


Abstract
Analysis of bisulfite sequencing data usually requires two tasks: to call methylated cytosines (mCs) in a sample, and to detect differentially methylated regions (DMRs) between paired samples. Although numerous tools have been proposed for mC calling, methods for DMR detection have been largely limited. Here, we present Bisulfighter, a new software package for detecting mCs and DMRs from bisulfite sequencing data. Bisulfighter combines the LAST alignment tool for mC calling, and a novel framework for DMR detection based on hidden Markov models (HMMs). Unlike previous attempts that depend on empirical parameters, Bisulfighter can use the expectation-maximization algorithm for HMMs to adjust parameters for each data set. We conduct extensive experiments in which accuracy of mC calling and DMR detection is evaluated on simulated data with various mC contexts, read qualities, sequencing depths and DMR lengths, as well as on real data from a wide range of biological processes. We demonstrate that Bisulfighter consistently achieves better accuracy than other published tools, providing greater sensitivity for mCs with fewer false positives, more precise estimates of mC levels, more exact locations of DMRs and better agreement of DMRs with gene expression and DNase I hypersensitivity. The source code is available at http://epigenome.cbrc.jp/bisulfighter.
INTRODUCTION
Cytosine methylation is an epigenetic modification that affects a wide range of biological processes such as gene expression, cell differentiation and carcinogenesis (1). Traditionally, methylation measurements have been focused on CpG dinucleotides in preselected sites (e.g. CpG islands). More recently, genome-wide profiling of methylation patterns including non-CpG contexts has been enabled by bisulfite sequencing, where unmethylated cytosines are converted and sequenced as thymines (2).
Analysis of bisulfite sequencing data usually requires two tasks: to call methylated cytosines (mCs) in a sample, and to detect differentially methylated regions (DMRs) between paired samples. The former involves alignment of bisulfite-converted reads to a reference genome, and estimation of the mC level (the ratio of mCs in a cell population) at each cytosine. The latter involves comparison of alignment results between paired samples, and grouping of differentially methylated cytosines (DMCs) at neighbor positions as a contiguous DMR. To date, numerous tools have been proposed for mC calling (3), whereas methods for DMR detection have been largely limited. In fact, it is only recently that BSmooth (4) has been reported as the first software package applicable to both mC calling and DMR detection.
Previous studies have attempted DMR detection by determining individual DMCs with statistical tests, and then chaining DMCs within a user-specified distance (4,5). However, such strategies depend on the choice of distance parameters, hindering automated analysis and possibly leading to biased conclusions. Moreover, fixed-length chaining criteria may be problematic for detecting DMRs whose lengths range from hundreds of base pairs as in small CpG islands, to millions of base pairs as in cancer aberrations (6).
BSmooth has been designed to deal with biological variability inferred from biological replicates (4). Although this strategy is expected to improve DMR detection, BSmooth always requires biological replicates, and thus cannot be applied to data sets without biological replicate information. Because current protocols for bisulfite sequencing are costly, it is prohibitively expensive to obtain sufficient biological replicates for both of two conditions to be compared. Even when many biological replicates are obtained in tissue preparation, some of them are often combined into one sample before library generation for sequencing experiments (5–8). This makes it impossible to establish the one-to-one correspondence between biological replicates and database entries [e.g. the SRX identifiers in the Sequence Read Archive (SRA)]. Additionally, there are cases where biological replicates are in principle difficult to obtain, such as retrospective studies using archival samples (9).
Another problem in previous studies has been the lack of experiments to benchmark mC calling and DMR detection in a systematic manner. For example, performance of existing methods has not been extensively evaluated for various mC contexts, read qualities, sequencing depths and DMR lengths. Furthermore, it is common that some methods are not included as competitors even though their implementations are publicly available.
Here, we present Bisulfighter, a new software package for analyzing bisulfite sequencing data. Bisulfighter uses LAST (10) for alignment procedures in mC calling, and a novel framework for DMR detection based on hidden Markov models (HMMs) that enable automated adjustment of DMC chaining criteria. Bisulfighter does not require biological replicates for DMR detection, and thus maintains applicability to data sets without biological replicate information. We conduct extensive experiments on simulated data as well as on real data, and demonstrate that Bisulfighter consistently achieves better accuracy than other published tools.
MATERIALS AND METHODS
Overview of Bisulfighter
Bisulfighter consists of the two modules that perform mC calling and DMR detection, respectively. The mC calling module in Bisulfighter aligns bisulfite-converted reads using LAST. Unlike most existing aligners, LAST can assess probability (or reliability) of each aligned read by taking into account information of read quality and multilocus mapping (11). We use these probabilities for filtering out unreliable alignments, and for weighting mC level estimates (Figure 1a). The DMR detection module in Bisulfighter uses a novel framework named ‘ComMet’ (a shortening of ‘comparative methylomics’), which is based on HMMs that capture probability distributions of distances among neighbor DMCs (Figure 1b). Unlike previous attempts that depend on empirical distance parameters, Bisulfighter can use the expectation-maximization algorithm for HMMs to adjust DMC chaining criteria automatically for each data set.
Figure 1.

Overview of Bisulfighter. (a) mC calling. Bisulfite-converted reads are aligned to a reference genome, and the mC level is estimated as a ratio of C–C matches. A major feature is the utilization of alignment probability for filtering out unreliable alignments, and for weighting mC level estimates. (b) DMR detection. Neighbor cytosines differentially methylated between paired samples are grouped as a DMR (UP or DOWN). A novel HMM-based framework enables automatic learning of chaining criteria, and detection of DMRs using likelihood ratio scores. Colors in the state transition track correspond to those in the state transition diagram at the top. NoCh: no change of methylation between paired samples.
Bisulfighter pools all biological replicates from one condition as one sample, and detects DMRs between two conditions by comparing a pair of two samples. Even if biological replicates are not available (i.e. only one measurement is available) from one condition, a sample can still be prepared from that measurement. Therefore, Bisulfighter is applicable to data sets without biological replicate information. Bisulfighter can address either single- or paired-end reads, produced from either whole-genome bisulfite sequencing (WGBS) or reduced representation bisulfite sequencing (RRBS).
mC calling
For the read mapping procedure in Bisulfighter, we use a local alignment program, LAST, as Frith et al. (10) have recently reported that LAST maps bisulfite-converted reads more accurately in shorter computation time compared with other alignment programs. However, they have focused only on binary classification of mCs, and have not addressed estimation of mC levels (10) (See the ‘Accuracy measure for mC calling' section for the definitions of these two problem settings). Therefore, in this study, we aim to advance Frith et al. (10) by proposing a new LAST-based method applicable to estimation of mC levels. After mapping reads followed by removing unreliable alignments with probability <0.9, we estimate the mC level for each cytosine by dividing the count of C–C matches (supporting mCs) by the count of all reads mapped at the same position.
LAST has several program options that might improve accuracy of mC calling. First, to equalize the sensitivity between C–C matches (mCs) and C–T mismatches (unmethylated cytosines), all Cs in reads might be virtually treated as Ts during alignment procedures. Second, in estimation of mC levels, the count of C–C matches might be weighted by alignment probability and/or read quality. To determine the default setting for Bisulfighter, we compared eight combinations of options: with or without equal treatment of Cs and Ts, with or without weighting by alignment probability and with or without weighting by read quality. We found that equal treatment of Cs and Ts improved accuracy, while the weighing schemes did not substantially contribute to accuracy (Supplementary Figure S1). Considering these results, we determined the method with the all options turned on as the Bisulfighter’s default setting.
DMR detection
For the DMR detection module in Bisulfighter, we designed ComMet, an HMM-based framework that captures distance distributions among neighbor DMCs. The motivation for using HMMs came from our observations of several real data sets (6–9,12–17) covering a wide range of biological processes and sequencing protocols summarized in Supplementary Table S1b. For most of the data sets, DMCs showed distance distributions distinct from the other cytosines whose methylation was not changed (Supplementary Figures S2a–j). Moreover, the differences between distance distributions were statistically significant (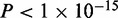 ), even for the data set where the differences were not apparent from visual inspection (Supplementary Figure S2k). We therefore took advantage of these distributions to adjust DMC chaining criteria.
), even for the data set where the differences were not apparent from visual inspection (Supplementary Figure S2k). We therefore took advantage of these distributions to adjust DMC chaining criteria.
ComMet has pairs of states for CpG positions and their interval positions (named ‘gap’), each of which has three types for the directions of differential methylation: hypermethylation (UP), hypomethylation (DOWN) and no change (NoCh). Supplementary Figure S3 shows the state transition diagram. Transition probabilities among UP, DOWN and NoCh states represent distinct distance distributions among DMCs. We implemented two variants of HMM architectures: the naive model and the dual model. The naive model has only one gap state, approximating a distance distribution by a single geometric distribution (Supplementary Figure S3a, also shown in Figure 1b). On the other hand, the dual model uses two gap states per direction, and thus can capture a complex distance distribution by a two-component geometric mixture (Supplementary Figure S3b). It is well known that there are at least two types of DMRs with high and low CpG densities, and they are expected to be modeled by two components in a geometric mixture, respectively.
Emission probabilities at CpG states represent how likely each CpG is differentially methylated or not. Given the alignment results of bisulfite-converted reads, we can observe at each CpG position the count of reads supporting mCs in each of two samples. If a CpG is differentially methylated, the counts can be considered to be taken from separate probability distributions that reflect the difference in mC levels between two samples. On the other hand, if CpG methylation is not changed, the counts should be the consequence of the common mC level. Therefore, we designed emission functions for CpG states as follows:
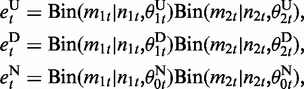 |
where Bin() is a binomial distribution, and mst and nst (s = 1,2) are the count of reads supporting mCs and the count of all aligned reads at the t-th CpG position in the s-th sample, respectively. θ is the occurrence probability of mC-supporting reads, which is computed by the maximum a posteriori estimation with pseudocount regularization (18):
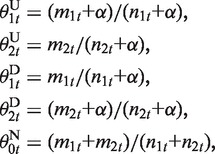 |
where α is the strength of regularization (fixed as α = 8 throughout this study). For gap states, we currently use no emission function.
ComMet enables us to use well-established learning algorithms for optimizing parameters in HMMs. ComMet first computes θ, and fixes emission probabilities. Then, transition probabilities are trained by the standard expectation-maximization algorithm (18). As observed in Supplementary Figure S2, distance distributions among DMCs are highly data-dependent, possibly reflecting underlying epigenetic modifiers for differential methylation. Additionally, sequencing protocols may impact distance distributions; we observed some features of distance distributions possibly specific to RRBS (Supplementary Figure S2ij). Therefore, we execute the learning procedure for each data set to be analyzed, rather than seeking a general training data set.
After parameter learning, ComMet detects DMRs based on log-likelihood ratio scores. The score for detecting a certain region as a DMR directed to dir (=UP or DOWN) is defined as follows:
where 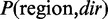 and
and 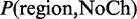 are probabilities of the region with the corresponding state transitions in HMMs. The region that maximizes this score can be computed by a simple dynamic programming (DP) algorithm. It should be noted that, in the DP algorithm, DMRs are extended by chaining CpGs, as long as their scores increase, even if the gains are fairly small. Therefore, small differences in distance distributions (Supplementary Figure S2) can still have a significant contribution to DMR detection, especially for determining boundaries of DMRs. As will be shown in the ‘Results’ section, this strategy successfully detects DMRs that reciprocally overlap with various lengths of true DMRs. ComMet controls the number of output DMRs by iterative procedures in which the portion of DP tables for previous DMRs is masked, and the next maximum-scoring region is detected from the remaining portion of DP tables.
are probabilities of the region with the corresponding state transitions in HMMs. The region that maximizes this score can be computed by a simple dynamic programming (DP) algorithm. It should be noted that, in the DP algorithm, DMRs are extended by chaining CpGs, as long as their scores increase, even if the gains are fairly small. Therefore, small differences in distance distributions (Supplementary Figure S2) can still have a significant contribution to DMR detection, especially for determining boundaries of DMRs. As will be shown in the ‘Results’ section, this strategy successfully detects DMRs that reciprocally overlap with various lengths of true DMRs. ComMet controls the number of output DMRs by iterative procedures in which the portion of DP tables for previous DMRs is masked, and the next maximum-scoring region is detected from the remaining portion of DP tables.
In some cases, analysis may be focused on individual DMCs rather than chained DMRs. For this purpose, ComMet also assesses posterior probabilities that each CpG is directed to UP, DOWN or NoCh by the standard forward-backward algorithm (18).
Benchmark
Summary
To evaluate accuracy of mC calling and DMR detection extensively, we used both simulated and real data sets as summarized in Supplementary Table S1. In the benchmark for mC calling, we simulated bisulfite-converted reads with various mC contexts, read qualities and sequencing depths, and evaluated accuracy of detected mCs using the information of true mCs. In the benchmark for DMR detection, we used simulated data with various DMR lengths and sequencing depths, and evaluated accuracy of detected DMRs for their overlap with true DMRs. Furthermore, we applied Bisulfighter to real data sets, and evaluated agreement of detected DMRs with gene expression, and DNase I hypersensitivity. These data sets were collected from a wide range of biological processes including pathogenesis and normal development, and consist of both single- and paired-end reads with various lengths (Supplementary Table S1b). We note that there are no gold standards (i.e. true biological mCs and DMRs) to benchmark mC calling and DMR detection, which is a limitation common to this and all previous studies. We attempt to address this problem to the extent possible by using multilateral evaluation based on a series of simulated and real data sets.
Simulation of bisulfite-converted reads
To generate bisulfite-converted reads for benchmark data, we used the human chromosome X (chrX) as a reference. Using DNemulator (10), we randomly assigned an mC level to each cytosine in the chrX with respect to its context (CpG, CHG or CGG where H stands for non-G nucleotide). We also assigned polymorphisms (substitutions and indels) in the chrX by referring to real allele frequencies obtained from ‘snp132Common.txt’ in the UCSC Genome Browser (http://genome.ucsc.edu/). We then randomly extracted sequence fragments from the chrX with unmethylated cytosines converted to thymines. To evaluate the effects of sequencing depth, we varied the number of generated reads to 1 million (M), 3M, 5M, 7M, 10M, 20M and 50M. We also varied read quality by simulating quality values in ‘SRR019072’ (Data set A; low quality) and ‘SRR094461’ (Data set B; high quality), which are files for bisulfite sequencing with Illumina platforms obtained from the SRA (http://www.ncbi.nlm.nih.gov/sra). These reads were single-end, 85 or 87 bp in length, and generated by a WGBS-like procedure (Supplementary Table S1a). In total, we prepared 42 data sets to benchmark mC calling (three mC contexts, seven sequencing depths and two read qualities).
Accuracy measure for mC calling
We evaluated accuracy of mC calling in two problem settings: binary classification of mCs, and estimation of mC levels. In binary classification, cytosines with non-zero mC levels were positives, while unmethylated cytosines were negatives. We evaluated the true-positive rate (TPR) and the false discovery rate (FDR) defined as 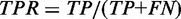 and
and 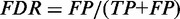 , where TP, FN and FP are the numbers of true positives, false negatives and false positives, respectively. Accuracy was considered to be good if a high TPR was obtained at a small FDR. In estimation of mC levels, we evaluated errors between estimated mC levels and simulated true values. Accuracy was considered to be good if the distribution of errors was concentrated at zero.
, where TP, FN and FP are the numbers of true positives, false negatives and false positives, respectively. Accuracy was considered to be good if a high TPR was obtained at a small FDR. In estimation of mC levels, we evaluated errors between estimated mC levels and simulated true values. Accuracy was considered to be good if the distribution of errors was concentrated at zero.
Simulation of DMRs
DMRs were simulated by preparing a pair of data sets, each of which has different assignments of mC levels for generating bisulfite-converted reads. We first generated reads using the chrX and DNemulator as mentioned in the above section. Then, we defined a certain genomic region as a DMR, and coordinately changed mC levels of all CpGs in the region to the maximum (UP) or the minimum (DOWN). After locating 200 DMRs for UP and DOWN, we again generated reads from changed assignments of mC levels, and made a pair of data sets before and after the change. To evaluate performance on various DMR lengths, we produced four versions: 50 bp, 500 bp, 5 kb and 50 kb. (Note that actual lengths were not exactly the same as these values, as we required the ends of a DMR to be closed by CpGs.) To investigate the negative influence of the smoothness assumption, we prepared another type of DMRs with independence of neighbor positions. Specifically, we changed mC levels of 10% of all CpGs independently to random directions (UP or DOWN). DMRs were defined as regions where neighbor CpGs were occasionally changed to the same direction. This procedure produced DMRs with the median length of 58 bp. For both types of DMRs, we generated reads with quality values in ‘SRR094461’, and sequencing depth was varied from 1M to 50M as in the above section.
Accuracy measure for DMR detection
We evaluated accuracy of DMR detection by the number of true positives among the top 100 detected DMRs. A true positive was defined as a true DMR that reciprocally overlapped with a detected DMR in a certain proportion of their lengths. For example, a true positive with 50% reciprocal overlap was counted only if the length of the overlapping region was larger than half the length of the true DMR, and half the length of the detected DMR. Similarly, we also defined true positives for 90 and 99% reciprocal overlaps.
Experiments on real data—gene expression
To validate the effectiveness of Bisulfighter for real data, we conducted experiments similar to the previous study (4) that evaluated agreement between gene expression and detected DMRs. For this purpose, we searched the SRA for studies in which both transcriptome profiling (RNA-Seq) and WGBS were performed on paired samples. We collected data from (6) (Carcinogenesis data set; breast cancer versus normal breast), and from (5) (Adipogenesis data set; mature fat cells versus adipose-derived stem cells). RNA-Seq reads were mapped to the human genome by TopHat (19), and gene expression was measured by fragments per kilobase of transcript per million mapped reads (FPKM) as computed by Cufflinks (20). Differential gene expression was measured by the fold change in FPKM. Differentially expressed genes (DEGs) were determined by the threshold of 5-fold FPKM change. The agreement between DEGs and detected DMRs was evaluated according to the previous study (4). Specifically, we focused on DEGs whose±5 kb regions around transcription start sites (TSSs) contained detected DMRs. The number of these overlapped DEGs was counted for the top 1000 or 3000 detected DMRs, and used as a measure of the agreement. For the baseline of accuracy, we calculated the expected number of overlapped DEGs when DMRs were randomly placed in the TSS windows (denoted by ‘random guessing’).
Experiments on real data—DNase I hypersensitivity
Although agreement with gene expression is useful to evaluate DMRs detected around TSSs, it neglects DMRs detected at regulatory elements distal to TSSs. Thus, we conducted additional experiments to evaluate agreement between DNase I hypersensitivity and detected DMRs. We collected WGBS data from (7) (Hematopoiesis data set; mature B cells versus hematopoietic stem cells), and from (8) (Fibroblast development data set; foreskin fibroblasts versus embryonic stem cells). For these cell types, the genome-wide information of DNase I hypersensitivity is available from the ENCODE project (http://ftp.ebi.ac.uk/pub/databases/ensembl/encode/integration_data_jan2011/byDataType/openchrom/jan2011). The file for each cell type provides the genomic locations of 150 bp regions that show the local maxima of DNase I hypersensitivity. We defined ‘differentially sensitive sites’ (DSSs) as those 150 bp regions present in only one side of paired cell types. The agreement between DSSs and detected DMRs was evaluated similarly to the experiment for DEGs. We focused on DSSs whose ±5 kb regions around the midpoints contained detected DMRs. The number of these overlapped DSSs was counted for the top 1000 or 3000 detected DMRs, and used as a measure of the agreement. We note that the 150 bp regions provided by the ENCODE project are just the fixed-length windows, and thus do not necessarily indicate exact boundaries of DSSs. Therefore, we did not use the 150 bp regions directly, but evaluated whether their extended regions contained the entire lengths of detected DMRs. This agreement measure is designed to penalize a method that outputs irrelevantly long or short DMRs. For example, when an irrelevantly long DMR is inferred producing overlaps with many DSSs, it does not contribute to the agreement measure because any DSS cannot contain its entire length. Moreover, when one true DMR is wrongly split into short segments producing multiple overlaps with one DSS, the agreement measure is not biased since each DSS is counted only once.
RESULTS
mC calling
We compared accuracy of mC calling between Bisulfighter and the comprehensive list of published tools whose implementations are publicly available: BatMeth (21), Bismark (22), BRAT (23), BS_Seeker (24), BSMAP (25), BSmooth (4), Lister (5), MethylCoder (26), RMAP (27) and Novoalign (http://www.novocraft.com). To ensure a fair comparison between Bisulfighter and the other tools, we optimized the options provided by these competitors (Supplementary Notes S1 and S2 for details). Typical results for CpG context and low-quality reads are shown in Figure 2.
Figure 2.

Benchmark for mC calling. (a and b) Binary classification of mCs. CpGs were called as mCs if nonzero mC levels were estimated. (a) Trade-off between the true-positive rate and the number of false positives for varying sequencing depths. (b) The true-positive rate and the FDR at the limited sequencing depth of 3M reads (shown as dots in a). For Bisulfighter, 3M reads were equivalent to the mean coverage of 2.4 among those CpGs with at least one aligned read. True-positive rates plateaued around 0.9 due to low quality of simulated reads. (c) Estimation of mC levels. Distributions of errors between estimated and true mC levels are shown as box plots (top; 25th–75th percentile), and histograms for BatMeth, RMAP and Bisulfighter (bottom). The complete results including non-CpG contexts, high-quality reads and higher sequencing depths are found in Supplementary Figures S5–S7.
At varying sequencing depth, Bisulfighter exhibited a greater true-positive rate than the other tools compared at the same number of false-positive mC calls (Figure 2a). The difference in true-positive rates was especially remarkable when only a small number of false positives were allowed. Because current protocols for bisulfite sequencing are expensive (2), performance on limited sequencing depth is of practical interest. Figure 2b shows that Bisulfighter maintained sensitive mC calling even when the mean CpG coverage was only 2.4. Again, the trade-off between sensitivity and specificity was superior to the other tools. For example, Bismark attained a true-positive rate comparable with Bisulfighter, but the number of false-positive mC calls for Bismark were about twice those for Bisulfighter. In Supplementary Table S2, we showed the computation time of each tool for simulated reads. The speed of Bisulfighter was comparable with the other tools.
Bisulfighter produced precise estimates of mC levels with small errors from simulated true values (Figure 2c). It is notable that Bisulfighter achieved the best accuracy, both in estimation of mC levels and in binary classification of mCs (Figure 2ab). Among the other tools, there were preferences such that BatMeth and RMAP performed better than Bismark in binary classification of mCs, but worse in estimation of mC levels. These results demonstrated the versatility of Bisulfighter. In Supplementary Figure S4, we evaluated estimation of mC levels at selected CpG sites above coverage threshold of 3×, 5× and 10×. We confirmed that our conclusion was largely consistent over the different choices of coverage threshold; Bisulfighter achieved better accuracy than the other tools, or at least comparable accuracy among the best-performing tools (Supplementary Figure S4a–c). In addition, for given coverage threshold, Bisulfighter achieved the larger number of captured CpG sites than the other tools (Supplementary Figure S4d).
In other conditions with non-CpG contexts and high-quality reads, Bisulfighter stably provided good performance (Supplementary Figures S5–S7). Consequently, we concluded that Bisulfighter is a promising tool for mC calling.
DMR detection
We compared accuracy of DMR detection between Bisulfighter and other methods found in previous studies: Smoothing (4) and Fisher (5). As mentioned in Introduction, BSmooth cannot be directly applied to data sets without biological replicate information, including those used in this study (5–8). To make a comparison with Bisulfighter, we implemented a modified version of BSmooth, named Smoothing, that pools all biological replicates as one sample, and thus does not require biological replicate information (Supplementary Note S1 for details).
For simulated data, Bisulfighter consistently achieved the best accuracy, while the performance of the other methods critically depended on various DMR lengths (Figure 3a). The superior accuracy was also seen under varying sequencing depths (Figure 3b and Supplementary Figure S8). As expected, the dual model achieved slightly better accuracy than the naive model in most cases. To further validate the ComMet framework in Bisulfighter, we asked how well the other methods determined individual DMCs before chaining them as DMRs. For comparison, DMCs for Bisulfighter were determined based on posterior probabilities computed by HMMs (the ‘Materials and Methods’ section). We observed that the difference in accuracy was not so large when determining individual DMCs (Supplementary Figure S9). Thus, the improvements in DMR detection were confirmed to be due to the DMC chaining phase by the ComMet framework.
Figure 3.

Benchmark for DMR detection. (a and b) Experiments on simulated data. (a) For various DMR lengths and the fixed sequencing depth of 50M reads, true positives with 50 (left), 90 (center) or 99% (right) reciprocal overlap are shown. Ind: simulation with independence of neighbor positions. (b) For varying sequencing depths and the fixed DMR length of 500 bp, true positives with 50% reciprocal overlap are shown. (c and d) Experiments on real data. (c) Agreement between detected DMRs and gene expression. Carcino: Carcinogenesis data set. Adipo: Adipogenesis data set. See the ‘Experiments on real data—gene expression’ section for details. (d) Agreement between detected DMRs and DNase I hypersensitivity. Hemato: Hematopoiesis data set. Fibro: Fibroblast development data set. See the ‘Experiments on real data—DNase I hypersensitivity’ section for details. dual, naive: results for the corresponding HMM architectures in Bisulfighter.
Another feature of Bisulfighter is that it does not assume smoothness of mC levels along a genomic sequence. In BSmooth, mC levels are preprocessed by smoothing techniques, assuming that mC levels at neighbor positions do not vary sharply (4). To evaluate to what extent accuracy depends on the smoothness assumption, we simulated DMRs with independence of neighbor positions (See the ‘Simulation of DMRs’ section). We observed that Bisulfighter maintained moderate accuracy, whereas the other methods did not (Figure 3a, Ind.). In addition, the performance of Smoothing was worse for shorter DMRs. These results indicate that methods based on the smoothness assumption are not effective when inherent smoothness is absent (independent DMCs) or weak (short DMRs).
Because the chrX used for simulating bisulfite-converted reads has a relatively low GC content, we conducted similar experiments with the GC-rich chromosome 19 (Supplementary Figure S10). Bisulfighter maintained better accuracy than existing methods in both mC calling and DMR detection. These results suggest that the performance of Bisulfighter is robust to various GC contents, as also seen below in our whole-genome evaluation based on real data.
Bisulfighter detected reasonable DMRs not only in simulated data but also in real data (Figure 3c). In the Carcinogenesis data set, Bisulfighter achieved better agreement between DEGs and detected DMRs than the other methods. In the Adipogenesis data set, Bisulfighter was similar to Fisher in accuracy, whereas Smoothing was no more accurate than random guessing. To validate that these results did not depend on the selected threshold of 5-fold expression change for determining DEGs, we varied the threshold from 2 to 10 (Supplementary Figure S11). We confirmed that our conclusion was largely consistent over the different choices of expression fold change; Bisulfighter achieved better agreement than the other methods in the Carcinogenesis data set, and similar accuracy to Fisher in the Adipogenesis data set. We note that the Adipogenesis data set seems to be a real example where the smoothness assumption is not fully satisfied, due to a number of short DMRs. The median length of the top 10 000 DMRs detected by Bisulfighter was much shorter in the Adipogenesis data set (234 bp), compared with that in the Carcinogenesis data set (5587 bp). Bisulfighter is free from the smoothness assumption, and therefore applicable to various lengths of DMRs occurred in a wide range of biological processes.
DMRs detected by Bisulfighter were further supported by agreement with DNase I hypersensitivity (Figure 3d). For both the Hematopoiesis data set and the Fibroblast development data set, Bisulfighter achieved better agreement than the other methods. In addition, we increased the resolution of DSSs from ±5 kb to±500 bp, and evaluated their agreement with detected DMRs separately for TSS-proximal and TSS-distal regions (Supplementary Figure S12). For the various definitions of DSSs, Bisulfighter achieved better agreement than the other methods for both the Hematopoiesis data set and the Fibroblast development data set. These results demonstrated that Bisulfighter can detect reasonable DMRs not only around TSSs but also at distal regulatory elements. We note that agreement for the Hematopoiesis data set was worse compared with that for the Fibroblast development data set, which might be due to imperfect matching of cell samples between DNase I hypersensitivity data and WGBS data; the ENCODE project and (7) have used slightly different subpopulations of hematopoietic stem cells. We emphasize that Bisulfighter consistently achieved the best accuracy among the other methods, in each of the Hematopoiesis data set and the Fibroblast development data set. Therefore, our conclusion is still valid even though the agreement measure involves some data set dependency.
DISCUSSION
In this article, we described Bisulfighter, a new software package for analyzing bisulfite sequencing data. In contrast to existing methods developed solely for mC calling or DMR detection, Bisulfighter is successfully applicable to both of these essential tasks. We presented the first systematic benchmark in which accuracy of mC calling and DMR detection was evaluated for various mC contexts, read qualities, sequencing depths and DMR lengths, as well as for real data from a wide range of biological processes including pathogenesis and normal development. Bisulfighter consistently outperforms existing methods.
We demonstrated that Bisulfighter can take full advantage of bisulfite sequencing, which, in contrast to microarray platforms, produces methylation measurements at whole-genome scale and single nucleotide resolution. Bisulfighter can identify DMRs even when their genomic locations and lengths are not prespecified. Moreover, Bisulfighter does not rely on the smoothness assumption, and thus it can detect biological events involving short DMRs and independent DMCs. As previously implied by the authors of BSmooth (4), our results suggest that smoothing techniques may collapse rich information of methylation signals obtained at single nucleotide resolution. There is increasing evidence that short DMRs and independent DMCs have biological significance, especially in regions whose methylation status has not been well-studied (e.g. gene bodies) (28). Bisulfighter will contribute to accelerating such studies, and expanding our knowledge of methylomes.
As a future direction, we are planning to improve Bisulfighter in the following aspects. First, since the weighting schemes did not substantially contribute to mC calling, the variations of the estimation formula in Figure 1a should be investigated. For example, mC calling may be improved by weighting the denominator n using pi and qi. Second, while the transition functions well captured distance distributions among neighbor DMCs, the emission functions still have room for improvement, especially in the handling of pseudocounts. The pseudocount terms can be removed by using beta-binomial distributions instead of binomial distributions. The parameters of a beta mixture used in beta-binomial distributions may be estimated by using a nonparametric Bayesian approach similar to that used in (29). Third, since the dual model achieved better accuracy than the naive model, more complex HMM architectures may further improve DMR detection. To determine optimal HMM architectures, it is useful to use a model selection approach such as factorized asymptotic Bayesian HMMs (30), instead of standard HMMs. Finally, although Bisulfighter was designed not to require biological replicates, the current approach has a limitation that it pools replicates even when they are available. It is thus desirable to extend Bisulfighter so that it incorporates biological variability inferred from available replicates, while maintaining its applicability to data sets without biological replicate information. For this purpose, it will be convenient to use existing frameworks developed for extending replicate-free methods to replicate-aware versions (31). The source code and the binaries of Bisulfighter are available at http://epigenome.cbrc.jp/bisulfighter.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: This work was supported by Japan Science and Technology Agency (JST) CREST Program; and The New Energy and Industrial Technology Development Organization (NEDO).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank M.C. Frith for many helpful suggestions about simulating and mapping bisulfite-converted reads.
REFERENCES
- 1.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 2.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 3.Bock C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012;13:705–719. doi: 10.1038/nrg3273. [DOI] [PubMed] [Google Scholar]
- 4.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hon GC, Hawkins RD, Caballero OL, Lo C, Lister R, Pelizzola M, Valsesia A, Ye Z, Kuan S, Edsall LE, et al. Global DNA hypomethylation coupled to repressive chromatin domain formation and gene silencing in breast cancer. Genome Res. 2012;22:246–258. doi: 10.1101/gr.125872.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR, et al. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol. Cell. 2011;44:17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–331. doi: 10.1101/gr.101907.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beyan H, Down TA, Ramagopalan SV, Uvebrant K, Nilsson A, Holland ML, Gemma C, Giovannoni G, Boehm BO, Ebers GC, et al. Guthrie card methylomics identifies temporally stable epialleles that are present at birth in humans. Genome Res. 2012;22:2138–2145. doi: 10.1101/gr.134304.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frith MC, Mori R, Asai K. A mostly traditional approach improves alignment of bisulfite-converted DNA. Nucleic Acids Res. 2012;40:e100. doi: 10.1093/nar/gks275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frith MC, Wan R, Horton P. Incorporating sequence quality data into alignment improves DNA read mapping. Nucleic Acids Res. 2010;38:e100. doi: 10.1093/nar/gkq010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl Acad. Sci. USA. 2012;109:10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tung J, Barreiro LB, Johnson ZP, Hansen KD, Michopoulos V, Toufexis D, Michelini K, Wilson ME, Gilad Y. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc. Natl Acad. Sci. USA. 2012;109:6490–6495. doi: 10.1073/pnas.1202734109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi S, Hong K, Liu R, Shen L, Inoue A, Diep D, Zhang K, Zhang Y. Tet1 controls meiosis by regulating meiotic gene expression. Nature. 2012;492:443–447. doi: 10.1038/nature11709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krivtsov AV, Figueroa ME, Sinha AU, Stubbs MC, Feng Z, Valk PJ, Delwel R, Dohner K, Bullinger L, Kung AL, et al. Cell of origin determines clinically relevant subtypes of MLL-rearranged AML. Leukemia. 2013;27:852–860. doi: 10.1038/leu.2012.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- 19.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim JQ, Tennakoon C, Li G, Wong E, Ruan Y, Wei CL, Sung WK. BatMeth: improved mapper for bisulfite sequencing reads on DNA methylation. Genome Biol. 2012;13:R82. doi: 10.1186/gb-2012-13-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harris EY, Ponts N, Le Roch KG, Lonardi S. BRAT-BW: efficient and accurate mapping of bisulfite-treated reads. Bioinformatics. 2012;28:1795–1796. doi: 10.1093/bioinformatics/bts264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen PY, Cokus SJ, Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedersen B, Hsieh TF, Ibarra C, Fischer RL. MethylCoder: software pipeline for bisulfite-treated sequences. Bioinformatics. 2011;27:2435–2436. doi: 10.1093/bioinformatics/btr394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith AD, Chung WY, Hodges E, Kendall J, Hannon G, Hicks J, Xuan Z, Zhang MQ. Updates to the RMAP short-read mapping software. Bioinformatics. 2009;25:2841–2842. doi: 10.1093/bioinformatics/btp533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012;44:1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 29.Zhang L, Meng J, Liu H, Huang Y. A nonparametric Bayesian approach for clustering bisulfate-based DNA methylation profiles. BMC Genomics. 2012;13(Suppl. 6):S20. doi: 10.1186/1471-2164-13-S6-S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimaki R, Hayashi K. Proceedings of the 29th International Conference on Machine Learning. Scotland, UK: 2012. Factorized asymptotic Bayesian hidden Markov models. 2012. [Google Scholar]
- 31.Li QH, Brown JB, Huang HY, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011;5:1752–1779. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.