

Abstract
Genomic imprinting is an epigenetic mechanism by which alleles of some specific genes are expressed in a parent-of-origin manner. It has been observed in mammals and marsupials, but not in birds. Until now, only a few genes orthologous to mammalian imprinted ones have been analyzed in chicken and did not demonstrate any evidence of imprinting in this species. However, several published observations such as imprinted-like QTL in poultry or reciprocal effects keep the question open. Our main objective was thus to screen the entire chicken genome for parental-allele-specific differential expression on whole embryonic transcriptomes, using high-throughput sequencing. To identify the parental origin of each observed haplotype, two chicken experimental populations were used, as inbred and as genetically distant as possible. Two families were produced from two reciprocal crosses. Transcripts from 20 embryos were sequenced using NGS technology, producing ∼200 Gb of sequences. This allowed the detection of 79 potentially imprinted SNPs, through an analysis method that we validated by detecting imprinting from mouse data already published. However, out of 23 candidates tested by pyrosequencing, none could be confirmed. These results come together, without a priori, with previous statements and phylogenetic considerations assessing the absence of genomic imprinting in chicken.
INTRODUCTION
Parental genomic imprinting is a process that leads to the differential expression of alleles depending on their parental origin. This phenomenon has been described in eutherian mammals, marsupials, plants and insects, but has never been observed in birds (1,2).
Even if not the only one (3), the main theory proposed to explain this phenomenon is the parental conflict hypothesis, also known as the kinship theory. Briefly, imprinting is leading to an imbalance of parental allele contribution, and the parental conflict hypothesis argues that the genes controlling the allocation of resources to the embryo should be particularly affected, with the maternal genome restricting the use of resources, allowing to preserve the mother and future progeny, and the paternal genome favoring growth of his offspring (4–8). Readers are referred to (9) for an in-depth discussion of the kinship theory. According to this theory, imprinting should be restricted to organisms in which maternal resources directly affect the embryo. It would therefore be unlikely to observe this phenomenon in oviparous animals (10), where the amount of resources is limited by the egg size, controlled by the mother. Genomic imprinting evolved both in angiosperm plants and placental mammals (11) but is also seen in insects, and it has even been shown that it arose several times during mammals' evolution due to different selective pressure at several loci (12). Regardless of the kinship theory, the appearance of genomic imprinting in nonmammalian animals is thus not implausible. Genomic imprinting was studied in birds, but until now, only the expression of genes known to be imprinted in mammals has been analyzed in chickens, most of them showing biallelic expression and thus, leading to the conclusion of the lack of imprinting in this species: several studies have been conducted on IGF2, known to be paternally expressed in mice and humans (13), all of them except one (14) leading to the conclusion that it was biallelically expressed (15–19). In this genomic region, the H19 imprinting center, controlling an imprinted cluster including IGF2, is shown to be absent from the chicken genome (19). Other genes known to be imprinted in mammals have been shown to be biallelically expressed in chicken, such as ASCL2/MASH2, M6PR/IGF2R, DLK1 and UBE3A (19–21). However, parent-of-origin QTLs have been detected in chicken and quail (22–24). Whereas some of them may finally not be considered as relevant to genomic imprinting due to linkage disequilibrium or bias generated by the experimental design (25,26), others appeared to be consistent, when using appropriate experimental animal population designs and methodology (25,27). Moreover, reciprocal effects, of great importance in poultry production, may be partly explained by imprinting. Together with several molecular features, such as the absence of DNMT3L (key factor of the establishment of DNA methylation) in the chicken genome (28) and the possible existence of imprinting mechanisms other than DNA methylation (29,30), these contradictory observations encourage to definitely answer the question (31).
Advances in sequencing technologies now allow to study genomic imprinting through whole transcriptome sequencing (32–34), avoiding any a priori choice of target genomic regions, such as the chicken homologous of imprinted regions in mammals. We therefore chose Next Generation Sequencing (NGS) technology to analyze imprinting in chicken, without any preconception on whether it exists. Chicken is a model species for numerous studies in birds, and the availability of inbred lines is a great advantage for our purpose: we set up an experimental design consisting in two families from two reciprocal crosses between chicken lines, chosen so as to allow the identification of the parental origin of each allele in embryonic offspring. We sequenced whole embryos transcriptomes, and the relative expression level of each parental allele was compared, for the informative loci, between both cross ways.
MATERIALS AND METHODS
Ethics statement
Chickens were bred at INRA, UE1295 Pôle d’Expérimentation Avicole de Tours, F-37380 Nouzilly, in accordance with European Union Guidelines for animal care. The farm is registered by the Ministry of Agriculture with the license number C37–175–1 for animal experimentation. The experiment was realized under authorization 37–002 delivered to D. Gourichon.
Chicken lines and crosses
To maximize the probability of identifying the parental origin of an allele in F1 individuals from two reciprocal crosses, we selected 2 chicken lines among 10 (Supplementary Table S1), available at INRA experimental farm (UE1295 PEAT, F-37380 Nouzilly, France). Genotyping was performed on 3–12 individuals from each population with an Illumina 57K Single Nucleotide Polymorphism (SNP) array [57 636 SNPs (35)]. SNPs were filtered (call freq > 0.9, GenTrainScore > 0.5, Minor Allele Frequency (MAF) > 0.01) and allele frequencies within each line calculated with plink, option—freq (36). Genetic similarities between lines (homozygozities for a given line) were then calculated (Figure 1) from the 52 189 filtered SNPs. The most genetically distant pair of lines together with the highest homozygosity each were selected, i.e. the Line 6 (37) and the Line R− (38). A male and a female from each line were reciprocally crossed, and 24 F1 embryos, 12 from each cross (6 males and 6 females per cross), were harvested from the same batch at embryonic day 4.5 (stage 26). Genomic DNA and total RNA were concurrently extracted from the same samples of crushed whole embryos without extraembryonic membranes (AllPrep DNA/RNA Mini Kit, Qiagen). RNA quality was measured by a BioAnalyzer (Agilent); all samples had a RNA Integrity Number ≥ 9.9.
Figure 1.

Experimental design. (A) Heatmap of genetic similarities between lines. Colors correspond to the inbred level of tested individuals (red is inbred). Lines 6 and R− (red arrows) were chosen to be as inbred and as genetically distant as possible. (B) Reciprocal cross between the two selected lines. In case of genomic imprinting, embryos are preferentially expressing one of their parents’ allele (this figure shows a case of paternally expressed gene). Detection of such event is possible with polymorphisms differentiating the lines.
Parental genomic DNA was extracted from blood samples through a high-salt extraction method (39).
Sequencing
Chicken data
RNA sequencing
Libraries with a mean insert size of 200 bp were prepared following Illumina instruction for RNA-Seq analysis, by selecting polyA+ fragments (TruSeq RNA Sample Prep Kit) from each sample. Samples were tagged to allow subsequent identification, amplified by polymerase chain reaction (PCR) and quantified by quantitative PCR (Agilent QPCR Library Quantification Kit) and then sequenced (paired ends, 100 bp) in triplicate on an Illumina HiSeq 2000 sequencer (Illumina, TruSeq PE Cluster Kit v3, cBot and TruSeq SBS Kit v3) by randomizing their position in six different sequencing lanes.
DNA sequencing
Parental DNA was sequenced on four lanes of Illumina HiSeq 2000 to allow the detection of SNPs discriminating the two lines. Library preparation (mean insert size of 300 bp), DNA quantification and sequencing (paired ends, 100 bp) were performed according to the manufacturer instructions (TruSeq DNA Sample Prep Kit Illumina, Agilent QPCR Library Quantification Kit, TruSeq PE Cluster Kit v3 cBot TruSeq SBS Kit v3).
Mouse data
RNA-seq sequences were collected from a previous study (40). They correspond to liver mRNA from 18 F1 males and females DBA/2J and C57BL/6J reciprocally crossed mice (two pools of nine mice). These crosses are termed as DXB and BXD in the article.
Computational analysis
When not specified, analyses were made with homemade Perl, Python and R scripts.
Genomic sequences analyses
Reads from DNA sequencing of parents were aligned on the last assembly of chicken genome (Galgal4) using the bwa program version 0.6.1, option aln (41).
Identification of SNP discriminating parents from both lines
Allelic counts for each base position on the genome were determined. These counts allowed selecting only loci discriminating parents, i.e. positions where both parents from each line were homozygous for distinct alleles.
The functional consequence of each SNP in each transcript was predicted using the Ensembl Variant Effect Predictor version 71 (42).
Transcriptome characterization
Chicken data
Tophat 2.0.5 software (43) was used to align transcript sequences from the F1 embryos on Galgal4 chicken assembly. Reads were first mapped allowing multiple alignments, with the intron length parameter set between 3 and 25 000bp to limit the number of false positive while discovering most junctions, the deletion length was limited to 1 instead of 3, and the mate inner distance in our case corresponded to 200. The option read-realign-edit-dist was set to 0 to map every read in all the Tophat mapping steps to get the best possible alignment. Finally, a micro exon search was applied. Other alignment options were set as default.
To remove potential PCR reads duplicates from the analysis, the program samtools rmdup (41) was run on each individual after alignment. Afterward, reads with potential multiple locations were discarded.
Samtools flagstat command (41) was used to generate statistics on aligned reads, notably the total number of reads mapped after applying our criteria.
To characterize the general level of expression in all the embryos, cufflinks software 2.0.0 was used on Galgal4.72 GTF file to quantify expression on known genes (44).
Mouse data
Sequencing reads were aligned with Tophat 2.0.5 on an artificial reference genome where all SNPs known between DBA/2J and C57BL/6J (45) were converted to ‘N’ to prevent the preferential mapping of the reference allele. In order not to affect to a large extend the alignment efficiency, no penalty was set for reads mapping on N in this new reference. A maximum of three mismatches was allowed for a read to be kept. Finally, a filter was applied to keep only reads uniquely mapped to the reference genome.
Identification of SNP with biased expression
Transcripts alignments were merged by parental cross for further analysis. Allelic counts allowed to select SNPs represented with at least 10 reads in each cross, with an inverted allelic ratio between both crosses (Fold Change ≥2.5).
To test the expression bias, a Fisher Exact Test was made on every transcribed loci and the significance threshold was fixed at False Discovery Rate (FDR) < 0.05 using the R package ‘qvalue’ (46).
Validation assays
Pyrosequencing
Twenty-three markers were tested on a Qiagen PyroMark Q24 sequencer. Primers were designed using PyroMark Assay Design software (Supplementary Table S2). PCR samples were produced using PyroMark PCR Kit (Qiagen). Runs were analyzed by PyroMark Q24 1.0.10 software with default analysis parameters.
Droplet Digital PCR
In total, three markers were tested on Bio-Rad QX100™ Droplet Digital PCR (ddPCR) System machine. Primers and probes were ordered at Sigma (Supplementary Table S3).
RESULTS
Constitution of an informative device
To detect genomic imprinting for a given gene, it is required that we are able to discriminate the parental origin of its expressed allele in the F1 embryos. The principal purpose of the lines' choice was thus to maximize the chances to identify from which parent the expressed allele had been transmitted to the embryos. To select parents as inbred (to reduce intra-population polymorphism) and as genetically distant (to maximize inter-population polymorphism) as possible, 3–12 animals from 10 chicken populations were genotyped on an Illumina 57K SNP array (35). Line R− and Line 6 were the two that fitted the best these criteria (Figure 1A) and were reciprocally crossed: one male from Line 6 was mated with a female from Line R− and one R− male was crossed with one female from Line 6 (Figure 1B). We selected 12 embryos from each reciprocal cross, with balanced ratio of six males and six females from each cross.
To detect SNPs discriminating the parents from both lines, genomic DNA from all four parents was sequenced. A total of 897 648 484 paired-ends reads (100 bp) were produced from an Illumina HiSeq 2000 sequencer and 813 407 461 (91%) aligned to the Galgal4 reference genome, which was thus covered at 98%, with a mean depth of 20×.
We found 2 298 622 SNPs discriminating the parents from both lines on genomic DNA, amounting to 2.1 ± 0.7 SNP/kb of sequence. When considering the number of annotated genes with at least one discriminating SNP, we found that 12 303 genes were represented. It corresponds to 72% of chicken annotated genes (Supplementary Table S4). Among the observed discriminating SNPs, 54.9% were localized in intergenic regions (Supplementary Figure S1), and 27.4% of the SNP detected in the coding region were nonsynonymous, which is similar to results previously obtained in chicken (47,48).
To have a better idea of their density in the potentially expressed regions, SNPs within exons were considered, that is 55 016 SNPs (i.e. 2% of detected SNPs). It corresponds to a density of 1.3 SNP/kb of coding regions, which is sufficient to have a reasonable screening of the exome. Nevertheless, we used the total set of available discriminating SNPs in the study, to include nonannotated regions in the analysis.
The fraction of these SNPs that will be observed in the RNA-Seq data represents the number of opportunities to link the allelic imbalance of transcripts detected in embryos with their parental origin.
Transcriptome characterization
To link the parental origin of the alleles to their expression pattern in embryos, we performed RNA-Seq on 12 embryos from each cross. We obtained reliable sequencing results from 20 embryos (SRA accession number SRP033603), 11 from Cross 1 (5 females and 6 males) and 9 from Cross 2 (4 females and 5 males); the other 4 were discarded because of sequencing tags problems.
From those 20 embryos, 1.33 × 109 and 6.07 × 108 sequences were produced from Cross 1 and Cross 2 individuals, respectively (200 Gb of sequence data altogether), corresponding on average to 121.1 million reads and 67.4 million reads for each individual in Cross 1 and Cross 2, respectively.
After alignment to the chicken assembly, removing of PCR duplicates and allowing only one possible hit, 74% of reads were mapped to the reference genome, which corresponds to 42% coverage of the genome (39% for Cross 1 and 35% for Cross 2, Supplementary Figure S2). We set a minimal depth of 10 reads to consider a region as covered, to avoid background of low expressed regions.
Regarding the general expression in chicken embryo, a minimal FPKM of 1 was expected (one fragment per kilobase per million reads) to call a gene as ‘expressed’. Among 17 108 genes already described in chicken (available from galgal4.72 GTF file), 11 416 genes, i.e. 67% of them, had a FPKM superior or equal to 1 in our data set.
Filters—bias in expression
RNA sequences were filtered to detect allelic imbalance expression at loci discriminating parents from both lines. Around 300 million bases were counted as expressed in both crosses, meaning that at least one read was observed at this position in each cross. To avoid bias in expression linked to a lack of depth, a threshold of 10 reads had to be reached in each cross (which corresponds to a minimal depth of 20 in total), leading to 123 532 053 positions, i.e. 40% of base positions expressed in both crosses.
Among these loci, 5 388 148 were polymorphic, and therefore potentially informative to test allelic imbalance. When crossing these expressed positions to discriminating SNPs (201 209 positions), we found that 7731 of 11 416 expressed genes (68%) were informative, i.e. containing at least one SNP discriminating parents of embryos in both crosses (Supplementary Table S4). A Fisher Exact Test was applied on these 5 million SNPs to test if the expression profile was different between Cross 1 and Cross 2. At a 5% FDR threshold, we observed 117 740 loci for which expression profiles were significantly different between Cross 1 and Cross 2, including positions with an allelic bias in only one of the crosses. Only positions where the allelic ratio was inverted between both crosses were potentially candidates, i.e. the minor allele in one cross is overrepresented in the reciprocal cross. Because the study was made on the whole embryo (a mixture of different tissues) a complete extinction of one allele was not necessarily expected, and we set a minimal fold change of 2.5 for this filter. A total of 4904 SNPs met these criteria. They could either result from polymorphisms in parental sequences or from an effective bias in expression. A further filtering was done removing the SNPs that, even if significant, were heterozygous in the parents and therefore not informative to trace back the allele parental origin. For that reason, linking these candidate SNPs to the parental origin of the allele was only possible at loci discriminating parents from both crosses. To do so, we crossed filtered data with the 2 298 622 discriminating SNPs detected in parallel on parents' genomic DNA. We checked that the detected allelic imbalance was similar for most of the pooled embryos in each cross, to avoid any bias due to a high monoallelic expression in few embryos (never observed in our candidates). At the end, we ended up with 79 potentially imprinted SNPs. These SNPs were mostly located in introns (51% of them); the others were upstream or downstream of annotated genes (31%), exons (6%), 3′-UTR (4%) or intergenic regions (8%) (Figure 2, Table 1). This distribution is similar to that of discriminating SNPs in sequences declared as expressed in both crosses, where 11% of these SNPs are located in known annotated transcripts (exons and UTR regions). This attribution was based on the known chicken annotation (Ensembl release 72, http://www.ensembl.org/Gallus_gallus/Info/Index). We considered expressed intergenic regions as unannotated regions.
Figure 2.

Genomic contexts of the candidate loci on the chicken genome.
Table 1.
Candidate positions, allelic counts and statistical results
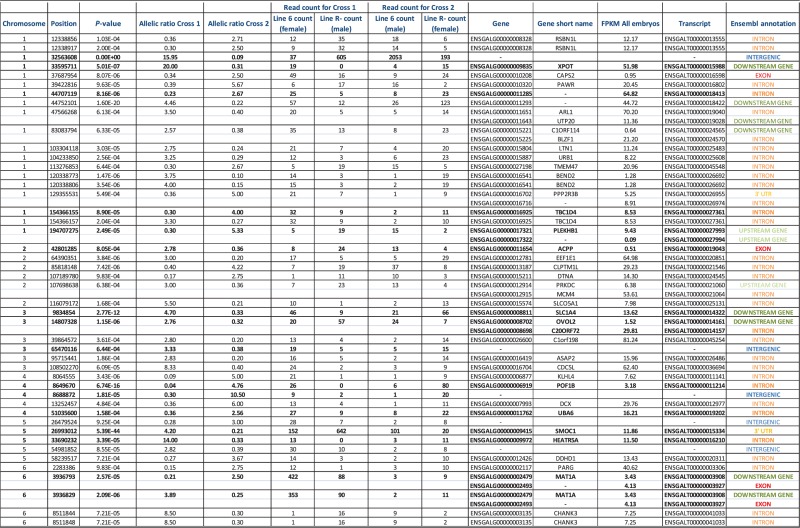 |
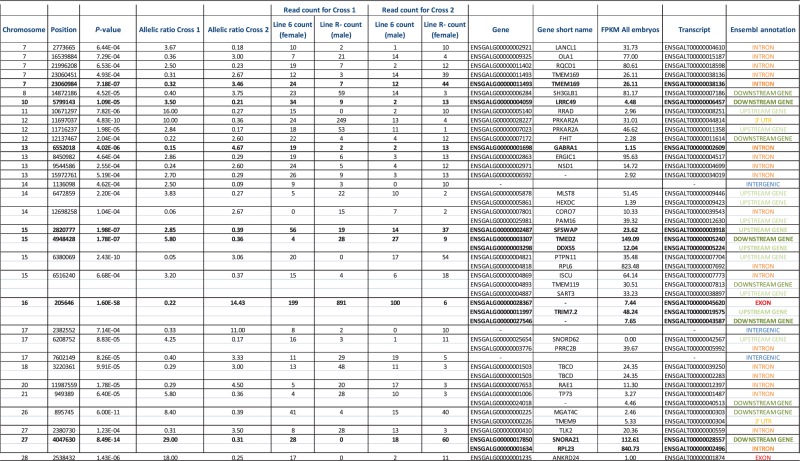 |
In bold, positions tested by pyrosequencing.
Allelic ratios = allele 1 counts/allele 2 counts, for each cross.
Validation assays
We tested 23 candidate SNPs to confirm the Illumina HiSeq results with pyrosequencing as an independent validation method (49,50). A total of 16 embryos (8 per cross) were tested. Tested candidates were chosen to get a panel of different situations highlighted after sequences analysis: intronic, exonic, regulatory regions and unannotated regions were looked at. Furthermore, candidates with different depth profiles were tested. To select the positions to be tested, we preferentially chose loci where SNPs surrounding the candidate position had an allelic profile similarly unbalanced: taking into account these additional positions—even if not passing our filters due to a lack of depth or an allelic ratio <2.5—ensure to avoid artifactual SNPs.
First, to avoid any bias due to PCR amplification of an allele or to genomic duplication in one cross, we checked that the pattern obtained by pyrosequencing on genomic DNA from the embryos truly corresponds to a heterozygous profile. Then we quantified the allelic ratio observed on cDNA.
Unfortunately, while confirming the heterozygous status of embryos in each case, we could not observe on cDNA any allelic imbalance corresponding to an imprinting profile (Figure 3).
Figure 3.

Comparison of results on a candidate gene obtained through HiSeq, droplet digital PCR and pyrosequencing. Colors are differentiating both alleles in sequencing results and ddPCR. (A) Hiseq counts of both alleles at the candidate locus (A allele in green, G allele in blue). (B) Pyrosequencing results from one sample of each cross (chosen as representative of average results). Analyses were made on the reverse strand. Allele A is overexpressed in both crosses (left: Cross 1 results from one embryo's cDNA, right: Cross 2 results from one embryo's cDNA).The highlighted peaks correspond to the SNP where the relative proportion is quantified. (C) Droplet digital PCR on the candidate locus from eight samples of each cross (A allele in green, G allele in blue).
Three markers were also tested by droplet digital PCR as another complementary quantitative approach. This confirmed the pyrosequencing results, further invalidating our HiSeq results (Figure 3).
Method validation with RNA sequencing data from mouse
As parental genomic imprinting is thought to be absent in chicken, we used as ‘positive control’ the mouse data set produced by Lagarrigue and collaborators (40) to detect imprinted genes in mouse liver, to validate the approach we proposed on chicken. In the mouse study, a reciprocal cross was made between DBA/2J (further called D) and C57BL/6J (further called B) mouse strains and RNA was sequenced from liver samples of 18 F1 mice.
Out of 285 166 513 reads generated by RNA-seq in both mouse crosses, 145 412 592 reads (51%) were mapped uniquely on an artificial reference genome where SNPs between both mouse strains were replaced by ‘N’. It corresponds to 74 733 541 reads for one cross (DXB) and 70 679 051 reads for the reciprocal cross (BXD).
These sequences were then filtered for bias in expression following the same pipeline as for chicken embryos. At a 5% FDR, we found 20 candidate SNPs with an inverted ratio between both crosses (minimum Fold Change of 2.5, Table 2). Of these, five are located in three genes known to be imprinted in mouse: H13 (33), Peg13 (51), both detected in the original study, and Snurf (52), not revealed by the first analysis. A fourth gene, Sgce, also detected by Lagarrigue et al., was identified when less stringent conditions on sequencing depth threshold were applied (i.e. when accepting nine reads in each cross).
Table 2.
Candidate position obtained from the mouse data set
| Chromosome | Position | Gene short name | Localization | Imprinting statusa | P-value Fisher exact test | Allelic ratio Cross 1 | Allelic ratio Cross 2 |
|---|---|---|---|---|---|---|---|
| 1 | 24619956 | Not annotated | 1.55e-117 | 8.64 | 0.004 | ||
| 2 | 152530393 | H13 | Exon | Maternal | 1.25e-62 | 3.91 | 0.30 |
| 4 | 60672634 | Mup2 or Mup11 | Exon | Unknown | 1.06e-26 | 792 | 0.31 |
| 4 | 142807839 | Not annotated | 2.70e-05 | 3.83 | 0.34 | ||
| 4 | 144508797 | Dhrs3 or AK147071 | Intron | Unknown | 4.44e-07 | 2.82 | 0.07 |
| 4 | 61746350 | Mup3 | Intron | Unknown | 1.22e-14 | 0.03 | 4 |
| 5 | 87427181 | Not annotated | 3.94e-26 | 0.31 | 103 | ||
| 7 | 31513450 | Gapdhs or Tmem147 | Intron | Unknown | 6.15e-15 | 19 | 0.13 |
| 7 | 67133548 | Snurf | Exon | Paternal | 5.64e-32 | 85 | 0.03 |
| 7 | 67131688 | Snurf | Exon | Paternal | 3.36e-40 | 0.01 | 47 |
| 11 | 83318343 | Taf15 | Exon | Unknown | 4.04e-08 | 10.5 | 0.37 |
| 11 | 98911391 | Igfbp4 | Intron | Unknown | 1.75e-55 | 5.11 | 0.01 |
| 11 | 98911393 | Igfbp4 | Intron | Unknown | 5.69e-73 | 0.12 | 90 |
| 12 | 105186516 | Serpina1e | Intron | Unknown | 1.86e-13 | 0.05 | 14 |
| 14 | 22266157 | Adk | Intron | Unknown | 2.17e-05 | 0.07 | 2.8 |
| 14 | 30847794 | Chdh | intron | Unknown | 1.33e-13 | 0.02 | 10 |
| 15 | 72637471 | Peg13 | Exon | Paternal | 1.77e-09 | 0.06 | 18 |
| 15 | 72640049 | Peg13 | Exon | Paternal | 1.11e-21 | 0.02 | 18 |
| 17 | 35534271 | Bat1a | Intron | Unknown | 4.63e-12 | 0.37 | 4.1 |
| 19 | 46726575 | D19Wsu16 or Opal1 | Intron | Unknown | 3.33e-05 | 0.09 | 3.4 |
aKnown imprinting status in mouse in the literature.
In bold, genes known to be imprinted in the literature.
Allelic ratios = allele 1 counts/allele 2 counts, for each cross.
These results confirm the efficiency of our pipeline to detect genomic imprinting from RNA-Seq data that allowed not only confirming three genes identified by Lagarrigue et al. but also demonstrating the presence of another imprinted gene, Snurf, which had not been detected in their study. The newly discovered loci have to be further analyzed.
DISCUSSION
This work represents, to our knowledge, the first transcriptome-wide investigation of genomic imprinting in birds. Previous studies in chicken were made on targeted genes, known to be imprinted in mammals, and they concluded to the nonexistence of this phenomenon in birds (15–18,20,53). To get free of any a priori, investigations should not be restricted to targeted loci but consider every single chicken gene. RNA sequencing technology is an approach meeting these expectations (54,55). Transcriptome sequencing has already been used with this intention in other species, especially in mouse (32,33,54). It not only allowed the confirmation of most mammalian imprinted genes but also the discovery of putative new ones.
We thus set up an experimental cross designed for the search of parental genomic imprinting in chicken, by using RNA-Seq on whole embryos from reciprocal crosses between chicken lines, as inbred and genetically distant as possible. Our results identified 79 potentially imprinted SNPs, mainly located in introns from distinct genes, not shown to be imprinted when analyzing their coding portion. It has been demonstrated that imprinted retrogenes could be hosted in introns of biallelically expressed genes in mouse (56), leading us to consider these loci as possibly interesting. But in the presence of genes truly imprinted, one could expect a higher number of candidate SNPs detected in known coding regions, and these results, with a large proportion of SNPs outside the coding regions, strengthened the need of experimental validations of these results.
These positions out of the coding sequence raise the question of the functional significance of the transcribed portion of the genome. There is a strong debate in the literature about the proportion of the genome that is ‘functional’ (57–60), following the ENCODE results, which assigned biochemical functions to 80.4% of the human genome, for which the cumulative coverage is 62.1% for processed transcripts (61). In our analysis, only ∼40% of the genome is considered to be transcribed, with a minimal coverage of 10 reads in the pool of all 20 embryos data at a single-base scale. This discrepancy may be partly explained by our selection of only polyadenylated mRNA. Nevertheless, the transcribed portion of the genome far exceeds its exonic subset, a more and more admitted characteristic of the genome (62,63).
Before validation, we ensured that candidate SNPs were not declared biased because of only one highly expressed embryo (Supplementary Figure S3). None of the candidate loci tested through validation experiments was confirmed as imprinted. These candidates being probably false positives, we thus validated our methodology by reanalyzing a mouse data set, as a ‘positive control’, where imprinting should be detected. By confirming the three previously identified imprinted genes [H13, Peg13 and Sgce (40)], and by discovering one more in these data, Snurf, previously known to be imprinted (52), we confirmed the reliability of the method. These four genes found imprinted in mouse are a severe underestimate of the total set of genes known to be imprinted in this species. This low number of genes is discussed in the original study by Lagarrigue and coworkers (40), notably highlighting the fact that only a fraction of imprinted genes are expressed in a given tissue at a precise developmental stage. Their study was the first, to our knowledge, to examine the parent-of-origin specific gene expression in adult liver in mouse, and there may be few imprinted genes in this tissue. The amount of data is probably the main reason for this low number of detected genes. Our chicken data set contains 10 times more sequences aligned and analyzed than the mouse data set. Considering that the mouse has a slightly higher number of annotated genes, expression can be better estimated in our chicken RNA-Seq sequences. With a higher sequencing depth, more genes could potentially have been detected. Nevertheless, two analysis methods from the same data set lead to the detection of imprinted genes in mouse. The differences between the results obtained here (four genes) and in the previous analysis (three genes) from the same data have two main origins: first, the minimal depth was originally set to 10 for each allele, which discarded Snurf, almost totally imprinted, as a candidate locus. Moreover, the original study did not take into account the noncoding region (explaining the discovery of only exonic imprinted SNPs in the first analysis). The biological reality of the newly discovered loci has to be determined, but this analysis confirmed that imprinting, when existing, is detected with our method. This validation is reinforced by the fact that we analyzed 200 Gb of chicken sequence (versus 3 Gb for the mouse study), leading to the assumption that we should have observed significant results if imprinting did exist in chicken. On the other hand, the use of whole embryos in our study (versus only liver samples in mouse) might have affected the final results. But our choice was, for the same amount of data, to reach a sufficient depth to reliably discover multitissue parental imprinting, without having to a priori select one single tissue. The choice of the embryo was determined by the high number of imprinted genes already known to be imprinted in the embryo, compared with single adult tissues (64). Moreover, others studies conducted on the whole embryo allowed the detection of imprinting in mouse (65). If imprinting does exist in chicken at this developmental stage, but is observed only in one tissue, or if a locus is paternally expressed in some tissues but maternally expressed in others (66), our device might have not allowed to detect such cases. While such profiles were already observed, most genes are imprinted in multiple tissue types (67). Nevertheless, it would be interesting to confirm the absence of imprinting in chicken by studying other tissues, extraembryonic membranes in particular: most of the known imprinted genes in mammals are expressed in the placenta (68,69), notably regulating nutritional resources to the embryo during development (70,71).
Another question concerning our model's choice was the embryonic stage of development. Stage 26 is a developmental stage from which a sufficient quantity of RNA can be extracted for RNA-Seq and validation experiments, and the expression of imprinted genes has already been described at the equivalent stage in the mouse embryo (64). It would also have been interesting to check the imprinting status of genes expressed in adult brain: by applying the kinship theory to a social context, the existence of imprinting would be possible in a species showing maternal care, as previously proposed (72). Incidentally, several genes have been shown to be imprinted in the mouse brain, notably related to social behavior (73). But the best bird lines to study would have been in this case from species with strong parental care behavior, and inbred chicken lines, despite their suitability for the RNA-Seq strategy used, are probably not the best model in this area.
Besides practical reasons as availability of inbred lines and of a reliable genome assembly, the choice of the chicken for this study has been dictated by several arguments: parent-of-origin QTLs have been detected in this species (27), and chicken is an example of farm animals used in crossbreeding, where genomic imprinting could be important for the utilization of reciprocal effects. It is also an important animal model in developmental biology and phylogenetics, and is used as a model for other avian species, especially in genetics and genomics (47).
A last comment about the device can be made about the sequenced transcripts: we limited the screening to polyA RNA, and thus we could not conclude on the potential imprinted status of noncoding RNAs, some of which are also known to be imprinted in mammals (74).
With a reliable analysis method but no evidence of imprinting in our chicken data set, our results seem to confirm the absence of imprinting in chicken, at least in the whole embryo at embryonic day 4.5. This analysis is a first step in the genome-wide search of genomic imprinting in birds. The absence of this phenomenon will be definitely confirmed—taking into account the difficulty of claiming the absence of such a multifarious mechanism—only when other studies will complete this first one: other transcribed parts of the genome (such as non-coding RNA), other tissues at other developmental stages must be analyzed, as well as other chicken lines and, most importantly, other bird species. But given previous work on several candidate genes (15–21), and absence of differential methylated regions according to the parental allele in the chicken genome (75), it gives a strong argument, through a genome-wide study, in agreement with previous studies. This absence would be in accordance with the most shared hypothesis, the kinship theory, which leads to the assumption that imprinting does not exist in oviparous species (10). Another theory concerning the evolution of genomic imprinting mechanisms is the ‘host defence’ hypothesis, assuming that imprinting appeared consequently to mechanisms aimed at silencing foreign DNA elements inserted into the genome, and evolved in parallel with accumulation of repeats in certain genomic regions (12,70,76,77). This is again not in favor of the existence of this phenomenon in chicken, a species with a low level of repeats and active transposable elements, compared with mammals (78).
The absence of this phenomenon would lead to alternative explanations of reciprocal effects. These effects represent ∼20% of the phenotypic variability of body weight in broiler and of egg viability in layers (79), and one cause could have been genomic imprinting. But other mechanisms can lead to such effects, as direct maternal effects, sex-linked genes or mitochondrial DNA transmission. Regarding QTLs detected as showing parent-of-origin effects, most of them can result from biases due to the animal device, as previously pointed out (25,26,80). Rowe and coworkers, in a paper describing a maternally expressed QTL obtained in a pedigree avoiding such biases, comment their results by stating they ‘have insufficient evidence to confirm that these are truly imprinted effects’. Taking into account the historical discussion about the absence of genomic imprinting in chicken and our results, it is possible that no QTL truly showing parent-of-origin effect does exist in chicken.
To understand why these false imprinted candidates were detected, we tried to investigate the possible biases and issues encountered during the analysis. This is not a unique situation in RNA sequencing data analysis, as some of the results from imprinting analysis through transcriptome sequencing had also been contested in mouse (81,82), highlighting many possible biases inherent to RNA sequencing data, from experiments to analyses. In particular, some alleles can be underrepresented through library preparation, several SNPs can appear to be artifactual and read alignment can be biased due to a better mapping of the reference allele (83). Because we were looking for inverted allelic ratios between reciprocal crosses in our study, this last issue could not explain our results because regions where the reference allele was preferentially mapped were avoided. We considered that the possible problem of artifactual SNPs was also absent from our data because we crossed the SNPs observed from the RNA-Seq data with the polymorphisms detected between the parents by genomic sequencing. Furthermore, all the candidate SNPs analyzed through other methods, as Sanger sequencing (data not shown) or pyrosequencing, confirmed the heterozygous status of the embryo's DNA. The possibility of underrepresentation of an allele due to the library preparation remains, even if less likely due to the use of reciprocal crosses: to generate such a bias, an allele should be ‘randomly’ retained in one cross, while the alternative allele should be ‘randomly’ kept in the other cross during the library preparations.
Likewise, there is a possible dependence between nucleotide frequency at a SNP and its position on sequence reads (84). This bias can originate from the sequencing platform or from a positional bias of the reads across the transcripts. In any case, correcting this bias can improve expression estimates (85). All 79 positions were studied for this putative bias, but only four of them do have a median position among reads in the 20% extremities (Figure 4). The sequencing depth may be another cause of the median deviation from the theoretical value of 50 (average position on the read). While location of a SNP on a read may randomly vary, lower expression level increases the probability of the median position of the SNP differing from this medium position. By removing candidate SNPs being in extremities of reads, we either remove low expressed loci or effectively biased loci. Excluding these four candidates is thus a conservative choice. But the unconfirmed allelic imbalance of the 75 left ones cannot be explained this way. Moreover, if we consider the proportion of forward and reverse reads for each candidate, it appears that no overrepresentation of one direction of sequencing was detected (no candidate SNP with a proportion of forward read <20% or >80%). This potential bias was thus not the source of false positive in our case.
Figure 4.

Candidate SNP positions among sequenced reads. Boxplot of SNP localization among reads for each candidate ordered by median. Orange color represents candidates with a median position in the 20% extremities of reads.
If the discovery of false-positive SNPs does not directly come from alignment or position bias, it can also be linked with analysis criteria. One of the main issues in such studies is the sequencing depth threshold to declare allelic imbalance. Studies made until now used different threshold in their analyses (32,33,86). This issue was underlined in many allelic imbalance studies using RNA sequences data, but until now, no consensus was made on a reliable threshold (32,86,87). We set ours to a minimum read depth of 10 in each cross, which corresponds to a minimal coverage of 20 reads for each locus considered in the analysis. This threshold was appropriate to highlight imprinted loci in the mouse data set. Thus, taking into account the lower sequencing depth in the mouse study compared with the chicken one, we should have detected imprinted loci in chicken if present: even a higher threshold would have brought to light such loci from our data set.
We thus looked at the candidates with regard to the general distribution of sequencing depth. Most of them have a low expression profile (Figure 5), which could lead to false-positive candidates. We can raise the hypothesis that at some low sequencing depth, i.e. at low expression levels, genes might express one allele preferentially, or that it is not technically possible to properly amplify both alleles when the gene is expressed under a certain limit. Under these conditions, the principal null hypothesis used in this study could be modified: instead of a 50:50 ratio between both alleles in transcripts of heterozygous embryos, the null hypothesis would depend on the expression rate of the genes. This hypothesis is in accordance with previous studies, highlighting the technical issues leading to allelic imbalance (86,88,89).
Figure 5.

Sequencing depth distribution for all SNPs (top), with classes containing candidate SNPs in gray, and for candidate SNPs (bottom), with tested SNPs in black.
By looking to possible biases in our analysis, we tried to understand why no genomic imprinting could be observed on the candidate SNPs that had been highlighted. This leads to several considerations: firstly, results' validation is required through a reliable quantitative method, as we did mainly by pyrosequencing; secondly, possible experimental and analytical biases should be considered in the final results interpretation and care should be taken regarding general sequencing depth, possible alignment biases and threshold choices, as previously underlined (82,90).
This study was designed to detect parental genomic imprinting in birds, with chicken as a model species. Our results show that, at least at this developmental stage, there is no genomic region imprinted at the whole embryo level. This does not exclude specific events occurring in a small subset of tissues or at different developmental stages that would most probably not have been detectable in the combination of tissues used in this work. Nevertheless, this study proposes a performing method to investigate the existence of genomic imprinting at a genome scale, without a priori choice of targets genes known to be imprinted in mammals. Our results support the fact that, while the evolution of imprinting has occurred in a convergent manner for several clades, from plants to mammals, it is probably missing in birds.
ACCESSION NUMBERS
NCBI_SRA accessions number: SRP033603.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Région Midi-Pyrénées (http://www.midipyrenees.fr/) and Animal Genetics Division (INRA, http://www.ga.inra.fr/en/) (to L.F.); French ‘Agence Nationale de la Recherche’ EpiBird grant [ANR-009-GENM-004, http://www.agence-nationale-recherche.fr/]. Funding for open access charge: INRA, UMR444 Laboratoire de Génétique Cellulaire, Castanet-Tolosan, F-31326, France.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for support by the PEAT team in managing animals. We thank GeT-PlaGe Genotoul platform for performing the HiSeq sequencing, and the Bio-Rad team for their help in the ddPCR analyses. We are grateful to Déborah Bourc'his, Jérôme Cavaillé, Hervé Acloque and Julie Demars for informal discussions throughout this work.
REFERENCES
- 1.Das R, Anderson N, Koran MI, Weidman JR, Mikkelsen TS, Kamal M, Murphy SK, Linblad-Toh K, Greally JM, Jirtle RL. Convergent and divergent evolution of genomic imprinting in the marsupial Monodelphis domestica. BMC Genomics. 2012;13:394. doi: 10.1186/1471-2164-13-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.da Rocha ST, Ferguson-Smith AC. Genomic imprinting. Curr. Biol. 2004;14:R646–R649. doi: 10.1016/j.cub.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Holman L, Kokko H. The evolution of genomic imprinting: costs, benefits and long-term consequences. Biol. Rev. Camb. Philos. Soc. 2013 doi: 10.1111/brv.12069. (doi: 10.1111/brv.12069; epub ahead of print, October 28, 2013) [DOI] [PubMed] [Google Scholar]
- 4.Haig D, Graham C. Genomic imprinting and the strange case of the insulin-like growth factor II receptor. Cell. 1991;64:1045–1046. doi: 10.1016/0092-8674(91)90256-x. [DOI] [PubMed] [Google Scholar]
- 5.Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- 6.Haig D. Parental antagonism, relatedness asymmetries, and genomic imprinting. Proc. Biol. Sci. 1997;264:1657–1662. doi: 10.1098/rspb.1997.0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haig D. The kinship theory of genomic imprinting. Ann. Rev. Ecol. Evol. Syst. 2000;31:9–32. [Google Scholar]
- 8.Ashbrook DG, Hager R. Empirical testing of hypotheses about the evolution of genomic imprinting in mammals. Front. Neuroanat. 2013;7:6. doi: 10.3389/fnana.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haig D. Coadaptation and conflict, misconception and muddle, in the evolution of genomic imprinting. Heredity (Edinb) 2013 doi: 10.1038/hdy.2013.97. (doi: 10.1038/hdy.2013.97; epub ahead of print, October 16, 2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwasa Y. The conflict theory of genomic imprinting: how much can be explained? Current Topics in Developmental Biology. 1998;40:255–293. doi: 10.1016/s0070-2153(08)60369-5. [DOI] [PubMed] [Google Scholar]
- 11.Feil R, Berger F. Convergent evolution of genomic imprinting in plants and mammals. Trends Genet. 2007;23:192–199. doi: 10.1016/j.tig.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Renfree MB, Suzuki S, Kaneko-Ishino T. The origin and evolution of genomic imprinting and viviparity in mammals. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013;368:20120151. doi: 10.1098/rstb.2012.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giannoukakis N, Deal C, Paquette J, Goodyer CG, Polychronakos C. Parental genomic imprinting of the human IGF2 gene. Nat. Genet. 1993;4:98–101. doi: 10.1038/ng0593-98. [DOI] [PubMed] [Google Scholar]
- 14.Koski LB, Sasaki E, Roberts RD, Gibson J, Etches RJ. Monoalleleic transcription of the insulin-like growth factor-II gene (Igf2) in chick embryos. Mol. Reprod. Dev. 2000;56:345–352. doi: 10.1002/1098-2795(200007)56:3<345::AID-MRD3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 15.Nolan CM, Killian JK, Petitte JN, Jirtle RL. Imprint status of M6P/IGF2R and IGF2 in chickens. Dev. Genes Evol. 2001;211:179–183. doi: 10.1007/s004270000132. [DOI] [PubMed] [Google Scholar]
- 16.O'Neill MJ, Ingram RS, Vrana PB, Tilghman SM. Allelic expression of IGF2 in marsupials and birds. Dev. Genes Evol. 2000;210:18–20. doi: 10.1007/pl00008182. [DOI] [PubMed] [Google Scholar]
- 17.Wang G, Yan B, Deng X, Li C, Hu X, Li N. Insulin-like growth factor 2 as a candidate gene influencing growth and carcass traits and its bialleleic expression in chicken. Sci. China C Life Sci. 2005;48:187–194. doi: 10.1007/BF02879672. [DOI] [PubMed] [Google Scholar]
- 18.Yokomine T, Kuroiwa A, Tanaka K, Tsudzuki M, Matsuda Y, Sasaki H. Sequence polymorphisms, allelic expression status and chromosome locations of the chicken IGF2 and MPR1 genes. Cytogenet. Genome Res. 2001;93:109–113. doi: 10.1159/000056960. [DOI] [PubMed] [Google Scholar]
- 19.Yokomine T, Shirohzu H, Purbowasito W, Toyoda A, Iwama H, Ikeo K, Hori T, Mizuno S, Tsudzuki M, Matsuda Y, et al. Structural and functional analysis of a 0.5-Mb chicken region orthologous to the imprinted mammalian Ascl2/Mash-Igf2-H19 region. Genome Res. 2005;15:154–165. doi: 10.1101/gr.2609605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin S, Han JY, Lee K. Cloning of avian Delta-like 1 homolog gene: the biallelic expression of Delta-like 1 homolog in avian species. Poult. Sci. 2010;89:948–955. doi: 10.3382/ps.2009-00572. [DOI] [PubMed] [Google Scholar]
- 21.Colosi DC, Martin D, More K, Lalande M. Genomic organization and allelic expression of UBE3A in chicken. Gene. 2006;383:93–98. doi: 10.1016/j.gene.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 22.Minvielle F, Kayang BB, Inoue-Murayama M, Miwa M, Vignal A, Gourichon D, Neau A, Monvoisin JL, Ito S. Microsatellite mapping of QTL affecting growth, feed consumption, egg production, tonic immobility and body temperature of Japanese quail. BMC Genomics. 2005;6:87. doi: 10.1186/1471-2164-6-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuiskula-Haavisto M, De Koning DJ, Honkatukia M, Schulman NF, Mâki-Tanila A, Vilkki J. Quantitative trait loci with parent-of-origin effects in chicken. Genet. Res. 2004;84:57–66. doi: 10.1017/s0016672304006950. [DOI] [PubMed] [Google Scholar]
- 24.Tuiskula-Haavisto M, Vilkki J. Parent-of-origin specific QTL—a possibility towards understanding reciprocal effects in chicken and the origin of imprinting. Cytogenet. Genome Res. 2007;117:305–312. doi: 10.1159/000103192. [DOI] [PubMed] [Google Scholar]
- 25.de Koning DJ, Bovenhuis H, van Arendonk JA. On the detection of imprinted quantitative trait loci in experimental crosses of outbred species. Genetics. 2002;161:931–938. doi: 10.1093/genetics/161.2.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sandor C, Georges M. On the detection of imprinted quantitative trait loci in line crosses: effect of linkage disequilibrium. Genetics. 2008;180:1167–1175. doi: 10.1534/genetics.108.092551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rowe SJ, Pong-Wong R, Haley CS, Knott SA, de Koning DJ. Detecting parent of origin and dominant QTL in a two-generation commercial poultry pedigree using variance component methodology. Genet. Sel. Evol. 2009;41:6. doi: 10.1186/1297-9686-41-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokomine T, Hata K, Tsudzuki M, Sasaki H. Evolution of the vertebrate DNMT3 gene family: a possible link between existence of DNMT3L and genomic imprinting. Cytogenet. Genome Res. 2006;113:75–80. doi: 10.1159/000090817. [DOI] [PubMed] [Google Scholar]
- 29.Lewis A, Mitsuya K, Umlauf D, Smith P, Dean W, Walter J, Higgins M, Feil R, Reik W. Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat. Genet. 2004;36:1291–1295. doi: 10.1038/ng1468. [DOI] [PubMed] [Google Scholar]
- 30.Peters J, Williamson CM. Control of imprinting at the Gnas cluster. Epigenetics. 2007;2:207–213. doi: 10.4161/epi.2.4.5380. [DOI] [PubMed] [Google Scholar]
- 31.Frésard L, Morisson M, Brun JM, Collin A, Pain B, Minvielle F, Pitel F. Epigenetics and phenotypic variability: some interesting insights from birds. Genet. Sel. Evol. 2013;45:16. doi: 10.1186/1297-9686-45-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregg C, Zhang J, Weissbourd B, Luo S, Schroth GP, Haig D, Dulac C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science. 2010;329:643–648. doi: 10.1126/science.1190830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Soloway PD, Clark AG. A survey for novel imprinted genes in the mouse placenta by mRNA-seq. Genetics. 2011;189:109–122. doi: 10.1534/genetics.111.130088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Miller DC, Harman R, Antczak DF, Clark AG. Paternally expressed genes predominate in the placenta. Proc. Natl Acad. Sci. USA. 2013;110:10705–10710. doi: 10.1073/pnas.1308998110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groenen MA, Megens HJ, Zare Y, Warren WC, Hillier LW, Crooijmans RP, Vereijken A, Okimoto R, Muir WM, Cheng HH. The development and characterization of a 60K SNP chip for chicken. BMC Genomics. 2011;12:274. doi: 10.1186/1471-2164-12-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bumstead N, Barrow PA. Genetics of resistance to Salmonella typhimurium in newly hatched chicks. Br. Poult. Sci. 1988;29:521–29. doi: 10.1080/00071668808417078. [DOI] [PubMed] [Google Scholar]
- 38.Bordas A, Tixier-Boichard M, Merat P. Direct and correlated responses to divergent selection for residual food intake in Rhode Island Red laying hens. Br. Poult. Sci. 1992;33:741–754. doi: 10.1080/00071669208417515. [DOI] [PubMed] [Google Scholar]
- 39.Roussot O, Feve K, Plisson-Petit F, Pitel F, Faure JM, Beaumont C, Vignal A. AFLP linkage map of the Japanese quail Coturnix japonica. Genet. Sel. Evol. 2003;35:559–572. doi: 10.1186/1297-9686-35-6-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lagarrigue S, Martin LJ, Hormozdiari F, Roux PF, Pan C, van Nas A, Demeure O, Cantor R, Ghazalpour A, Eskin E, et al. Analysis of allele specific expression in mouse liver by RNA-seq: a comparison with “Cis”-eQTL identified using genetic linkage. Genetics. 2013;195:1157–1166. doi: 10.1534/genetics.113.153882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dabney A, Storey JD. qvalue: Q-value estimation for false discovery rate control. 2009 R package, v1.24.0 ed. http://CRAN.R-project.org/package=qvalue. [Google Scholar]
- 47.Burt DW, White SJ. Avian genomics in the 21st century. Cytogenet. Genome Res. 2007;117:6–13. doi: 10.1159/000103159. [DOI] [PubMed] [Google Scholar]
- 48.Kranis A, Gheyas AA, Boschiero C, Turner F, Yu L, Smith S, Talbot R, Pirani A, Brew F, Kaiser P, et al. Development of a high density 600K SNP genotyping array for chicken. BMC Genomics. 2013;14:59. doi: 10.1186/1471-2164-14-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun A, Ge J, Siffert W, Frey UH. Quantification of allele-specific G-protein beta3 subunit mRNA transcripts in different human cells and tissues by Pyrosequencing. Euro. J. Hum. Genet. 2005;13:361–369. doi: 10.1038/sj.ejhg.5201334. [DOI] [PubMed] [Google Scholar]
- 50.Wang H, Elbein SC. Detection of allelic imbalance in gene expression using pyrosequencing. Methods Mol. Biol. 2007;373:157–176. doi: 10.1385/1-59745-377-3:157. [DOI] [PubMed] [Google Scholar]
- 51.Smith RJ, Dean W, Konfortova G, Kelsey G. Identification of novel imprinted genes in a genome-wide screen for maternal methylation. Genome Res. 2003;13:558–569. doi: 10.1101/gr.781503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Le Meur E, Watrin F, Landers M, Sturny R, Lalande M, Muscatelli F. Dynamic developmental regulation of the large non-coding RNA associated with the mouse 7C imprinted chromosomal region. Dev. Biol. 2005;286:587–600. doi: 10.1016/j.ydbio.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 53.Yokomine T, Shirohzu H, Purbowasito W, Toyoda A, Iwama H, Ikeo K, Hori T, Mizuno S, Tsudzuki M, Matsuda Y, et al. Structural and functional analysis of a 0.5-Mb chicken region orthologous to the imprinted mammalian Ascl2/Mash2-Igf2-H19 region. Genome Res. 2005;15:154–165. doi: 10.1101/gr.2609605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Babak T, DeVeale B, Armour C, Raymond C, Cleary MA, van der Kooy D, Johnson JM, Lim LP. Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol. 2008;18:1735–1741. doi: 10.1016/j.cub.2008.09.044. [DOI] [PubMed] [Google Scholar]
- 55.Babak T. Identification of imprinted Loci by transcriptome sequencing. Methods Mol. Biol. 2012;925:79–88. doi: 10.1007/978-1-62703-011-3_6. [DOI] [PubMed] [Google Scholar]
- 56.Monk D, Arnaud P, Frost JM, Wood AJ, Cowley M, Martin-Trujillo A, Guillaumet-Adkins A, Iglesias Platas I, Camprubi C, Bourc'his D, et al. Human imprinted retrogenes exhibit non-canonical imprint chromatin signatures and reside in non-imprinted host genes. Nucleic Acids Res. 2011;39:4577–4586. doi: 10.1093/nar/gkq1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doolittle WF. Is junk DNA bunk? A critique of ENCODE. Proc. Natl Acad. Sci. USA. 2013;110:5294–5300. doi: 10.1073/pnas.1221376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E. On the immortality of television sets: “function” in the human genome according to the evolution-free gospel of ENCODE. Genome Biol. Evol. 2013;5:578–590. doi: 10.1093/gbe/evt028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niu DK, Jiang L. Can ENCODE tell us how much junk DNA we carry in our genome? Biochem. Biophys. Res. Commun. 2013;430:1340–1343. doi: 10.1016/j.bbrc.2012.12.074. [DOI] [PubMed] [Google Scholar]
- 60.Eddy SR. The ENCODE project: missteps overshadowing a success. Curr. Biol. 2013;23:R259–R261. doi: 10.1016/j.cub.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 61.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clark MB, Amaral PP, Schlesinger FJ, Dinger ME, Taft RJ, Rinn JL, Ponting CP, Stadler PF, Morris KV, Morillon A, et al. The reality of pervasive transcription. PLoS Biol. 2011;9:e1000625. doi: 10.1371/journal.pbio.1000625. discussion e1001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stamatoyannopoulos JA. What does our genome encode? Genome Res. 2012;22:1602–1611. doi: 10.1101/gr.146506.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schulz R, Woodfine K, Menheniott TR, Bourc'his D, Bestor T, Oakey RJ. WAMIDEX: a web atlas of murine genomic imprinting and differential expression. Epigenetics. 2008;3:89–96. doi: 10.4161/epi.3.2.5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulz R, Menheniott TR, Woodfine K, Wood AJ, Choi JD, Oakey RJ. Chromosome-wide identification of novel imprinted genes using microarrays and uniparental disomies. Nucleic Acids Res. 2006;34:e88. doi: 10.1093/nar/gkl461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Garfield AS, Cowley M, Smith FM, Moorwood K, Stewart-Cox JE, Gilroy K, Baker S, Xia J, Dalley JW, Hurst LD, et al. Distinct physiological and behavioural functions for parental alleles of imprinted Grb10. Nature. 2011;469:534–538. doi: 10.1038/nature09651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prickett AR, Oakey RJ. A survey of tissue-specific genomic imprinting in mammals. Mol. Genet. Genomics. 2012;287:621–630. doi: 10.1007/s00438-012-0708-6. [DOI] [PubMed] [Google Scholar]
- 68.Barbaux S, Gascoin-Lachambre G, Buffat C, Monnier P, Mondon F, Tonanny MB, Pinard A, Auer J, Bessieres B, Barlier A, et al. A genome-wide approach reveals novel imprinted genes expressed in the human placenta. Epigenetics. 2012;7:1079–1090. doi: 10.4161/epi.21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat. Rev. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- 70.Ferguson-Smith AC. Genomic imprinting: the emergence of an epigenetic paradigm. Nat. Rev. 2011;12 doi: 10.1038/nrg3032. [DOI] [PubMed] [Google Scholar]
- 71.Gutierrez-Marcos JF, Constancia M, Burton GJ. Maternal to offspring resource allocation in plants and mammals. Placenta. 2012;33(Suppl. 2):e3–e10. doi: 10.1016/j.placenta.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 72.Ubeda F, Gardner A. A model for genomic imprinting in the social brain: juveniles. Evolution. 2010;64:2587–2600. doi: 10.1111/j.1558-5646.2010.01015.x. [DOI] [PubMed] [Google Scholar]
- 73.Curley JP. Is there a genomically imprinted social brain? Bioessays. 2011;33:662–668. doi: 10.1002/bies.201100060. [DOI] [PubMed] [Google Scholar]
- 74.Peters J, Robson JE. Imprinted noncoding RNAs. Mamm. Genome. 2008;19:493–502. doi: 10.1007/s00335-008-9139-4. [DOI] [PubMed] [Google Scholar]
- 75.Li Q, Li N, Hu X, Li J, Du Z, Chen L, Yin G, Duan J, Zhang H, Zhao Y, et al. Genome-wide mapping of DNA methylation in chicken. PLoS One. 2011;6:e19428. doi: 10.1371/journal.pone.0019428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barlow DP. Methylation and imprinting: from host defense to gene regulation? Science. 1993;260:309–310. doi: 10.1126/science.8469984. [DOI] [PubMed] [Google Scholar]
- 77.Renfree MB, Hore TA, Shaw G, Marshall Graves JA, Pask AJ. Evolution of genomic imprinting: insights from marsupials and monotremes. Annu. Rev. Genomics Hum. Genet. 2009;10:241–262. doi: 10.1146/annurev-genom-082908-150026. [DOI] [PubMed] [Google Scholar]
- 78.Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MA, Delany ME, et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 79.Fairfull RW. In: Poultry Breeding and Genetics. Crawford RD, editor. Amsterdam: Elsevier Science Publishers; 1990. pp. 913–933. [Google Scholar]
- 80.Lawson HA, Cheverud JM, Wolf JB. Genomic imprinting and parent-of-origin effects on complex traits. Nat. Rev. Genet. 2013;14:609–617. doi: 10.1038/nrg3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kelsey G, Bartolomei MS. Imprinted Genes … and the Number Is? PLoS Genetics. 2012;8:e1002601. doi: 10.1371/journal.pgen.1002601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.DeVeale B, van der Kooy D, Babak T. Critical evaluation of imprinted gene expression by RNA-Seq: a new perspective. PLoS Genet. 2012;8:e1002600. doi: 10.1371/journal.pgen.1002600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Degner JF, Marioni JC, Pai AA, Pickrell JK, Nkadori E, Gilad Y, Pritchard JK. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2009;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hansen KD, Brenner SE, Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Res. 2010;38:e131. doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heap GA, Yang JH, Downes K, Healy BC, Hunt KA, Bockett N, Franke L, Dubois PC, Mein CA, Dobson RJ, et al. Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing. Hum. Mol. Genet. 2010;19:122–134. doi: 10.1093/hmg/ddp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, Clark AG. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One. 2008;3:e3839. doi: 10.1371/journal.pone.0003839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heinrich V, Stange J, Dickhaus T, Imkeller P, Kruger U, Bauer S, Mundlos S, Robinson PN, Hecht J, Krawitz PM. The allele distribution in next-generation sequencing data sets is accurately described as the result of a stochastic branching process. Nucleic Acids Res. 2012;40:2426–2431. doi: 10.1093/nar/gkr1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nothnagel M, Wolf A, Herrmann A, Szafranski K, Vater I, Brosch M, Huse K, Siebert R, Platzer M, Hampe J, et al. Statistical inference of allelic imbalance from transcriptome data. Hum. Mutat. 2011;32:98–106. doi: 10.1002/humu.21396. [DOI] [PubMed] [Google Scholar]
- 90.Coolon JD, Stevenson KR, McManus CJ, Graveley BR, Wittkopp PJ. Genomic imprinting absent in Drosophila melanogaster adult females. Cell Rep. 2012;2:69–75. doi: 10.1016/j.celrep.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.