267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muller-Decker K. Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: pharmacological, genetic, and clinical evidence. Cancer Metastasis Rev. 2011;30(3–4):343–361. doi: 10.1007/s10555-011-9306-z. [DOI] [PubMed] [Google Scholar]
- 85.Muller-Decker K, Neufang G, Berger I, Neumann M, Marks F, Furstenberger G. Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis. Proc Natl Acad Sci U S A. 2002;99(19):12483–12488. doi: 10.1073/pnas.192323799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cooks T, et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer cell. 2013;23(5):634–646. doi: 10.1016/j.ccr.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grivennikov S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis. 2003;24(3):353–362. doi: 10.1093/carcin/24.3.353. [DOI] [PubMed] [Google Scholar]
- 89.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138(6):2101–2114.e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 90.Wroblewski LE, Peek RM, Peek RM, Jr, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. 2010;23(4):713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tu S, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14(5):408–419. doi: 10.1016/j.ccr.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bachelor MA, Bowden GT. UVA-mediated activation of signaling pathways involved in skin tumor promotion and progression. Semin Cancer Biol. 2004;14(2):131–138. doi: 10.1016/j.semcancer.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 93.Yaar M, Gilchrest BA. Photoageing: mechanism, prevention and therapy. Br J Dermatol. 2007;157(5):874–887. doi: 10.1111/j.1365-2133.2007.08108.x. [DOI] [PubMed] [Google Scholar]
- 94.Hu B, et al. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell. 2012;149(6):1207–1220. doi: 10.1016/j.cell.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Freedman ND, Silverman DT, Hollenbeck AR, Schatzkin A, Abnet CC. Association between smoking and risk of bladder cancer among men and women. JAMA. 2011;306(7):737–745. doi: 10.1001/jama.2011.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Reynolds P. Smoking and breast cancer. J Mammary Gland Biol Neoplasia. 2013;18(1):15–23. doi: 10.1007/s10911-012-9269-x. [DOI] [PubMed] [Google Scholar]
- 97.Salem AF, et al. Cigarette smoke metabolically promotes cancer, via autophagy and premature aging in the host stromal microenvironment. Cell Cycle. 2013;12(5):818–825. doi: 10.4161/cc.23722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoshimoto S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 99.Campisi J, Andersen JK, Kapahi P, Melov S. Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol. 2011;21(6):354–359. doi: 10.1016/j.semcancer.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9(2):81–94. doi: 10.1038/nrc2560. [DOI] [PubMed] [Google Scholar]
- 101.Kuilman T, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133(6):1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 102.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 103.Hoenicke L, Zender L. Immune surveillance of senescent cells — biological significance in cancer- and non-cancer pathologies. Carcinogenesis. 2012;33(6):1123–1126. doi: 10.1093/carcin/bgs124. [DOI] [PubMed] [Google Scholar]
- 104.Kang TW, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 105.Xue W, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445(7128):656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bechtel W, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu M, Polyak K. Microenvironmental regulation of cancer development. Curr Opin Genet Dev. 2008;18(1):27–34. doi: 10.1016/j.gde.2007.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu M, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37(8):899–905. doi: 10.1038/ng1596. [DOI] [PubMed] [Google Scholar]
- 109.Jiang L, et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008;68(23):9900–9908. doi: 10.1158/0008-5472.CAN-08-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weinberg RA. Coevolution in the tumor microenvironment. Nat Genet. 2008;40(5):494–495. doi: 10.1038/ng0508-494. [DOI] [PubMed] [Google Scholar]
- 111.Bar J, Moskovits N, Oren M. Involvement of stromal p53 in tumor-stroma interactions. Semin Cell Dev Biol. 2010;21(1):47–54. doi: 10.1016/j.semcdb.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fukino K, Shen L, Matsumoto S, Morrison CD, Mutter GL, Eng C. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64(20):7231–7236. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 113.Hill R, Song Y, Cardiff RD, Van Dyke T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123(6):1001–1011. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 114.Kurose K, Gilley K, Matsumoto S, Watson PH, Zhou XP, Eng C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32(3):355–357. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 115.Matsumoto N, Yoshida T, Yamashita K, Numata Y, Okayasu I. Possible alternative carcinogenesis pathway featuring microsatellite instability in colorectal cancer stroma. Br J Cancer. 2003;89(4):707–712. doi: 10.1038/sj.bjc.6601141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Paterson RF, et al. Molecular genetic alterations in the laser-capture-microdissected stroma adjacent to bladder carcinoma. Cancer. 2003;98(9):1830–1836. doi: 10.1002/cncr.11747. [DOI] [PubMed] [Google Scholar]
- 117.Tuhkanen H, et al. Genetic alterations in the peritumoral stromal cells of malignant and borderline epithelial ovarian tumors as indicated by allelic imbalance on chromosome 3p. Int J Cancer. 2004;109(2):247–252. doi: 10.1002/ijc.11733. [DOI] [PubMed] [Google Scholar]
- 118.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 119.Driskell RR, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504(7479):277–281. doi: 10.1038/nature12783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koumas L, King AE, Critchley HO, Kelly RW, Phipps RP. Fibroblast heterogeneity: existence of functionally distinct Thy 1(+) and Thy 1(–) human female reproductive tract fibroblasts. Am J Pathol. 2001;159(3):925–935. doi: 10.1016/S0002-9440(10)61768-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Koumas L, Smith TJ, Feldon S, Blumberg N, Phipps RP. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. Am J Pathol. 2003;163(4):1291–1300. doi: 10.1016/S0002-9440(10)63488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Phipps RP, et al. Characterization of two major populations of lung fibroblasts: distinguishing morphology and discordant display of Thy 1 and class II MHC. Am J Respir Cell Mol Biol. 1989;1(1):65–74. doi: 10.1165/ajrcmb/1.1.65. [DOI] [PubMed] [Google Scholar]
- 123.Sorrell JM, Baber MA, Caplan AI. Site-matched papillary and reticular human dermal fibroblasts differ in their release of specific growth factors/cytokines and in their interaction with keratinocytes. J Cell Physiol. 2004;200(1):134–145. doi: 10.1002/jcp.10474. [DOI] [PubMed] [Google Scholar]
- 124.Chang HY, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99(20):12877–12882. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rinn JL, Bondre C, Gladstone HB, Brown PO, Chang HY. Anatomic demarcation by positional variation in fibroblast gene expression programs. PLoS Genet. 2006;2(7):e119. doi: 10.1371/journal.pgen.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rinn JL, et al. A dermal HOX transcriptional program regulates site-specific epidermal fate. Genes Dev. 2008;22(3):303–307. doi: 10.1101/gad.1610508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sorrell JM, Baber MA, Caplan AI. Clonal characterization of fibroblasts in the superficial layer of the adult human dermis. Cell Tissue Res. 2007;327(3):499–510. doi: 10.1007/s00441-006-0317-y. [DOI] [PubMed] [Google Scholar]
- 128.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 129.Cunha GR, Hayward SW, Wang YZ, Ricke WA. Role of the stromal microenvironment in carcinogenesis of the prostate. Int J Cancer. 2003;107(1):1–10. doi: 10.1002/ijc.11335. [DOI] [PubMed] [Google Scholar]
- 130.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 131.Hemminki A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 132.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9(8):563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Miyoshi H, Nakau M, Ishikawa TO, Seldin MF, Oshima M, Taketo MM. Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 2002;62(8):2261–2266. [PubMed] [Google Scholar]
- 134.Katajisto P, et al. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat Genet. 2008;40(4):455–459. doi: 10.1038/ng.98. [DOI] [PubMed] [Google Scholar]
- 135.Vaahtomeri K, et al. Lkb1 is required for TGFβ-mediated myofibroblast differentiation. J Cell Sci. 2008;121(pt 21):3531–3540. doi: 10.1242/jcs.032706. [DOI] [PubMed] [Google Scholar]
- 136.Hinz B, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180(4):1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bhowmick NA, et al. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303(5659):848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 138.Placencio VR, et al. Stromal transforming growth factor-β signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res. 2008;68(12):4709–4718. doi: 10.1158/0008-5472.CAN-07-6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li X, et al. Prostate tumor progression is mediated by a paracrine TGF-β/Wnt3a signaling axis. Oncogene. 2008;27(56):7118–7130. doi: 10.1038/onc.2008.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Achyut BR, et al. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-β signaling. PLoS Genet. 2013;9(2):e1003251. doi: 10.1371/journal.pgen.1003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Furusawa C, Kaneko K. A dynamical-systems view of stem cell biology. Science. 2012;338(6104):215–217. doi: 10.1126/science.1224311. [DOI] [PubMed] [Google Scholar]
- 142.Kicheva A, Cohen M, Briscoe J. Developmental pattern formation: insights from physics and biology. Science. 2012;338(6104):210–212. doi: 10.1126/science.1225182. [DOI] [PubMed] [Google Scholar]
- 143.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10(7):445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 144.Potter JD. Morphogens, morphostats, microarchitecture and malignancy. Nat Rev Cancer. 2007;7(6):464–474. doi: 10.1038/nrc2146. [DOI] [PubMed] [Google Scholar]
- 145.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Demehri S, Turkoz A, Kopan R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell. 2009;16(1):55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27(38):5115–5123. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hu B, et al. Control of hair follicle cell fate by underlying mesenchyme through a CSL-Wnt5a-FoxN1 regulatory axis. Genes Dev. 2010;24(14):1519–1532. doi: 10.1101/gad.1886910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20(19):2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- 150.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3(11):859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 151.Rittie L, Fisher GJ. UV-light-induced signal cascades and skin aging. Ageing Res Rev. 2002;1(4):705–720. doi: 10.1016/S1568-1637(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 152.Castel D, Mourikis P, Bartels SJ, Brinkman AB, Tajbakhsh S, Stunnenberg HG. Dynamic binding of RBPJ is determined by Notch signaling status. Genes Dev. 2013;27(9):1059–1071. doi: 10.1101/gad.211912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Johnson JE, Macdonald RJ. Notch-independent functions of CSL. Curr Top Dev Biol. 2011;97:55–74. doi: 10.1016/B978-0-12-385975-4.00009-7. [DOI] [PubMed] [Google Scholar]
- 154.Marsh T, Pietras K, McAllister SS. Fibroblasts as architects of cancer pathogenesis. Biochim Biophys Acta. 2013;1832(7):1070–1078. doi: 10.1016/j.bbadis.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Polanska UM, Orimo A. Carcinoma-associated fibroblasts: non-neoplastic tumour-promoting mesenchymal cells. J Cell Physiol. 2013;228(8):1651–1657. doi: 10.1002/jcp.24347. [DOI] [PubMed] [Google Scholar]
- 156.Goruppi S, Dotto GP. Mesenchymal stroma: primary determinant and therapeutic target for epithelial cancer. Trends Cell Biol. 2013;23(12):593–602. doi: 10.1016/j.tcb.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 158.Kaaks R. Nutrition, insulin, IGF-1 metabolism and cancer risk: a summary of epidemiological evidence. Novartis Found Symp. 2004;262:247–260. [PubMed] [Google Scholar]
- 159.Schmitt AR, Bordeaux JS. Solar keratoses: Photodynamic therapy, cryotherapy, 5-fluorouracil, imiquimod, diclofenac, or what? Facts and controversies. Clin Dermatol. 2013;31(6):712–717. doi: 10.1016/j.clindermatol.2013.05.007. [DOI] [PubMed] [Google Scholar]