

Abstract
The tyrosine hydroxylase (TH) gene encodes a monoxygenase that catalyzes the rate limiting step in dopamine biosynthesis. A hallmark of Parkinson’s disease (PD) is the loss of dopaminergic neurons in the substantia nigra. Consistent with the essential role of TH in dopamine homeostasis, missense mutations in both alleles of TH have been associated with severe Parkinsonism-related phenotypes including infantile Parkinsonism. It has been speculated for a long time that genetic variants in the TH gene modify adult-onset PD susceptibility but the answer has not been clear. Genetic variants (both sequence variations and structural variations) can be classified into three categories based on their relative frequency in population: common variants (polymorphisms), rare variants and mutations. Each of these factors has a different mode in influencing the genetic risk and often requires different approaches to decipher their contributions to the disease. In the past few years, the revolutionary advances in genomic technology have allowed systematic evaluations of these genetic variants in PD, such as the genome-wide association study (GWAS, to survey common variants), copy number variation analysis (to detect structural variations), and massive parallel next generation sequencing (to detect rare variants and mutations). In this review, we have summarized the latest evidence on TH genetic variants in PD, including our ongoing effort of using whole exome sequencing to search for rare variants in PD patients.
Keywords: Rare variant, copy number variations, Parkinson’s disease, genetic association, tyrosine hydroxylase polymorphism, next generation sequencing, genome wide association study
1. INTRODUCTION
Parkinson’s disease (PD) is a major neurodegenerative disorder [1]. Although the molecular mechanism underlying the development of PD remains largely unknown, it is clear that PD is under strong genetic influences. For the Mendelian forms of PD, exonic mutations in the genes PARK2, SNCA, PARK7(DJ-1), PINK1, ATP 13A2 and LRRK2 have been identified as the genetic causes. However, the Mendelian forms of PD comprise less than 10% of all PD cases. The majority of PD cases are believed to result from a complex interplay between environmental and genetic factors. Several environmental and genetic risk factors, as well as gene-environment interactions in PD have been reported by us and others [2–14]. Environmental-wise, cigarette smoking, coffee drinking, use of on-steroid anti-inflammatory drugs and pesticide exposure have been identified as environmental modifiers of PD [15–17]. Genetic-wise, genome-wide linkage and association studies have identified susceptibility loci for the common non-Mendelian form of PD. These loci include genomic regions containing SNCA, MAPT, BST1, HLA, GAK and LRRK2 [18–22]. Despite the progress that has been made, the known environmental and genetic risk factors cannot account for all the PD cases, warranting further efforts to uncover additional contributing factors missed in previous studies.
PD is characterized by the development of a combination of classic cardinal features including tremor at rest, rigidity, bradykinesia, and postural instability. The disease is hallmarked by the loss of dopaminergic neurons in the substantia nigra pars compacta [23, 24]. Indeed, a dramatic response to levodopa, the metabolic precursor of dopamine is an important diagnostic indicator for PD. The tyrosine hydroxylase (TH; EC 1.14.16.2) gene encodes a monoxygenase that catalyzes the conversion of L-tyrosine to L-dihydroxyphenylalanine (L-DOPA), which is the rate limiting step in dopamine biosynthesis. Consistent with the essential role of TH in dopamine homeostasis, missense mutations in TH have been associated with severe Parkinsonism-related phenotypes, such as Segawa’s syndrome, L-DOPA responsive infantile Parkinsonism, or L-DOPA responsive dystonia (DRD), in recessive form, i.e. both alleles are affected [25, 26]. It has been speculated for a long time that heterozygous missense mutations, i.e. one allele is affected, or genetic variants with less damaging effects than the missense mutations in the TH gene could modify susceptibility for the common form of PD. The answer, however, has not been clear. In this review, we will discuss the current understandings on the genetic variants in TH and their correlations with PD.
2. GENETIC STRUCTURE AND REGULATION THE TH GENE
Human TH has 14 exons and is located in a gene-rich region on the short arm of chromosome 11 in band 15.5 [27, 28]. TH is physically close to the genes encoding insulin (INS) and insulin-like growth factor 2 (IGF-2). This genomic region is also known as the IGF2-INS-TH gene cluster [29]. The TH transcript is subject to alternative splicing at the 5′ end producing four different isoforms of the protein (hTH1, hTH2, hTH3, hTH4) with different N-terminal ends [30]. All four isoforms of TH are expressed in healthy human brain and all are found to be markedly decreased in substantia nigra in the postmortem brain of PD patients comparing to age-matched non-affected people [31]. Among the four isoforms, hTH1 is the most conserved isoform across species and is the most abundant isoform in the brain. Given that hTH1 is the most studied isoform in the literature, all the amino acid positions referred in this review are based upon the codon in hTH1, which encodes 498 amino acids [31–33]. Through alternative splicing, other TH isoforms have inframe insertion between the first and second exon, which results in slightly larger TH proteins (502, 525, 529 amino acids for hTH2, hTH3, hTH4, respectively) [34]. The first 164 amino acids from the N-terminus constitute the regulatory domain modifying enzyme activity through phosphorylation while the rest of the protein constitutes a catalytic domain which starts from the 165th amino acid toward the C-terminus [30]. The last 20 amino acids at the C-terminus are thought to facilitate tetramerization of the enzyme [35]. The variations among the four TH isoforms are all located within the regulatory domain.
Cellular TH enzyme activity is regulated by a wide variety of mechanisms ranging from DNA methylation, transcriptional regulation, to numerous posttranslational events. In short-term regulations, the catalytic activity of TH protein is inhibited by catecholamines (CA). This CA-mediated inhibition can be relieved by phosphorylation of the serine residues (ser19, ser31 and ser 40) in the regulatory domain of TH [31, 36]. In the short-term regulation, only the balance within the pool of existing CA and TH enzyme is altered, respectively through degradation of CA or phosphorylation of TH [37, 38]. In long term regulations, prolonged or repetitive impulses in the dopaminergic cells activate TH gene transcription, increase stability of the TH mRNA and augment translation; together leading to increased levels of TH protein [38, 39]. As such, genetic variations in the regulatory elements in TH promoter, such as response element for cAMP-responsive element binding protein and Nurr1, as well as genetic variations in the 3′UTR, which is essential for mRNA stability, could contribute to the inter-individual variation on TH level.
3. GENOMIC VARIATIONS IN THE TH GENE
Genetic variants (both sequence variations and structural variations) can be classified into three categories based on their relative frequency in population: common variants (polymorphisms), rare variants and private mutations. The different frequencies of these variants in the population require different experimental strategies to detect them. In addition, each of them has different modes on influencing the genetic risk and often demands different analytic approaches to decipher its contributions to diseases [40, 41]. In the past a few years, the revolutionary advances in genomic technology has allowed systematic evaluations of these genetic variants in complex diseases, such as the GWAS to survey common variants, high throughput array-based copy number variation analysis to detect structural variations [42], and massive parallel next generation sequencing to identify rare variants and mutations. In the following sections, we will discuss studies on TH genetic variants in each of the three classes.
3.1. Disease Causing Mutations in the TH Gene and Infantile Parkinsonism
TH has been established as a recessive gene causing TH deficiency, which is associated with a broad phenotypic spectrum ranging from the milder DRD to infantile Parkinsonism or progressive infantile encephalopathy at the severe end [43]. In total, 41 disease causing point mutations in TH have been reported and recorded in the Human Genome Mutation Database [44]. Among them, 33 are missense mutations leading to an amino acid change, 3 are nonsense mutations leading to a premature stop codon, 2 are frame shifting single base deletion, and 3 are promoter mutations affecting TH transcription (Fig. 1). Of interest is that all these disease causing missense mutations are clustered in the catalytic domain and none is found in the regulatory domain of the enzyme (Fig. 1b). On the contrary, a few missense mutations have been reported in the regulatory domain but none of them has been demonstrated a high penetrance on disease phenotype (Table 1). This observation is consistent with the importance of the catalytic domain in maintaining essential functions of tyrosine dydroxylase. In addition, 30 of the 33 amino acid changes caused by these missense mutations are predicated to have damaging effects on the protein product by the Polyphen2 program [45] (Table 1).
Fig. 1.
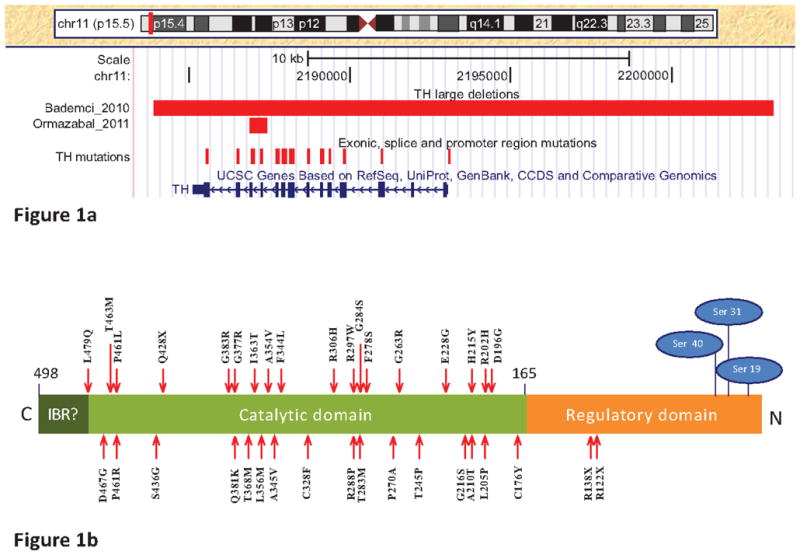
Genomic variations in the TH gene. a) Display of the genomic variations in TH using the UCSC [50] genome browser (NCBI37/hg19). The “TH mutations” track displays the genomic position of disease causing missense, nonsense, frameshift and promoter region mutations as the vertical red line. The red horizontal bar displays the start and end position of the genomic deletions in TH reported by Bademci et al., 2010 and Ormazabal et al., 2011; b). Protein structure and missense/nonsense mutations in the TH protein. Point mutations are depicted with red arrows, phosphorylation sites of the serine residues activating the enzyme in the regulatory domain are depicted in blue ovals. The nomenclature of point mutation is based on the TH isoform 1 with 498 amino acids (NM_199292.2). Fig. (1b) is modified from Hufton et al. 1995[30]. (IBR=Intersubunit binding region)
Table 1.
Missense and Nonsense Variations in TH
| Variant | Description | Predicated Function# | Population Frequency* |
|---|---|---|---|
| A6T | Rare variant | Benign | 0.46% |
| V81M | Common variant | Benign | 34% |
| R122X | Disease causing mutation | Nonsense | N/A |
| V136M | Rare variant | Benign | 0.02% |
| R138X | Disease causing mutation | Nonsense | N/A |
| C176Y | Disease causing mutation | Probably damaging | N/A |
| D196G | Disease causing mutation | Probably damaging | N/A |
| R202H | Disease causing mutation | Probably damaging | 0.01% |
| L205P | Disease causing mutation | Benign | N/A |
| A210T | Disease causing mutation | Probably damaging | N/A |
| H215Y | Disease causing mutation | Benign | N/A |
| G216S | Disease causing mutation | Probably damaging | N/A |
| E228G | Disease causing mutation | Probably damaging | N/A |
| T245P | Disease causing mutation | Possibly damaging | N/A |
| G263R | Disease causing mutation | Probably damaging | N/A |
| P270A | Disease causing mutation | Probably damaging | N/A |
| F278S | Disease causing mutation | Probably damaging | N/A |
| T283M | Disease causing mutation | Probably damaging | N/A |
| G284S | Disease causing mutation | Probably damaging | N/A |
| R288P | Disease causing mutation | Probably damaging | N/A |
| R297W | Disease causing mutation | Probably damaging | N/A |
| R306H | Disease causing mutation | Probably damaging | N/A |
| C328F | Disease causing mutation | Probably damaging | N/A |
| F344L | Disease causing mutation | Probably damaging | N/A |
| A345V | Disease causing mutation | Probably damaging | N/A |
| A354V | Disease causing mutation | Possibly damaging | N/A |
| L356M | Disease causing mutation | Probably damaging | N/A |
| I363T | Disease causing mutation | Probably damaging | N/A |
| T368M | Disease causing mutation | Probably damaging | N/A |
| G377R | Disease causing mutation | Probably damaging | N/A |
| Q381K | Disease causing mutation | Possibly damaging | N/A |
| G383R | Disease causing mutation | Probably damaging | N/A |
| Q428X | Disease causing mutation | Nonsense | N/A |
| S436G | Disease causing mutation | Possibly damaging | N/A |
| P461R | Disease causing mutation | Probably damaging | N/A |
| P461L | Disease causing mutation | Probably damaging | N/A |
| T463M | Disease causing mutation | Probably damaging | N/A |
| D467G | Disease causing mutation | Benign | N/A |
| L479Q | Disease causing mutation | Probably damaging | N/A |
The first mutation in TH was found in the study of the Segawa syndrome characterized by progressive dystonia with diurnal fluctuation [46]. Through linkage analysis of microsatellite markers and partial sequencing of TH exons (exon 1–8 and 11–12), the Q381K mutation was identified in one Caucasian family with two affected and one unaffected children. Both affected children are homozygotes while the unaffected sibling and both parents are heterozygotes for the Q381K. Subsequently, seven missense mutations were reported in four families with infantile Parkinsonism. The group who identified the Q381K mutation first reported the L205P mutation in a Greek family with a female whose symptoms of Parkinsonism manifested at 3 months of age [47]. DNA sequencing of TH exons revealed a homozygous L205P mutation in the aforementioned female. Both parents and the brother of the affected female carry the heterozygous L205P mutation and were non-symptomatic at the time of examination. In addition to severe Parkinsonian symptoms in early infancy, the female also had symptoms of a dysfunction of specific sympathetic neurons in the peripheral nervous system (ptosis). Both dopaminergic and noradrenergic neurons also seemed to be affected in this patient. Unlike the Segawa syndrome patient, there was no diurnal variability in symptoms in the patient with the homozygous L205P mutation. Although the L205P mutation is predicted to be a benign amino acid substitution by Polyphen2, in vitro expression of the mutant TH with L205P demonstrated that the mutant protein has very low homospecific activity as compared to wild type TH [47]. Later, Swaans et al. reported four TH missense mutations in two families with infantile Parkinsonism [48]. In family A, two affected brothers had displayed motor symptoms at the age of 2 and 5 years old, and were not able to walk by the age of 5 and 9 years old. Sequencing TH exons revealed that the unaffected mother and the two affected brothers have heterozygous R306H mutation whereas the unaffected father and the two affected brothers have heterozygous T463M mutations, suggesting that the two affected children are compound heterozygote of the TH missense mutations. In family B, a patient had impaired motor development which began at age of 2. The patient carries heterozygous T245P and T283M mutations in the TH gene. It is not clear whether the two mutations are in cis- or trans- state as none of the parents or siblings were available for DNA testing. Given that TH is a recessive gene for TH deficiency, it is likely that the patient is a compound heterozygote of the two mutations [48]. The fourth family was reported by Diepold and colleagues. A female patient of 14 months of age started refusing to walk, development of intention tremor, fisting of hands, and the inability to hold her head or sit. after experiencing a brief viral infection [49]. The symptoms were somewhat alleviated by 19 months of age, but were later exacerbated after another viral infection. By the age of 2 years and 4 months, she suffered severe bradykinesia, hypertonic limbs with cogwheel rigidity, and dystonic posturing of her hands. Sequencing analysis found that the patient carries heterozygous H215Y and D467G mutations, which were transmitted from her mother and father, respectively [49]. In contrast to other mutations identified in other patients with infantile Parkinsonism, both H215Y and D467G are predicted coding tolerable amino acid change in the protein, i.e. having less deleterious effect on the enzyme activity. It is worth noting that the symptoms in this case were induced and worsen after viral infections suggesting a gene-environmental interaction.
In all above patients with TH mutations, administering a low to moderate dose of levodopa, sometimes in combination with a decarboxylase inhibitor eliminated the symptoms including Segawa syndrome and infantile Parkinsonism promptly. Interestingly, the dosage of levodopa treatment seemed to be correlated with the severity of mutations. For example, the Q381K mutation and the L205P mutation led to a decrease of TH activity to 15% and 1.5% of the wild type TH, respectively, in an in vitro model [46, 47]. The dosage of levodopa treatment administrated to relieve symptoms in patients carrying the Q381K mutation and the L205P mutation were 2mg/kg body weight one time and five times daily, respectively [46, 47]. Therefore, accurate genetic diagnostic will facilitate the management of these devastating symptoms effectively.
3.2. Common Variants in the TH Gene and Parkinson’s Disease
It is clear that individuals carrying deleterious TH mutations in both alleles can develop Parkinsonism symptoms at very young age. These mutations, however, are extremely rare; they have only been seen in a few families. Thus, several attempts have been made to examine if common variants with less deleterious effects modulate the susceptibility to adult onset PD. There are mainly two approaches to study association between common genetic variant and disease: GWAS and candidate gene study.
GWAS
Propelled by the international HapMap project and ultra high-throughput genotyping technology, GWAS has become a powerful approach to dissect the genetic basis of complex diseases. This approach uses a dense array of up to 1,000,000 single nucleotide polymorphisms (SNPs) that are printed or synthesized on a microarray (chip) to genotype hundreds and thousands of individuals. Estimates suggest that a well-designed array with 600,000 SNPs will capture ~93% of the common genetic variations in the human genome at an r2 of 0.7 within a Caucasian population. During the past a few years, GWAS has identified hundreds of “loci of interest” for many common complex diseases, including obesity, type I and type II diabetes, inflammatory bowel disease, prostate cancer, breast cancer, colorectal cancer, coronary artery disease, Alzheimer’s disease and PD [51–64]. Five independent GWAS on the common form of PD have been reported; four were conducted on non-Hispanic Caucasians and one was conducted on Japanese individuals. After correcting for multiple testing, genome-wide significant results were found in the genomic regions near SNCA, MAPT, BST1, HLA, GAK and LRRK2 [18–22, 65].
Within the TH gene, three SNPs (rs2072056, rs6356, and rs10743152) are either included in the GWAS SNP array or can be imputed using the HapMap reference panel. None of the GWAS, including the one conducted by us at the Hussman Institute for Human Genomics (HIHG) at the University of Miami, reported significant association with PD susceptibility at these SNPs. The negative finding, however, could be due to a lack of statistical power to detect a mild genetic effect in any one particular GWAS. To increase the power in the association analysis, we performed a joint analysis of our GWAS data set with two additional PD GWAS that were deposited in the database of Genotypes and Phenotypes (dbGAP), which is publically accessible; the National Institute of Neurological Disorders and Stroke (NINDS) and the joint dataset from the Progeni/GenePD studies that was genotyped at the Center for Inherited Disease Research (CIDR) [18, 65]. The final data set for the joint analysis include a combined sample size of 1752 cases and 1745 controls. Table 2 displays the allele frequency of the three TH SNPs in each of the three GWAS data sets and the p-value for association with PD risk in the combined data set. Even with improved power in the joint analysis, no evidence for association was found in any GWAS SNPs in TH.
Table 2.
The TH Gene and PD Susceptibility in GWAS and Joint Analysis
| GWAS Data Set | Minor Allele Frequency | P-Value in Combined Dataset | Major Allele | Minor Allele | |
|---|---|---|---|---|---|
| Case | Control | ||||
| rs2070762 | 0.87 | A | G | ||
| HIHG | 0.483 | 0.504 | |||
| CIDR | 0.494 | 0.497 | |||
| NINDS | 0.521 | 0.448 | |||
| rs6356 | 0.84 | T | C | ||
| HIHG | 0.383 | 0.374 | |||
| CIDR | 0.360 | 0.353 | |||
| NINDS | 0.356 | 0.375 | |||
| rs10743152 | 0.1 | T | C | ||
| HIHG | 0.401 | 0.384 | |||
| CIDR | 0.381 | 0.376 | |||
| NINDS | 0.386 | 0.314 | |||
Minor allele frequency of each TH SNP that has been examined in three GWAS, i.e. HIHG, CEDR and NINDS, were listed in cases and controls separately. The P value is generated using an additive model in the combined GWAS dataset of HIHG, CIDR, and NINDS. The Major and minor allele for each SNP is also listed.
Candidate gene study
Contrary to the “null-hypothesis” strategy in a GWAS, candidate gene study selects one or a few biological plausible genes to examine based on a priori knowledge. Since a candidate gene study involves much less genotyping compared to a GWAS, it is more affordable to researchers with limited resources. In addition, the threshold to claim statistical significance in a candidate gene study is much lower than that used in a GWAS due to less statistical tests performed. Given the pivotal role of TH in dopamine biosynthesis, several candidate gene studies have examined the relationship between common genetic variations in TH and adult onset PD susceptibility (Table 3).
Table 3.
The TH Gene and PD Susceptibility in Candidate Gene Study
| Study | Polymorphism | Description of Data Set | Results | ||
|---|---|---|---|---|---|
| Case | Control | Population | |||
| Plante-Bordeneuve, 1994 | HUMTH01 | 44 | 50 | England | Negative |
| Kunugi, 1998 | rs6356 | 99 | 161 | Japan | Negative |
| Mizuta, 2006 | rs2070762 | 190 | 190 | Japan | Negative |
| Sutherland, 2008 | HUMTH01, rs680 (IGF2), rs689 (INS) | 215 | 215 | Australia | Positive (mainly driven by rs680) |
| Punia, 2010 | 10 tagSNPs including rs6356 | 339 | 334 | North India | Negative |
Summary of candidate gene studies on the association between TH SNPs and PD susceptibility. The table lists the polymorphism(s) examined, number of cases and controls, countries where the study was carried out in each study. No study found a significant association between TH polymorphism and PD susceptibility.
The first PD genetic study on TH was conducted in England. One polymorphism, a tetra-nucleotide repeat marker HUMTH01 in the first intron of TH was examined in 44 individuals with sporadic PD, 48 individuals with familial PD and 89 of their unaffected relatives, and 50 population control subjects. No evidence for association or linkage at HUMTH01 was found in this small sample set [66]. To expand the study on TH and PD, Kunugi et al. examined the HUMTH01, the L205P mutation (causative TH variant for infantile PD) and rs6356 (a common missense mutation examined in GWAS) in 90 Japanese PD cases and 161 population controls [67]. The L205P mutation was not present in this sample set. The HUMTH01 and rs6356 were found to be highly correlated with each other (in high degree of linkage disequilibrium). Again, this study failed to show an association between TH and PD susceptibility. Later, Mizuta et al. conducted a large-scale candidate gene study in PD involving 1800 Japanese subjects (sample set contained approximately equal number of sporadic PD cases and controls) [68]. In total, 268 SNPs in 121 candidate genes, with one intronic SNP (rs2070762) in TH, were surveyed. The strongest association with PD was found in SNCA (P value =5.0 × 10−10), which is consistent with the GWAS findings. The TH SNP, however, is not significantly associated with PD in this data set. Finally, Sutherland and colleagues took a haplotyping approach to determine whether common genetic variation in the IGF2-INS-TH cluster influences the risk of idiopathic PD [69]. The subjects included in this study are Caucasian recruited from Brisbane, Australia. SNPs rs680 in IGF2, rs689in INS, and the tetra nucleotide repeat marker HUMTH01 in TH, were genotyped in 215 cases and 215 age- and gender-matched controls. The three polymorphisms together define 20 haplotypes. Among them, 8 common haplotypes account for >90% of the genetic variation in the population. The IGF2-INS-TH*6 haplotype was significantly less frequent in the PD cases (P value = 0.001). Additional analysis of the each individual polymorphisms, however, revealed that the A allele of rs680 in IGF2 is responsible for the protective effect of the haplotype, dwindling the importance of the TH variant on PD susceptibility. Recently, a study conducted in a North India PD cohort evaluated six genes that are involved in dopamine synthesis and metabolism, including TH. The study design includes 339 sporadic PD patients and 334 age and gender-matched controls. Within TH, only the missense SNP rs6356 was tested. A highly significant haplotype was found within the dopamine β-hydroxylase (DBH), but no evidence for association was found at rs6356 [70].
Within TH, rs6356 is the most examined SNP in the association studies. Nucleotide variation at this SNP leads to amino acid change between Met and Val at codon 81. As a missense SNP, rs6356 has been postulated to be a variant with functional consequence and therefore more likely to confer genetic risk. Closer examination at rs6356 reveals that the SNP is located in the regulatory domain of TH, a region that is not as functionally critical as the catalytic domain where all disease causing mutations cluster. In addition, the amino acid at codon 81 is not well conserved and the amino acid substitution is predicted to be benign according to PolyPhen2 (Table 1). The well-tolerated change at rs6356 is consistent with its high frequency in the population (Table 1). Given the little function impact at rs6356, may be it is not so surprising that no study found evidence for association at rs6356.
In summary, neither GWAS nor the candidate gene study approach provides convincing evidence for association between common TH polymorphisms and adult-onset PD. Both GWAS and candidate gene study, however, have limitations. Both of them are based on a tag SNP approach using representative markers across the genome or candidate gene to capture the majority of common genetic variations in the population. Such approaches are cost-efficient for screening large numbers of genes and individuals but could miss the actual functional variants underlying the phenotypic variations. In fact, although GWAS has led to novel findings in a number of complex diseases, the associated SNPs only account for a small fraction of the phenotypic variations, which have been referred to as the “missing heritability”. One of the theories for this phenomenon is that rare variants, in contrast to common variants, are not well-represented in the GWAS and they could account for, at least partially, the “missing heritability” in GWAS of complex diseases such as PD [40, 71]. Given the limitations embedded in the tagSNP approach, other strategies need to be taken to fully characterize the contribution of TH variants to the genetic susceptibility of PD.
3.3. Rare Variants in the TH Gene and Parkinson’s Disease
Rare variants have a population frequency less than 1–2%. The low frequency of rare variants makes them hard to be detected and catalogued. In addition, despite that rare variants are believed to have bigger effect size than the common variants, single-locus association test does not provide sufficient statistical power in analyzing rare variants due to its low allele frequency. The recent advance in next-generation sequencing technologies enables us to move beyond the “tagSNP” approaches implemented in GWAS and most candidate gene studies to exhaustively catalogue sequence variants and identify the causal variants leading to disease phenotypes. To accelerate genetic understandings on heart, lung and blood disorders, the National Heart, Lung and Blood Institute has funded a large Exome Sequencing Project aiming to extensively investigate exonic variants that could have been overlooked by GWAS. An internet based Exome Variant Server (EVS) has been developed to release whole exome data derived from 5379 individuals included in ESP cohorts [72]. Accompanied with the rapid advances in sequencing technology, new statistical methods have also been developed to analyze rare variants in complex diseases. Some of them simply examine if rare variants identified in cases are also seen in controls. Recently, the importance of evaluating cumulative effect of rare variants within a gene or a biological pathway has been recognized and several statistical programs have been developed to evaluate the multiple rare variants in aggregate.
In an effort to systematically investigate the contribution of exonic rare variants in PD, we have initiated a whole exome sequencing project in a cohort of well-characterized PD patients and unrelated controls. We have used the Agilent SureSelect exome kit to enrich genomic sequences of coding exons as listed in the Consensus Coding Sequences collection, exon-intron boundaries, and nearly all microRNA genes. The Agilent kit covers ~50Mb of genomic sequence comprising ~200,000 exons and exon-intron junctures. After the initial screening of 164 PD cases and 50 controls, we identified a heterozygous missense variation in TH in one case but not in any controls. The variation leads to an amino acid change from valine to metionine at codon 136 (V136M). The PD patient has an age-at-onset (AAO) of 54 and is from a multiplex family with 5 affected within one generation. DNA samples are available for testing in 5 affected and 2 unaffected siblings/cousins. Sanger sequencing of the 7 family members confirmed the V136M variation in the index patient screened in the whole exome sequencing but not in any other family members. We noted that all the five affected and one unaffected family members also carry one allele of the LRRK2 L1795F missense variation. Interestingly, the patient carrying the V136M has the youngest age-at-onset. In another candidate gene study on rare variants, Hertz et al. examined exonic variations in PARK2, TH, and GTP cyclohydrolase I (GCH1) in 87 Danish patients with earlyonsetPD (AAO <40, or AAO<50 if first degree relative is affected too) [26]. Within TH, only a heterozygous missense variant (A6T) was found in one patient. Interestingly again, the patient also has a heterozygous duplication of exon 11 in PARK2 [26].
With the limited data, it is not clear if A6T and V136M can significantly increase the risk of developing PD. Although both of them cause amino acid change in TH, there are several notable differences between the two rare variants and the previously discussed disease causing mutations in TH. First, both A6T and V136M are located within the regulatory domain where variations are believed to have a milder effect on the enzyme activity while all the disease causing mutations are located within the catalytic domain. Secondly, both rare variants are predicted to cause benign amino acid substitution while most of the diseases causing mutations are predicated to cause damaging amino acid substitution or have been demonstrated to have functional consequences on enzyme activity in vitro (Table 1). Thirdly, none of the disease causing missense mutations, except for the R202H, are found in more than 10,000 chromosomes recorded in the EVS database, with tR202H being found only once. In contrast, A6T and V136M have slight higher allele frequency (0.46% and 0.02%, respectively in the EVS database, Table1). Therefore, it is likely that the two rare variants themselves are not sufficient to significantly increase the risk for developing PD. It remains unclear, however, if they could serve as a modifier for genetic risk conferred by heterozygous variations in other genes, such as LRRK2 and PARK2, or have cumulative effect with other genetic variants on PD susceptibility. Future large scale, systematic studies are needed to fully understand the question.
3.4. Copy Number Variations in the TH Gene and Parkinson’s Disease
In recent years, it has been recognized that DNA copy number variations (CNVs) represent a major source of genome variations, accounting for more nucleotide variation per human genome than SNPs [73–75]. CNVs have been associated with several complex phenotypes, including autism [76, 77] osteoporosis [78], schizophrenia [79], psoriasis [80], autoimmune diseases [81, 82], bipolar disorder [83] and body mass index [84]. In PD, whole gene copy number gain in SNCA and duplication/deletion of one or multiple exons in PARK2 have been identified as causal genetic variations [85]. CNVs, however, in general are underrepresented in genetic studies because it has been cost and labor prohibitive to characterize CNV in large number of samples. CNV genotyping is technically more challenging than SNP genotyping, partially due to the quantitative rather than the qualitative nature of CNV assays. In the past, CNV is genotyped by PCR-based methods, such as semiquantitative PCR, quantitative PCR, and multiple ligation-dependent probe amplification (MLPA). For large recurrent deletions, direct PCR over the fixed breakpoints could be developed for genotyping. Many CNVs, however, are non-recurrent (as seen in TH and PARK2). They vary in size and breakpoint location even when they involve the same gene, thus requiring multiple probes (reactions) to comprehensively evaluate CNVs in one particular gene. As a result, identifying and characterizing previously unknown CNVs are even more challenging. Lately, SNP-array used in the GWAS and high resolution comparative genomic hybridization -array offer a high-throughput approach for genome-wide interrogation of CNVs.
A commercially available MLPA kit (P099-B2, MRC-Holland, Amsterdam, Netherlands) has been designed to survey CNVs in genes related to Dopa-responsive dystonia, including TH [86]. Limited by the MLPA technology, probes can only be designed to target exon 1, 3, 4, 8, 12 and 14 of TH, missing the other eight exons of TH. Using this kit, Liu et al. has screened 16 DRD patients (10 spordic patients and two pedigrees including six patients)87. No CNV was found in the TH gene in this small study [87]. Using the same MLPA kit, Ormazabal et al. recently reported a DRD patient with a deletion in the TH gene that encompasses several exons [88]. A heterozygous exon 12 deletion was first identified by MLPA. Additional analysis with long range PCR over breakpoints confirmed the deletion encompassing a segment of 716 bp (c.1197+25_1391del) including exon 12 and part of exon 13. The patient also has a heterozygous mutation (c.1 - 70G>A) at promoter region in another allele. The promoter mutation is believed to be the underlying cause for DRD in other patients [89].
In our GWAS on PD, we have used the Illumina BeadChips (Illumina, Inc) for a genome-wide SNP genotyping of 635 PD cases and 642 unrelated controls. The probe signal intensity and b-allele frequency data generated from the SNP-array were used for a genome-wide CNV analysis. The SNP-array data indicated a 34 kilobase deletion in the genomic region harboring TH in one PD patient but not in any controls. The deletion was defined by seven SNPs, starting at rs2070762 and ending at rs3922756. Using multiple qPCR assays targeting the deleted region delimited by the GWAS, we confirmed the deletion and narrowed the deletion to 17 kb over the entire TH gene. Re-sequencing all the exons of the remaining copy of the TH gene in the patient revealed no exonic mutation. The patient had an age-at-onset of 54 years, with no evidence for dystonia, and was responsive to L-DOPA. It is intuitive to propose that the hemizygous deletion of TH leads to a lower level of TH activity and thus increases PD risk [42]. Neither parent has DNA available for testing but both were reported to have been without any clinical symptoms of PD, well into their 70’s and 80’s. We cannot exclude the possibility that the entire TH deletion is a de nova mutation.
Our analysis of GWAS data suggests that the entire TH deletion is a rare event. It is possible, however, that smaller deletions in TH exist but were missed in the GWAS study due to the limited resolution of SNP-array for CNV. The probe density in a SNP-Array is a few kilobases on average and multiple probes are required to define a CNV. Therefore, SNP-array is useful in identifying large CNVs such as the 17 Kb entire TH deletion but will miss smaller deletion such as the 766 bp TH deletion involving only two exons. High resolution study is needed to comprehensively evaluate the contribution of CNVs in TH to PD.
4. INTERACTION BETWEEN TH AND OTHER PARKINSON’S DISEASE RELATED GENES
As discussed earlier, multiple genes have been identified as genetic risk factors for PD. The molecular mechanisms underlying the association between these genes and PD are not completely understood. Interestingly, the protein products of some of these genes interact with the TH protein and modulate the enzyme activity, thus providing a plausible mechanism for these risk genes to be involved in PD pathogenesis. These biological interactions also raise the question whether variants in TH could modify PD risk via increasing the genetic burden through an accumulative or synergistic effect with other risk genes.
In SNCA, gain-of-function misssense mutations (A30P, E46K and A53T) and copy number gain of the entire gene have been identified as the cause for early-onset familial PD [90–93]. Despite that aggregates of α-synuclein is a major component of Lewy bodies (a neuropathological hallmark of PD); the function of α-synuclein is unclear. Immunoprecipitation and immunoelectron microscopy have demonstrated a direct interaction between α-synuclein and tyrosine hydroxylase in brain homogenates and MN9D dopaminergic cells. Importantly, over-expressing α-synuclein in MN9D cells reduced the tyrosine hydroxylase enzyme activity which was accompanied with diminished dopamine synthesis and lower dopamine levels [94, 95]. In addition, it was shown that α-synuclein suppresses tyrosine hydroxylase enzyme activity without affecting the TH protein level, probably via inhibiting TH phosphorylation. Thus, SNCA could contribute to PD development by serving as a negative regulator of dopamine biosynthesis, which is consistent with the gain-of-function mechanism of identified SNCA mutations.
In DJ-1, multiple loss-of-function missense mutations have been identified as being responsible for early-onset familial PD [96]. DJ-1 plays a role in cellular protection against oxidative stress and regulates mitochondrial functions [97]. Recently, it has been reported that DJ-1 binds directly to the TH protein and activates the enzyme [98]. Mutant DJ-1 with missense mutations identified in PD patients, such as M26I, E64D and L166P, binds to TH protein but does not increase enzyme activity. Rather, the mutant DJ-1 blocks the TH activity induced by wild type DJ-1. Furthermore, several studies have shown that DJ-1 is involved in the transcriptional regulation of TH gene through different mechanisms in different species. In the mouse cells, DJ-1 increases TH gene expression by activating the transcription factor Nurr1 [99], while it has been shown that TH expression in human neuronal progenitor cells does not require Nurr1 [100]. Interestingly, in human cells, it has been found that DJ-1 serves as a positive regulator of TH by suppressing a transcriptional co-repressor polypyrimidine tract-binding protein-associated splicing factor [101, 102]. Nonetheless, DJ-1 could contribute to PD risk by acting as a positive regulator of dopamine biosynthesis, which is consistent with the loss-of-function mechanism of DJ-1 mutations.
5. SUMMARY
Encoding the enzyme catalyzing the rate limiting step of dopamine synthesis, TH is a theoretical susceptibility gene for PD pathogenesis. Indeed, recessive missense mutations in TH have been identified as a genetic cause for infantile Parkinsonism. The implication of these mutations in the adult-onset PD, however, is trivial due to the extreme rare presence of them. These disease-causing mutations have only been seen in a few families with infantile Parkinsonism but not in the population. On the other hand, several studies have examined association between common variants (minor allele frequency is greater than 5% in the population) in TH and adult onset PD. No evidence for association was found supporting a role of common TH variants in PD susceptibility. In comparison to disease-causing mutations, these common variants do not seem to have significant functional consequences, including a missense SNP rs6356. Recently, there are several cases in which rare genetic variants (minor allele frequency lesser than 1% in the population), such as a whole gene deletion and missense substitutions, are speculated to increase the risk of adult onset PD.
To fully understand the genetic risk conferred by TH, systematic study on rare variants, including point sequence variations and CNVs, are desired. With the rapid advances in genomic technology, such as next generation sequencing technology for sequence variations and Nanostring® for CNV, comprehensive genomic study can be performed to thoroughly evaluate rare variants. In particular, large scale whole exome sequencing and whole genome sequencing will exhaustively survey all the functional variants in TH as well as other genes through the human genome. This will not only allow for evaluation of the collective effects of multiple rare variants in TH, but also the epistasis between TH and other genes, especially the ones that interact with TH.
Acknowledgments
This work was supported by National Institutes of Health grant NS071674 (JMV) and Florida Biomedical Research Program grant NIR 1KN-14 (LW).
ABBREVIATIONS
- AAO
Age-at-onset
- CA
Catecholamines
- CGH
Comparative genomic hybridization
- CIDR
Center for Inherited Disease Research
- CNVs
Copy number variations
- dbGAP
Database of Genotypes and Phenotypes
- DRD
L-DOPA responsive dystonia
- EVS
Exome Variant Server
- GWAS
Genome-wide association study
- IGF-2
Insulin-like growth factor 2
- IND
Insulin
- L-DOPA
L-dihydroxyphenylalanine
- MLPA
Multiple ligation-dependent probe amplification ()
- NINDS
National Institute of Neurological Disorders and Stroke
- PD
Parkinson’s disease
- SNPs
Single nucleotide polymorphisms
- TH
Tyrosine Hydroxylase
Footnotes
CONFLICTS OF INTEREST
Declared none.
References
- 1.Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann NY Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 2.Hancock DB, Martin ER, Fujiwara K, Stacy MA, Scott BL, Stajich JM, Jewett R, Li YJ, Hauser MA, Vance JM, Scott WK. NOS2A and the modulating effect of cigarette smoking in Parkinson’s disease. Ann Neurol. 2006;60:366–373. doi: 10.1002/ana.20915. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Oliveira SA, Xu P, Martin ER, Stenger JE, Scherzer CA, Hulette C, Scott WK, Small GW, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Jankovic J, Goetz CG, Mastaglia F, Middleton LT, Roses AD, Saunders AM, Welsh-Bohmer KA, Schmechel DE, Gullans SR, Haines JL, Gilbert JR, Vance JM, Pericak-Vance MA. Glutathione S-transferase omega-1 modifies age-at-onset of Alzheimer disease and Parkinson disease. Hum Mol Genet. 2003;12:3259–3267. doi: 10.1093/hmg/ddg357. [DOI] [PubMed] [Google Scholar]
- 4.Oliveira S, Li Y, XJQ, Zuchner S, Noureddine MA, Hauser MA, TS, SE, Pericak-Vance MA, JMV Novel genes associated with risk and age-at-onset in parkinson disease identified through iterative association mapping. American Society of Human Genetics; Toronto, Canada. October 26–30: 2004; 2005. [Google Scholar]
- 5.Oliveira SA, Li YJ, Noureddine MA, Zuchner S, Qin X, Pericak-Vance MA, Vance JM. Identification of Risk and Age-at-Onset Genes on Chromosome 1p in Parkinson Disease. Am J Hum Genet. 2005;77:252–264. doi: 10.1086/432588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizuta I, Tsunoda T, Satake W, Nakabayashi Y, Watanabe M, Takeda A, Hasegawa K, Nakashima K, Yamamoto M, Hattori N, Murata M, Toda T. Calbindin 1, fibroblast growth factor 20, and alpha-synuclein in sporadic Parkinson’s disease. Hum Genet. 2008;124:89–94. doi: 10.1007/s00439-008-0525-5. [DOI] [PubMed] [Google Scholar]
- 7.van der Walt JM, Noureddine MA, Kittappa R, Hauser MA, Scott WK, McKay R, Zhang F, Stajich JM, Fujiwara K, Scott BL, Pericak-Vance MA, Vance JM, Martin ER. Fibroblast growth factor 20 polymorphisms and haplotypes strongly influence risk of Parkinson disease. Am J Hum Genet. 2004;74:1121–1127. doi: 10.1086/421052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Walt JMMN, PT, JB, JS, BS, MS, WKS, EM, YJL, MH, Pericak-Vance MAJMV. Metabolic stress induces cell death in Parkinson disease cybrids and fibroblasts. American Society of Human Genetics 2005 Annual Meeting. 2005 [Google Scholar]
- 9.McCulloch CC, Kay DM, Factor SA, Samii A, Nutt JG, Higgins DS, Griffith A, Roberts JW, Leis BC, Montimurro JS, Zabetian CP, Payami H. Exploring gene-environment interactions in Parkinson’s disease. Hum Genet. 2008;123:257–265. doi: 10.1007/s00439-008-0466-z. [DOI] [PubMed] [Google Scholar]
- 10.Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Kruger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, Lynch T, Mellick GD, Papapetropoulos S, Parsian A, Quattrone A, Riess O, Tan EK, Van Broeckhoven C. Genetic Epidemiology of Parkinson’s Disease (GEO-PD) Consortium Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- 11.Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum Mol Genet. 2006;15:1151–1158. doi: 10.1093/hmg/ddl030. [DOI] [PubMed] [Google Scholar]
- 12.Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. alpha-Synuclein and Parkinson disease susceptibility. Neurology. 2007;69:1745–1750. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- 13.Hancock DB, Martin ER, Vance JM, Scott WK. Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics. 2008;9:249–262. doi: 10.1007/s10048-008-0137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hancock DB, Martin ER, Fujiwara K, Stacy MA, Scott BL, Stajich JM, Jewett R, Li YJ, Hauser MA, Vance JM, Scott WK. NOS2A and the modulating effect of cigarette smoking in Parkinson’s disease. Ann Neurol. 2006;60:366–373. doi: 10.1002/ana.20915. [DOI] [PubMed] [Google Scholar]
- 15.Elbaz A, Clavel J, Rathouz PJ, Moisan F, Galanaud JP, Delemotte B, Alperovitch A, Tzourio C. Professional exposure to pesticides and Parkinson disease. Ann Neurol. 2009;66:494–504. doi: 10.1002/ana.21717. [DOI] [PubMed] [Google Scholar]
- 16.Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, Ascherio A. Nonsteroidal antiinflammatory drug use and the risk of Parkinson’s disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- 17.Hernan MA, Takkouche B, Caamano-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol. 2002;52:276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 18.Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodriguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- 19.Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet. 2010;74:97–109. doi: 10.1111/j.1469-1809.2009.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 22.Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010 doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.NAGATSU T, LEVITT M, UDENFRIEND S. Tyrosine Hydroxylase. the Initial Step in Norepinephrine Biosynthesis. J Biol Chem. 1964;239:2910–2917. [PubMed] [Google Scholar]
- 24.Haavik J, Toska K. Tyrosine hydroxylase and Parkinson’s disease. Mol Neurobiol. 1998;16:285–309. doi: 10.1007/BF02741387. [DOI] [PubMed] [Google Scholar]
- 25.Furukawa Y, Graf WD, Wong H, Shimadzu M, Kish SJ. Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase (TH) gene mutations. Neurology. 2001;56:260–263. doi: 10.1212/wnl.56.2.260. [DOI] [PubMed] [Google Scholar]
- 26.Hertz JM, Ostergaard K, Juncker I, Pedersen S, Romstad A, Moller LB, Guttler F, Dupont E. Low frequency of Parkin, Tyrosine Hydroxylase, and GTP Cyclohydrolase I gene mutations in a Danish population of early-onset Parkinson’s Disease. Eur J Neurol. 2006;13:385–390. doi: 10.1111/j.1468-1331.2006.01249.x. [DOI] [PubMed] [Google Scholar]
- 27.Craig SP, Buckle VJ, Lamouroux A, Mallet J, Craig I. Localization of the human tyrosine hydroxylase gene to 11p15: gene duplication and evolution of metabolic pathways. Cytogenet Cell Genet. 1986;42:29–32. doi: 10.1159/000132246. [DOI] [PubMed] [Google Scholar]
- 28.Saccone S, Federico C, Solovei I, Croquette MF, Della Valle G, Bernardi G. Identification of the gene-richest bands in human prometaphase chromosomes. Chromosome Res. 1999;7:379–386. doi: 10.1023/a:1009220131225. [DOI] [PubMed] [Google Scholar]
- 29.O’Malley KL, Rotwein P. Human tyrosine hydroxylase and insulin genes are contiguous on chromosome 11. Nucleic Acids Res. 1988;16:4437–4446. [PMC free article] [PubMed] [Google Scholar]
- 30.Hufton SE, Jennings IG, Cotton RG. Structure and function of the aromatic amino acid hydroxylases. Biochem J. 1995;311(Pt 2):353–366. doi: 10.1042/bj3110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakashima A, Hayashi N, Kaneko YS, Mori K, Sabban EL, Nagatsu T, Ota A. Role of N-terminus of tyrosine hydroxylase in the biosynthesis of catecholamines. J Neural Transm. 2009;116:1355–1362. doi: 10.1007/s00702-009-0227-8. [DOI] [PubMed] [Google Scholar]
- 32.Lewis DA, Melchitzky DS, Haycock JW. Four isoforms of tyrosine hydroxylase are expressed in human brain. Neuroscience. 1993;54:477–492. doi: 10.1016/0306-4522(93)90267-j. [DOI] [PubMed] [Google Scholar]
- 33.Grima B, Lamouroux A, Boni C, Julien JF, Javoy-Agid F, Mallet J. A single human gene encoding multiple tyrosine hydroxylases with different predicted functional characteristics. Nature. 1987;326:707–711. doi: 10.1038/326707a0. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi K, Kaneda N, Ichinose H, Kishi F, Nakazawa A, Kurosawa Y, Fujita K, Nagatsu T. Structure of the human tyrosine hydroxylase gene: alternative splicing from a single gene accounts for generation of four mRNA types. J Biochem. 1988;103:907–912. doi: 10.1093/oxfordjournals.jbchem.a122386. [DOI] [PubMed] [Google Scholar]
- 35.Haavik J, Toska K. Tyrosine hydroxylase and Parkinson’s disease. Mol Neurobiol. 1998;16:285–309. doi: 10.1007/BF02741387. [DOI] [PubMed] [Google Scholar]
- 36.Haycock JW. Species differences in the expression of multiple tyrosine hydroxylase protein isoforms. J Neurochem. 2002;81:947–953. doi: 10.1046/j.1471-4159.2002.00881.x. [DOI] [PubMed] [Google Scholar]
- 37.Xu L, Chen X, Sun B, Sterling C, Tank AW. Evidence for regulation of tyrosine hydroxylase mRNA translation by stress in rat adrenal medulla. Brain Res. 2007;1158:1–10. doi: 10.1016/j.brainres.2007.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lenartowski R, Goc A. Epigenetic, transcriptional and posttranscriptional regulation of the tyrosine hydroxylase gene. Int J Dev Neurosci. 2011;29:873–883. doi: 10.1016/j.ijdevneu.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Tank AW, Xu L, Chen X, Radcliffe P, Sterling CR. Posttranscriptional regulation of tyrosine hydroxylase expression in adrenal medulla and brain. Ann NY Acad Sci. 2008;1148:238–248. doi: 10.1196/annals.1410.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raychaudhuri S. Mapping rare and common causal alleles for complex human diseases. Cell. 2011;147:57–69. doi: 10.1016/j.cell.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bademci G, Edwards TL, Torres AL, Scott WK, Zuchner S, Martin ER, Vance JM, Wang L. A rare novel deletion of the tyrosine hydroxylase gene in Parkinson disease. Hum Mutat. 2010;31:E1767–E1771. doi: 10.1002/humu.21351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furukawa Y, Kish SJ, Fahn S. Dopa-responsive dystonia due to mild tyrosine hydroxylase deficiency. Ann Neurol. 2004;55:147–148. doi: 10.1002/ana.10820. [DOI] [PubMed] [Google Scholar]
- 44.Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ludecke B, Dworniczak B, Bartholome K. A point mutation in the tyrosine hydroxylase gene associated with Segawa’s syndrome. Hum Genet. 1995;95:123–125. doi: 10.1007/BF00225091. [DOI] [PubMed] [Google Scholar]
- 47.Ludecke B, Knappskog PM, Clayton PT, Surtees RA, Clelland JD, Heales SJ, Brand MP, Bartholome K, Flatmark T. Recessively inherited L-DOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum Mol Genet. 1996;5:1023–1028. doi: 10.1093/hmg/5.7.1023. [DOI] [PubMed] [Google Scholar]
- 48.Swaans RJ, Rondot P, Renier WO, Van Den Heuvel LP, Steenbergen-Spanjers GC, Wevers RA. Four novel mutations in the tyrosine hydroxylase gene in patients with infantile parkinsonism. Ann Hum Genet. 2000;64:25–31. doi: 10.1017/S0003480000007922. [DOI] [PubMed] [Google Scholar]
- 49.Diepold K, Schutz B, Rostasy K, Wilken B, Hougaard P, Guttler F, Romstad A, Birk Moller L. Levodopa-responsive infantile parkinsonism due to a novel mutation in the tyrosine hydroxylase gene and exacerbation by viral infections. Mov Disord. 2005;20:764–767. doi: 10.1002/mds.20416. [DOI] [PubMed] [Google Scholar]
- 50.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA SEARCH collaborators; kConFab; AOCS Management Group. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stacey SN, Manolescu A, Sulem P, Rafnar T, Gudmundsson J, Gudjonsson SA, Masson G, Jakobsdottir M, Thorlacius S, Helgason A, Aben KK, Strobbe LJ, Albers-Akkers MT, Swinkels DW, Henderson BE, Kolonel LN, Le Marchand L, Millastre E, Andres R, Godino J, Garcia-Prats MD, Polo E, Tres A, Mouy M, Saemundsdottir J, Backman VM, Gudmundsson L, Kristjansson K, Bergthorsson JT, Kostic J, Frigge ML, Geller F, Gudbjartsson D, Sigurdsson H, Jonsdottir T, Hrafnkelsson J, Johannsson J, Sveinsson T, Myrdal G, Grimsson HN, Jonsson T, von Holst S, Werelius B, Margolin S, Lindblom A, Mayordomo JI, Haiman CA, Kiemeney LA, Johannsson OT, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat Genet. 2007;39:865–869. doi: 10.1038/ng2064. [DOI] [PubMed] [Google Scholar]
- 54.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N, Mardis ER, McPherson JD, Minna JD, Morgan MB, Nadel M, Orringer MB, Osborne JR, Ozenberger B, Ramos AH, Robinson J, Roth JA, Rusch V, Sasaki H, Shepherd F, Sougnez C, Spitz MR, Tsao MS, Twomey D, Verhaak RG, Weinstock GM, Wheeler DA, Winckler W, Yoshizawa A, Yu S, Zakowski MF, Zhang Q, Beer DG, Wistuba II, Watson MA, Garraway LA, Ladanyi M, Travis WD, Pao W, Rubin MA, Gabriel SB, Gibbs RA, Varmus HE, Wilson RK, Lander ES, Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helgadottir A, Thorleifsson G, Manolescu A, Gretarsdottir S, Blondal T, Jonasdottir A, Jonasdottir A, Sigurdsson A, Baker A, Palsson A, Masson G, Gudbjartsson DF, Magnusson KP, Andersen K, Levey AI, Backman VM, Matthiasdottir S, Jonsdottir T, Palsson S, Einarsdottir H, Gunnarsdottir S, Gylfason A, Vaccarino V, Hooper WC, Reilly MP, Granger CB, Austin H, Rader DJ, Shah SH, Quyyumi AA, Gulcher JR, Thorgeirsson G, Thorsteinsdottir U, Kong A, Stefansson K. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 56.Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. 2007;8:657–662. doi: 10.1038/nrg2178. [DOI] [PubMed] [Google Scholar]
- 57.Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 58.Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, McCarthy MI, Hattersley AT Wellcome Trust Case Control Consortium (WTCCC) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 61.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 62.Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Tres A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, Isaacs SD, Aben KK, Graif T, Cashy J, Ruiz-Echarri M, Wiley KE, Suarez BK, Witjes JA, Frigge M, Ober C, Jonsson E, Einarsson GV, Mayordomo JI, Kiemeney LA, Isaacs WB, Catalona WJ, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K. Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet. 2007;39:631–637. doi: 10.1038/ng1999. [DOI] [PubMed] [Google Scholar]
- 63.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, Spain S, Lubbe S, Walther A, Sullivan K, Jaeger E, Fielding S, Rowan A, Vijayakrishnan J, Domingo E, Chandler I, Kemp Z, Qureshi M, Farrington SM, Tenesa A, Prendergast JG, Barnetson RA, Penegar S, Barclay E, Wood W, Martin L, Gorman M, Thomas H, Peto J, Bishop DT, Gray R, Maher ER, Lucassen A, Kerr D, Evans DG, Schafmayer C, Buch S, Volzke H, Hampe J, Schreiber S, John U, Koessler T, Pharoah P, van Wezel T, Morreau H, Wijnen JT, Hopper JL, Southey MC, Giles GG, Severi G, Castellvi-Bel S, Ruiz-Ponte C, Carracedo A, Castells A, Forsti A, Hemminki K, Vodicka P, Naccarati A, Lipton L, Ho JW, Cheng KK, Sham PC, Luk J, Agundez JA, Ladero JM, de la Hoya M, Caldes T, Niittymaki I, Tuupanen S, Karhu A, Aaltonen L, Cazier JB, Campbell H, Dunlop MG, Houlston RS CORGI Consortium; EPICOLON Consortium. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10pl4 and 8q23.3. Nat Genet. 2008;40:623–630. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 64.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, Barnetson RA, Theodoratou E, Cetnarskyj R, Cartwright N, Semple C, Clark AJ, Reid FJ, Smith LA, Kavoussanakis K, Koessler T, Pharoah PD, Buch S, Schafmayer C, Tepel J, Schreiber S, Volzke H, Schmidt CO, Hampe J, Chang-Claude J, Hoffmeister M, Brenner H, Wilkening S, Canzian F, Capella G, Moreno V, Deary IJ, Starr JM, Tomlinson IP, Kemp Z, Howarth K, Carvajal-Carmona L, Webb E, Broderick P, Vijayakrishnan J, Houlston RS, Rennert G, Ballinger D, Rozek L, Gruber SB, Matsuda K, Kidokoro T, Nakamura Y, Zanke BW, Greenwood CM, Rangrej J, Kustra R, Montpetit A, Hudson TJ, Gallinger S, Campbell H, Dunlop MG. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–637. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH. PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Plante-Bordeneuve V, Davis MB, Maraganore DM, Marsden CD, Harding AE. Tyrosine hydroxylase polymorphism in familial and sporadic Parkinson’s disease. Mov Disord. 1994;9:337–339. doi: 10.1002/mds.870090312. [DOI] [PubMed] [Google Scholar]
- 67.Kunugi H, Kawada Y, Hattori M, Ueki A, Otsuka M, Nanko S. Association study of structural mutations of the tyrosine hydroxylase gene with schizophrenia and Parkinson’s disease. Am J Med Genet. 1998;81:131–133. [PubMed] [Google Scholar]
- 68.Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, Nagai Y, Oka A, Inoko H, Fukae J, Saito Y, Sawabe M, Murayama S, Yamamoto M, Hattori N, Murata M, Toda T. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson’s disease. Hum Mol Genet. 2006;15:1151–1158. doi: 10.1093/hmg/ddl030. [DOI] [PubMed] [Google Scholar]
- 69.Sutherland G, Mellick G, Newman J, Double KL, Stevens J, Lee L, Rowe D, Silburn P, Halliday GM. Haplotype analysis of the IGF2-INS-TH gene cluster in Parkinson’s disease. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:495–499. doi: 10.1002/ajmg.b.30633. [DOI] [PubMed] [Google Scholar]
- 70.Punia S, Das M, Behari M, Mishra BK, Sahani AK, Govindappa ST, Jayaram S, Muthane UB, KTB, Juyal RC. Role of polymorphisms in dopamine synthesis and metabolism genes and association of DBH haplotypes with Parkinson’s disease among North Indians. Pharmacogenet Genom. 2010;20:435–441. doi: 10.1097/FPC.0b013e32833ad3bb. [DOI] [PubMed] [Google Scholar]
- 71.Johansen CT, Wang J, Lanktree MB, Cao H, McIntyre AD, Ban MR, Martins RA, Kennedy BA, Hassell RG, Visser ME, Schwartz SM, Voight BF, Elosua R, Salomaa V, O’Donnell CJ, Dallinga-Thie GM, Anand SS, Yusuf S, Huff MW, Kathiresan S, Hegele RA. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Exome Variant Server NHLBI Exome Sequencing Project (ESP) http://evs.gs.Washington.edu/EVS/
- 73.Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, Montgomery S, Tavare S, Deloukas P, Dermitzakis ET. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cook EH, Jr, Scherer SW. Copy-number variations associated with neuropsychiatric conditions. Nature. 2008;455:919–923. doi: 10.1038/nature07458. [DOI] [PubMed] [Google Scholar]
- 75.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Zhang J, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sebat J. Major changes in our DNA lead to major changes in our thinking. Nat Genet. 2007;39:S3–5. doi: 10.1038/ng2095. [DOI] [PubMed] [Google Scholar]
- 77.Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009 doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Deng FY, Zhao LJ, Pei YF, Sha BY, Liu XG, Yan H, Wang L, Yang TL, Recker RR, Papasian CJ, Deng HW. Genome-wide copy number variation association study suggested VPS13B gene for osteoporosis in Caucasians. Osteoporos Int. 2010;21:579–587. doi: 10.1007/s00198-009-0998-7. [DOI] [PubMed] [Google Scholar]
- 79.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K, Kahn RS, Linszen DH, van Os J, Wiersma D, Bruggeman R, Cahn W, de Haan L, Krabbendam L, Myin-Germeys I. Genetic Risk and Outcome in Psychosis (GROUP) Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Cid R, Riveira-Munoz E, Zeeuwen PL, Robarge J, Liao W, Dannhauser EN, Giardina E, Stuart PE, Nair R, Helms C, Escaramis G, Ballana E, Martin-Ezquerra G, den Heijer M, Kamsteeg M, Joosten I, Eichler EE, Lazaro C, Pujol RM, Armengol L, Abecasis G, Elder JT, Novelli G, Armour JA, Kwok PY, Bowcock A, Schalkwijk J, Estivill X. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McKinney C, Merriman ME, Chapman PT, Gow PJ, Harrison AA, Highton J, Jones PB, McLean L, O’Donnell JL, Pokorny V, Spellerberg M, Stamp LK, Willis J, Steer S, Merriman TR. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheumatoid arthritis. Ann Rheum Dis. 2008;67:409–413. doi: 10.1136/ard.2007.075028. [DOI] [PubMed] [Google Scholar]
- 82.Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, Heward JM, Gough SC, de Smith A, Blakemore AI, Froguel P, Owen CJ, Pearce SH, Teixeira L, Guillevin L, Graham DS, Pusey CD, Cook HT, Vyse TJ, Aitman TJ. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. 2007;39:721–723. doi: 10.1038/ng2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang S, Wang K, Gregory B, Berrettini W, Wang LS, Hakonarson H, Bucan M. Genomic landscape of a three-generation pedigree segregating affective disorder. PLoS One. 2009;4:e4474. doi: 10.1371/journal.pone.0004474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Willer CJ, Speliotes EK, Loos RJ, Li S, Lindgren CM, Heid IM, Berndt SI, Elliott AL, Jackson AU, Lamina C, Lettre G, Lim N, Lyon HN, McCarroll SA, Papadakis K, Qi L, Randall JC, Roccasecca RM, Sanna S, Scheet P, Weedon MN, Wheeler E, Zhao JH, Jacobs LC, Prokopenko I, Soranzo N, Tanaka T, Timpson NJ, Almgren P, Bennett A, Bergman RN, Bingham SA, Bonnycastle LL, Brown M, Burtt NP, Chines P, Coin L, Collins FS, Connell JM, Cooper C, Smith GD, Dennison EM, Deodhar P, Elliott P, Erdos MR, Estrada K, Evans DM, Gianniny L, Gieger C, Gillson CJ, Guiducci C, Hackett R, Hadley D, Hall AS, Havulinna AS, Hebebrand J, Hofman A, Isomaa B, Jacobs KB, Johnson T, Jousilahti P, Jovanovic Z, Khaw KT, Kraft P, Kuokkanen M, Kuusisto J, Laitinen J, Lakatta EG, Luan J, Luben RN, Mangino M, McArdle WL, Meitinger T, Mulas A, Munroe PB, Narisu N, Ness AR, Northstone K, O’Rahilly S, Purmann C, Rees MG, Ridderstrale M, Ring SM, Rivadeneira F, Ruokonen A, Sandhu MS, Saramies J, Scott LJ, Scuteri A, Silander K, Sims MA, Song K, Stephens J, Stevens S, Stringham HM, Tung YC, Valle TT, Van Duijn CM, Vimaleswaran KS, Vollenweider P, Waeber G, Wallace C, Watanabe RM, Waterworth DM, Watkins N, Witteman JC, Zeggini E, Zhai G, Zillikens MC, Altshuler D, Caulfield MJ, Chanock SJ, Farooqi IS, Ferrucci L, Guralnik JM, Hattersley AT, Hu FB, Jarvelin MR, Laakso M, Mooser V, Ong KK, Ouwehand WH, Salomaa V, Samani NJ, Spector TD, Tuomi T, Tuomilehto J, Uda M, Uitterlinden AG, Wareham NJ, Deloukas P, Frayling TM, Groop LC, Hayes RB, Hunter DJ, Mohlke KL, Peltonen L, Schlessinger D, Strachan DP, Wichmann HE, McCarthy MI, Boehnke M, Barroso I, Abecasis GR, Hirschhorn JN Wellcome Trust Case Control Consortium. Genetic Investigation of ANthropometric Traits Consortium Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat. 2010;31:763–780. doi: 10.1002/humu.21277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Anonymous. MRC-Holland, MLPA. www.mrc-holland.com.
- 87.Liu X, Zhang SS, Fang DF, Ma MY, Guo XY, Yang Y, Shang HF. GCH1 mutation and clinical study of Chinese patients with dopa-responsive dystonia. Mov Disord. 2010;25:447–451. doi: 10.1002/mds.22976. [DOI] [PubMed] [Google Scholar]
- 88.Ormazabal A, Serrano M, Garcia-Cazorla A, Campistol J, Artuch R, Castro de Castro P, Barredo-Valderrama E, Armstrong J, Toma C, Cormand B. Deletion in the tyrosine hydroxylase gene in a patient with a mild phenotype. Mov Disord. 2011;26:1558–1560. doi: 10.1002/mds.23564. [DOI] [PubMed] [Google Scholar]
- 89.Verbeek MM, Steenbergen-Spanjers GC, Willemsen MA, Hol FA, Smeitink J, Seeger J, Grattan-Smith P, Ryan MM, Hoffmann GF, Donati MA, Blau N, Wevers RA. Mutations in the cyclic adenosine monophosphate response element of the tyrosine hydroxylase gene. Ann Neurol. 2007;62:422–426. doi: 10.1002/ana.21199. [DOI] [PubMed] [Google Scholar]
- 90.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 91.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 92.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 93.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 94.Leong SL, Cappai R, Barnham KJ, Pham CL. Modulation of alpha-synuclein aggregation by dopamine: a review. Neurochem Res. 2009;34:1838–1846. doi: 10.1007/s11064-009-9986-8. [DOI] [PubMed] [Google Scholar]
- 95.Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neuro sci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 97.Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med. 2009;47:1354–1361. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 98.Ishikawa S, Taira T, Niki T, Takahashi-Niki K, Maita C, Maita H, Ariga H, Iguchi-Ariga SM. Oxidative status of DJ-1-dependent activation of dopamine synthesis through interaction of tyrosine hydroxylase and 4-dihydroxy-L-phenylalanine (L-DOPA) decarboxylase with DJ-1. J Biol Chem. 2009;284:28832–28844. doi: 10.1074/jbc.M109.019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu L, Sun X, Liu Y, Zhao H, Zhao S, Yang H. DJ-1 upregulates tyrosine hydroxylase gene expression by activating its transcriptional factor Nurr1 via the ERK1/2 pathway. Int J Biochem Cell Biol. 2012;44:65–71. doi: 10.1016/j.biocel.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 100.Jin H, Romano G, Marshall C, Donaldson AE, Suon S, Iacovitti L. Tyrosine hydroxylase gene regulation in human neuronal progenitor cells does not depend on Nurr1 as in the murine and rat systems. J Cell Physiol. 2006;207:49–57. doi: 10.1002/jcp.20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ishikawa S, Taira T, Takahashi-Niki K, Niki T, Ariga H, Iguchi-Ariga SM. Human DJ-1-specific transcriptional activation of tyrosine hydroxylase gene. J Biol Chem. 2010;285:39718–39731. doi: 10.1074/jbc.M110.137034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhong N, Kim CY, Rizzu P, Geula C, Porter DR, Pothos EN, Squitieri F, Heutink P, Xu J. DJ-1 transcriptionally up-regulates the human tyrosine hydroxylase by inhibiting the sumoylation of pyrimidine tract-binding protein-associated splicing factor. J Biol Chem. 2006;281:20940–20948. doi: 10.1074/jbc.M601935200. [DOI] [PubMed] [Google Scholar]