


Abstract
DNA damage is tightly associated with various biological and pathological processes, such as aging and tumorigenesis. Although detection of DNA damage is attracting increasing attention, only a limited number of methods are available to quantify DNA lesions, and these techniques are tedious or only detect global DNA damage. In this study, we present a high-sensitivity long-run real-time PCR technique for DNA-damage quantification (LORD-Q) in both the mitochondrial and nuclear genome. While most conventional methods are of low-sensitivity or restricted to abundant mitochondrial DNA samples, we established a protocol that enables the accurate sequence-specific quantification of DNA damage in >3-kb probes for any mitochondrial or nuclear DNA sequence. In order to validate the sensitivity of this method, we compared LORD-Q with a previously published qPCR-based method and the standard single-cell gel electrophoresis assay, demonstrating a superior performance of LORD-Q. Exemplarily, we monitored induction of DNA damage and repair processes in human induced pluripotent stem cells and isogenic fibroblasts. Our results suggest that LORD-Q provides a sequence-specific and precise method to quantify DNA damage, thereby allowing the high-throughput assessment of DNA repair, genotoxicity screening and various other processes for a wide range of life science applications.
INTRODUCTION
Cells continuously experience multiple types of DNA modifications that occur spontaneously as byproducts of normal cellular metabolism resulting in oxidative lesions. Other endogenous sources comprise collapsing replication forks or spontaneous deamination of cytosine. DNA damage may also be generated by exogenous stimuli such as high-energy radiation or genotoxic chemicals. If proper recognition and repair fails, DNA modifications can cause mutations, which may in some cases contribute to or even trigger severe physiologic dysfunctions such as cancer and hereditary diseases. Thus, development of methods to track and sequence-specifically quantify DNA modifications can help to disentangle disease mechanisms underlying genetic alterations.
Most conventional methods detect DNA damage either in a global, sequence-independent manner, such as the single-cell gel electrophoresis (comet) assay and TUNEL staining, or are restricted to one or a few defined types of DNA lesions (1). Standard immunostaining protocols employ, e.g. the detection of thymine dimers or γH2AX foci as a marker for DNA double-strand breaks (2–4) (see Supplementary Table S1 for a list of frequently applied methods). Genotoxic insults cause a variety of different DNA modifications in living cells, i.e. pyrimidine dimers, oxidation products and single or double-strand breaks. Many conventional DNA-damage detection methods therefore miss a major part of the introduced lesions, while detecting only a defined type of modification. On the other hand, sequence-unspecific methods fail to identify predisposed loci for DNA damage or to quantify DNA damage selectively, e.g. in cancer-relevant genes.
Among the various naturally formed DNA alterations, several nucleotide modifications interfere with DNA-dependent DNA polymerase and inhibit DNA synthesis. For this reason, PCR-based approaches constitute a suitable technique for DNA-damage quantification, in which the extent of template amplification is inversely proportional to the lesion frequency within a given DNA sequence. Additional advantages of PCR-based methods are even more evident: first, they are sequence-specific, thereby allowing the identification of differential lesion incidence for selected genomic loci. Second, analyses can be carried out in standard microtiter formats employing commercially available reagents, enabling high-throughput genotoxicity or DNA-repair analyses, while minimizing labor and cost efforts per sample.
Despite these advantages, previously reported PCR-based DNA-damage assays are mostly quantitative (q)PCR measurements. Although these assays allow the detection of certain, yet mostly undefined nucleotide modifications, they need to quantify DNA concentrations prior and after the PCR using fluorescence intensity determination of DNA dyes or error-prone band densitometry (5–7). In addition, qPCR approaches need to match the linear range of PCR amplification, which is limited to a restricted number of cycles and depends both on the quantity of DNA lesions and the efficiency of DNA amplification (5–7). QPCR approaches therefore require labor-intensive determination of amplification efficiencies of the individual samples and normalization procedures, making them less suitable for high-throughput analyses.
In contrast to qPCR, real-time (rt)PCR analyses employing established primers and additives can be applied without the need for the setup of experimental conditions. A limitation of previous rtPCR-based methods, however, remains the DNA probe length. So far, the amplicon size of rtPCR approaches was limited to several hundred base pairs (8). In addition, conventional fluorescent dyes used for rtPCR, most commonly SYBR Green, substantially inhibit the polymerase-driven elongation reaction in a concentration-dependent manner by intercalation into double-stranded (dsDNA) (9). Contrariwise, if lesions are uniformly distributed within the cellular genome, longer PCR-template sequences carry statistically more polymerase-inhibitory DNA modifications, resulting in impaired numbers of unmodified template sequences that can successfully be amplified. In rtPCR approaches, longer amplicons therefore exhibit a delayed exponential phase of amplification, which is commonly expressed as crossing point (Cp) or threshold (Ct) cycle number. Consequently, as probe length and DNA lesions incidence within the probe sequence correlate in a linear manner, longer DNA probes enhance the sensitivity of DNA-damage quantification.
In this study, we present a novel long-run rtPCR technique for DNA damage quantification (LORD-Q) that circumvents former restrictions in sensitivity caused by limited probe length. We report that this method enables the amplification of DNA sequences >3 kb under quantitative rtPCR conditions. As a consequence, the increase of probe length favors the detection of low DNA-damage quantities. In contrast to recently published rtPCR-based methods that are less sensitive and restricted to the detection of DNA damage in abundant mitochondrial genomes (8–10), LORD-Q allows the accurate quantification of DNA damage in both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) probes. As selected examples of its broad applicability, we demonstrate that LORD-Q can be used to determine cell type-specific DNA-damage frequencies, to monitor DNA repair capacities, or to compare DNA vulnerabilities of different genomic loci. LORD-Q is therefore a valuable method for a wide range of life science applications.
MATERIALS AND METHODS
Cell culture and genotoxic stress induction
The human T lymphocyte Jurkat cell line was obtained from ATCC and grown in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum (FCS; PAA Laboratories). For DNA-damage induction, cells were exposed to the indicated doses of the following genotoxic agents: bleomycin (Medac, 20 min), UVC radiation (254 nm; Stratalinker UV Crosslinker 2400, Stratagene), cisplatin (Sigma-Aldrich, 16 h) or etoposide (Sigma-Aldrich, 16 h) either in serum-free or standard media. After extensive washing cells were immediately harvested on ice or allowed to repair DNA damage for 2 h at 37°C and 5% CO2. For DNA-repair kinetics, human induced pluripotent stem (hiPS) cells derived from K7 fibroblasts were treated with 6 mJ/cm2 (indicated ‘low dose’) or 10 mJ/cm2 (‘high dose’) UV light, and 6 µM (‘low dose’) or 10 µM (‘high dose’) bleomycin (20 min), respectively. K7 fibroblasts were accordingly treated with 4 mJ/cm2 (‘low dose’) or 8 mJ/cm2 (‘high dose’) UVC light, and 4 µM (‘low dose’) or 8 µM (‘high dose’) bleomycin for 20 min. UVC irradiation was carried out in cell culture dishes (10 cm diameter) at identical medium volumes (10 ml).
DNA isolation
Total cell DNA was purified using DNeasy Blood and Tissue Kit (Qiagen) from cells kept under standard or DNA-damaging conditions. DNA purity was determined by spectrometric analysis (Nanodrop 1000, Peqlab). Isolated DNA with high purity (A260/A280 >1.8) was stored at −20°C.
Enzymatic digestion of isolated DNA
For quantification of defined lesion rates, 10 µg DNA isolated from untreated Jurkat cells was used for enzymatic digestion. The restriction endonuclease AleI was identified as single cutter for both the employed mtDNA (3724 bp) and the p53 (3075 bp) probe using the NEBcutter V2.0 software (http://tools.neb.com/NEBcutter2). Digestion was carried out at 37°C for 2 h in a 20-µl volume using 10 units AleI (Fermentas) followed by a thermal inactivation step (10 min, 65°C). For the analysis of DNA-lesion frequency both digested and undigested DNA was then diluted to 10 ng/µl and mixed at different ratios (as indicated in Supplementary Table S2) prior to LORD-Q analysis.
LORD-Q assay
Real-time PCR analysis was carried out on the LightCycler 480 II system (Roche) in 96-well plates. The PCR amplification was monitored by real-time measurement of the intercalation of the saturating fluorescent dye ResoLight (Roche) into dsDNA employing KAPA2G Fast Hot Start ReadyMix kit (Peqlab). For each nuclear or mitochondrial genomic region, HPLC-purified oligonucleotides (Sigma-Aldrich) were employed to detect DNA damage in long (3–4 kb) probes, while nested short (40–70 bp) amplicons served as internal undamaged reference.
For the LORD-Q method, we selected oligonucleotides meeting all required standards in PCR efficiency and specificity (11). Primers used for amplification of the mitochondrial and nuclear loci are listed in Supplementary Table S3. To exclude accidental co-amplification of nuclear mitochondrial sequences (numts) (12), we used validated mtDNA primers that do not anneal to any numts DNA present in the genome database. All primers, including mtDNA probe primers, yielded a single PCR product, as confirmed by agarose gel electrophoresis. The PCR efficiencies of all amplicons were calculated using standard template dilution series of 50, 25, 12.5 and 6.25 ng whole-cell DNA per reaction (Supplementary Figure S1A). The rtPCR reaction mix consisted of 1× KAPA2G Fast Hot Start ReadyMix, 0.05× LightCycler 480 ResoLight dye, 500 nM of each forward (sense) and reverse (antisense) primer for the long and short amplicon, and 50 ng of template DNA in a total volume of 20 µl per well. Cycling conditions were as follows: a pre-incubation phase of 5 min at 95°C was followed by up to 50 cycles of 10 s at 95°C, 10 s at 60°C and 1 s at 72°C (small amplicons or synthetic single-stranded oligonucleotide templates carrying a modified nucleotide) or 2:15 min at 72°C (large amplicons). To compare the accuracy and efficacy of the LORD-Q method with an established semi-long-run rtPCR-based approach, we employed a previously published protocol (8) using SYBR Green, Taq polymerase and established oligonucleotides to amplify mtDNA fragments of a maximum length of 974 bp.
LORD-Q data analysis
Data analysis was based on the measurement of Cp values derived from the rtPCR runs of the short and long fragments. Isolated DNA from the genotoxin-treated samples and mock-treated references (Ref) served as template. Sample and reference DNA was analyzed side-by-side. For each genomic region, a refined version of the common 2ΔΔCt method (17) was used in to calculate the detected DNA lesion rate per 10 kb according to the following equation:
 |
In this formula, EL and ES are the amplification efficiencies of the long and short probe, Cp values are determined via LightCycler 480 software, a is the number of base pairs of the long fragment and n is the number of control (reference) samples. EL and ES were determined by amplification of serial DNA dilutions (50–6.25 ng/reaction) and linear regression analyses.
For each condition rtPCR was carried out in triplicate and the mean Cp values for the long and the small amplicons were used to quantify lesions in genomic or mtDNA.
QPCR-based DNA-damage quantification
DNA-damage analysis via qPCR was carried out as previously described by Santos et al. (5). Briefly, DNA from Jurkat cells exposed to the indicated doses of bleomycin was quantified by comparison to a standard curve of known DNA concentrations (0–100 ng/µl). A dilution of a fluorescent dsDNA dye (SYBR Green I) was added to 2 µl of the standard or sample DNA, and the fluorescence of the respective wells was determined in 96-well ELISA plates using a Tecan infinite 2000 plate reader. For qPCR, 15 ng per sample were used as template and a 8.9-kb mtDNA fragment was amplified as described (5). For each sample, a second PCR was run that contained 7.5 ng of the template DNA (‘50% control’). The PCR was terminated after 30–35 cycles and the dsDNA concentration was determined by fluorescence measurement as described above. ‘Fifty-percent controls’ were used to validate that the PCR had been stopped within the exponential phase. Following subtraction of the background fluorescence (PCR without template), DNA-damage rates per 10 kb were calculated by the formula ‘–ln(A) × (10 kb/8.9 kb)’, where A is the relative amplification (fluorescence sample/fluorescence control). All PCRs were carried out in duplicate and background values (without template) were determined in quadruplicates.
Comet assay
The comet assay was performed with minor modifications as described in (13,14). Briefly, Jurkat cells were exposed to the indicated doses of bleomycin and incubated for 20 min at 37°C and 5% CO2. After incubation cells were kept on ice and measured for viability using Trypan blue staining. Cells were then mixed with 0.7% low-melting-point agarose (SeaKem), plated on microscopical slides (Trevigen) and lysed over night in lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, 1% sodium lauryl sarcosinate, pH 7.5). After electrophoresis analysis of DNA damage was performed by fluorescence microscopy using Komet software version 6 (Andor Technology), determining the median Olive tail moment (percentage of DNA in the tail × tail length) of at least 50 cells per sample.
Derivation and culture of primary human dermal fibroblast lines
The 6-mm thick-skin punch biopsies were obtained from the volar surface of the forearm of healthy volunteers (termed ‘K7’ and ‘K22’) after informed consent and approval of the Ethics Committee of the University of Tübingen. The biopsies were cut into 4-mm2 pieces and cultured in a 6-well plate under sterile conditions. A dense outgrowth of cells appeared after 7–14 days, which were then passaged by trypsinization and grown under standard conditions in RPMI-1640 medium supplemented with 10% FCS, 2 mM L-glutamine and penicillin/streptomycin.
Retroviral induction and culture of hiPS cells
hiPS cell lines were generated by transducing dermal fibroblasts with retroviruses expressing hOCT4, hSOX2, hKLF4 and hc-MYC reprogramming factors (Addgene) as described (15). After 5 days cells were seeded either on BD Matrigel matrix (BD Biosciences) or on MEF feeder layer (LHR Biosciences) and grown in human embryonic-stem-cell medium containing Knockout DMEM (Invitrogen), 20% Serum Replacement (Invitrogen), 2 mM L-glutamine, 1% non-essential amino acids, penicillin/streptomycin, 25 µM β-mercaptoethanol, 5 ng/ml basic FGF (PeproTech) and 10% TeSR-1 medium (Stem Cell Technologies). Medium was changed daily and hiPS cells were passaged every 5 to 6 days using collagenase/dispase (Roche).
Embryoid body-derived in vitro differentiation
hiPS cells were harvested employing collagenase/dispase, singularized with Accutase (Millipore) and cultured in AggreWell plates (Stemcell Technologies) for 2 days in embryonic-stem-cell medium lacking β-mercaptoethanol and basic FGF. To induce undirected differentiation, the formed embryoid bodies were cultured on matrigel-coated culture dishes with either endodermal/mesodermal differentiation media (Knockout DMEM supplemented with 20% FCS, 2 mM L-glutamine, 1% non-essential amino acids, penicillin/streptomycin and 25 µM β-mercaptoethanol) or ectodermal differentiation medium composed of Neurobasal Medium (Gibco), DMEM F12 (Invitrogen), BSA (Carl Roth), L-glutamine, non-essential amino acids, penicillin/streptomycin, 25 µM β-mercaptoethanol, 10 µM Y-27632 (Wako), 1× B-27 (Invitrogen) and 0.5× N2 (R&D Systems) for 8 days at 37°C and 5% CO2. The medium was replaced daily. Samples were fixed with Accustain (Sigma), permeabilized with 1% Triton X-100 (Carl Roth) and immunostained with mouse anti-α-fetoprotein (1:100, Sigma-Aldrich), mouse anti-α-smooth muscle actin (1:200, Abcam) or mouse anti-β3-tubulin antibody (1:500, Sigma-Aldrich) and Alexa Fluor 546-conjugated anti-mouse secondary antibody (1:1000; Invitrogen). Nuclei were counterstained with DAPI (Invitrogen).
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (ChIP) assays were performed as described (16). Briefly, 107 cells were cross-linked and lysed, and DNA was sheared into fragments of 300–800 bp employing a sonicator (Bioruptor, Diagenode). Extracts were pre-cleared with salmon sperm-blocked protein A agarose (Upstate) and subjected to ChIP analysis with anti-acetylated histone H3K9/K14 antibody (Merck-Millipore). Real-time PCR was carried out in triplicate using SYBR Green rtPCR kit (Fermentas). Relative enrichment was calculated by the 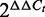 method (17).
method (17).
Oligonucleotides, RNA isolation and Reverse transcription-PCR
All primers and oligonucleotides carrying single DNA modifications were purchased from Sigma-Aldrich; their sequences are listed in Supplementary Tables S3–S5. Total RNA was isolated using the RNeasy kit (Qiagen) and quantified by spectroscopic analysis (Nanodrop 1000, Peqlab). cDNA was synthesized from 1 µg total RNA using Transcriptor First Strand cDNA Synthesis Kit (Roche). cDNA from hES H9 cells was kindly provided by Dr Marek Los (Linköping, Sweden). qRT-PCR was carried out using the SYBR Green real-time PCR Kit (Fermentas) employing the primers listed in Supplementary Table S5 and analyzed via LightCycler 480 II Real-Time PCR system (Roche). Gene-expression levels of hiPS cells were displayed as relative to the mRNA level of human fibroblasts and the expression of GAPDH and β-actin.
Flow cytometry
FACS analysis was performed employing a BD LSR II instrument (BD Biosciences) and BD FACS Diva software. FACS staining was carried out using human and mouse pluripotent stem cell analysis kits (BD Biosciences) containing PerCP-Cy5.5 mouse anti-Oct4, PE mouse anti-SSEA-1 and Alexa Fluor 647-coupled mouse anti-SSEA-4 and corresponding isotype control antibodies. Control beads were used for compensation, and cellular debris and doublets were excluded by suitable gating.
Statistics and receiver-operating characteristic analysis
Data are expressed as mean ± SD of at least three independent experiments. Receiver-operating characteristic (ROC) analyses were used to evaluate the sensitivity of DNA-damage detection of LORD-Q in comparison with a semi-long-run rtPCR method (8). Briefly, Jurkat cells were incubated with 1, 10, 100 and 2500 nM bleomycin for 20 min at 37°C under serum-free conditions and the isolated DNA of treated (true positives) and untreated (true negatives) Jurkat cells was analyzed side-by-side by the two aforementioned rtPCR methods using specific oligonucleotides for mtDNA [semi-long-run rtPCR: AL4.F (CTGTTCTTTCATGGGGAAGC), AS2.R (974 bp amplicon); LORD-Q: CL5.F, AS2.R (3723 bp), see Supplementary Table S3]. Both methods employed AS2.F and AS2.R (67-bp amplicon) as internal reference. ROC analysis was performed with OriginPro 8 software (OriginLab). ΔCp values were used to determine ROC curves and areas under the curve (AUC)
RESULTS
A real-time PCR-based assay for sequence-specific mtDNA- and nDNA-damage quantification
In order to develop a sensitive rtPCR-based method for DNA damage detection using elongated DNA amplicons, we first tried to overcome the challenge of size restriction of the DNA template in conventional rtPCR approaches, which usually aim to amplify sequences of a few hundreds of base pairs. After testing numerous PCR components such as DNA polymerases, DNA-intercalating dyes and additives, we finally chose a combination of the high-fidelity and rapid KAPA2G Fast DNA polymerase and the second-generation fluorescent DNA dye ResoLight. ResoLight, originally designed for high-resolution melting analyses, can be applied at very low concentrations and therefore leads to an only minor inhibition of template amplification (18).
In contrast to the conventional Taq DNA polymerases and the standard SYBR Green I dye, the combination of KAPA2G polymerase and ResoLight dye allowed the proper amplification of fragments of up to 4 kb (Supplementary Figure S1A). We therefore termed this method LORD-Q for long-run rtPCR-based DNA-damage quantification. In LORD-Q, long DNA fragments (∼3–4 kb) serve as damage sensors, whereas short-nested amplicons (∼50 bp) are used as an internal reference to counterbalance template concentration deviations (Figure 1A). Various primer pair candidates and template sequences were screened and selected to achieve consistent primer efficiencies. While the conventional 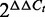 method for DNA quantification presumes amplification efficiencies of 100% and a template duplication for each PCR cycle (17), LORD-Q uses experimentally determined amplification efficiencies (Supplementary Table S3) for the calculation of DNA-damage rates. This is of major importance since amplification efficiencies of long PCR products (>1 kb) are significantly <100%. Thus, by employing experimentally determined amplification efficiencies for DNA-damage calculation, propagated errors caused by inaccurate bases in the underlying exponential calculation formula are minimized (see Materials and methods section).
method for DNA quantification presumes amplification efficiencies of 100% and a template duplication for each PCR cycle (17), LORD-Q uses experimentally determined amplification efficiencies (Supplementary Table S3) for the calculation of DNA-damage rates. This is of major importance since amplification efficiencies of long PCR products (>1 kb) are significantly <100%. Thus, by employing experimentally determined amplification efficiencies for DNA-damage calculation, propagated errors caused by inaccurate bases in the underlying exponential calculation formula are minimized (see Materials and methods section).
Figure 1.

LORD-Q allows the sequence-specific detection and accurate quantification of lesions in long DNA probes of mitochondrial and nuclear genomes. (A) Schematic illustration of the LORD-Q assay. Whole-cell DNA, either damaged or intact, is isolated (left). Undamaged template sequences of 3–4 kb-length are successfully amplified, whereas several types of DNA lesions or modifications impair or abrogate the PCR elongation reaction, as detected by reduced fluorescence (FL). Short fragments of 40–70 bp serve as reference templates (middle). Increasing DNA lesion/modification rates lead to elevated ΔCp values (right). (B) DNA damage induced by different stimuli can be quantified by LORD-Q. Jurkat cells were incubated with the indicated concentrations of bleomycin (20 min), cisplatin (16 h), etoposide (16 h) and different doses of UVC light. Both mtDNA and nDNA (Col1a1 locus) DNA lesions were quantified by LORD-Q (n = 3, mean ± SD). (C) Effect of bleomycin on cell death and mtDNA damage. Jurkat cells were stimulated with the indicated concentrations of bleomycin. After 20 min, half of the cells were harvested and analyzed by LORD-Q, while the remaining half was washed and further incubated under standard culture conditions. After 6 h cell viability was determined by flow cytometry and annexin V-FITC/propidium iodide staining (n = 3, mean ± SD). (D) Effect of the caspase inhibitor Q-VD-OPH on mtDNA and nDNA lesion rates. Jurkat cells were incubated with 100 µM cisplatin for 16 h (left) or irradiated with UVC (20 mJ/cm2, right) in the presence or absence of 10 µM Q-VD-OPH.
To investigate whether LORD-Q is suitable for DNA-damage quantification, we first treated human Jurkat T lymphocytes with different dosages of the genotoxic stimuli UVC light (254 nm), cisplatin, etoposide and bleomycin. As shown in Figure 1B, all stimuli induced DNA damage in a dose-dependent manner. Interestingly, comparative analysis of mitochondrial and nuclear probes revealed that UV irradiation and cisplatin treatment introduced similar lesion rates in both the mtDNA and nDNA, whereas short-term incubation with bleomycin, a redox-based genotoxic agent or overnight treatment with the topoisomerase II inhibitor etoposide primarily damaged the mitochondrial and nuclear genome, respectively (Figure 1B).
Next, we examined whether LORD-Q allowed the detection of DNA-damage rates induced by sublethal genotoxic stimulation. For this purpose Jurkat cells were exposed to different doses of bleomycin for 20 min and then either directly harvested and analyzed by LORD-Q or further cultured for 6 h under standard culture conditions prior to cell death measurement. We found that only high concentrations of bleomycin slightly compromised cell viability 6 h after drug incubation (Figure 1C), whereas virtually no cell-death induction was detectable at earlier time points and drug concentrations of 10 µM or less. However, DNA damage was robustly quantifiable after 20 min of incubation with sublethal dosages of bleomycin (Figure 1C). In addition, caspase inhibition by addition of Q-VD-OPH affected the DNA-lesion rate only after long-term exposure (16 h) to cisplatin, but not after short-term exposure to UVC radiation (Figure 1D) or bleomycin treatment (data not shown). These results indicate that early genotoxic events can be monitored by LORD-Q at sublethal conditions independent of the induction of apoptotic cell death, which might otherwise have biased the detected DNA damage.
LORD-Q is more sensitive than qPCR-based DNA-damage quantification and the comet assay
We further explored whether our method is able to accurately quantify defined lesion rates and how its sensitivity compares to other conventionally used DNA-damage quantification methods. To establish a standard curve with a known number of DNA lesions, we digested total cellular DNA with the restriction endonuclease AleI that was predicted to insert a single cut within both the amplified mtDNA and nDNA (p53 locus) probes. The digested DNA was then mixed with undigested DNA at different ratios, and the expected lesion rates were calculated from the uncut portion of the DNA employing the LORD-Q formula (Supplementary Table S2). Following LORD-Q analysis of the DNA mixtures, the expected lesion rates were blotted against the experimentally detected damage incidence. Importantly, for both the mtDNA and nDNA, the detected lesion rates were akin to the expected amount of DNA damage (Figure 2A), indicating a high accuracy of the LORD-Q method.
Figure 2.

Validation of the accuracy and sensitivity of the LORD-Q method. (A) Correlation of calculated and detectable DNA lesions. Whole-cell DNA was mixed with DNA that had been digested by the restriction endonuclease AleI introducing a single cut in both examined DNA loci). For both analyzed mtDNA (left) and nDNA (p53 locus, right) the lesion rates detected by LORD-Q were plotted against the calculated (expected) DNA lesions (n = 4, mean ± SD). (B) Comparison of LORD-Q with other methods of DNA damage detection. Jurkat cells were exposed for 20 min to the indicated concentrations of bleomycin under cell culture conditions. The extent of DNA damage was determined in parallel by the LORD-Q (left) and qPCR (middle) assay or by comet assay (right). (n = 3–4, mean ± SD). *P < 0.05, **P < 0.01 and ***P < 0.001.
Next, we directly compared the sensitivity of LORD-Q with previously established approaches, such as the PCR-based method developed by Santos et al. (5) and the classical comet assay. To this end, Jurkat cells were exposed to different concentrations of bleomycin and the samples were subsequently analyzed by the three methods for DNA-damage detection in side-by-side experiments. The qPCR method and the comet assay significantly detected DNA lesions after exposure to bleomycin concentrations of ≥1 µM and ≥500 nM, respectively (Figure 2B). Importantly, employing the LORD-Q we were able to detect DNA lesions at bleomycin concentrations of ≥10 nM (Figure 2B), indicating a strongly improved sensitivity of the LORD-Q method.
LORD-Q is highly accurate and recognizes defined nucleotide modifications
In order to verify the accuracy of the method, we further compared the sensitivity of LORD-Q with a semi-long-run rtPCR method that uses standard rtPCR components (8). Due to a lower efficiency of the employed DNA polymerase and the inhibitory effect of the SYBR Green dye, the semi-long-run method merely allows amplification of sequences of up to 1-kb length. In addition, this method is restricted to the detection DNA damage in abundant mtDNA but not in the nuclear genome (8). Analysis of DNA derived from Jurkat cells treated with very low doses (10 nM) of bleomycin revealed that mtDNA damage of 0.3 lesions per 10 kb could be robustly and significantly (n = 15, P = 1.2E – 6) detected by the LORD-Q method (Figure 3A). We then carried out ROC analyses, which determine the sensitivity and specificity (by plotting true positive against false positive events) as AUC of 1.0 for perfect discrimination between the treated and the control group and 0.5 for randomized results (19). ROC analyses revealed a significantly higher sensitivity of LORD-Q, in particular at low concentrations of bleomycin (AUC: 100 nM: 1.0, AUC 10 nM: 0.934), as compared to the previously described semi-long-run method (AUC: 100 nm: 0.901, AUC 10 nM: 0.5) (Figure 3A and B).
Figure 3.

Comparison of sensitivity of the semi-long-run rtPCR method and the LORD-Q assay by ROC analysis. Jurkat cells were treated with the indicated concentrations of bleomycin for 20 min in serum-free medium. Real-time PCR experiments were carried out in triplicate (n = 15 per concentration and experiment). (A) Quantification of detectable DNA lesions per 10 kb by the LORD-Q method. (B) Representative ROC analyses for LORD-Q after treatment of Jurkat cells with the indicated concentrations of bleomycin (Bleo, left panel), and averaged ROC AUC values for the semi-long-run rtPCR method and LORD-Q (right panel). (C) Effect of different DNA lesions and nucleotide modifications on relative template amplification rates. Synthetic DNA oligonucleotides with inserted single-base modifications were amplified applying LORD-Q (n = 4, mean ± SD). *P < 0.05, **P < 0.01 and ***P < 0.001.
We further investigated which types of DNA modification are detectable by LORD-Q. Besides single- and double-strand breaks that physically dissect the single-stranded DNA-template sequence, polymerase inhibition may also be provoked by chemical alterations of DNA bases. For this purpose, various synthetic oligonucleotides carrying a single modified base (Supplementary Table S3) were used as templates. As shown in Figure 3C, oligonucleotides harboring a thymine dimer or an abasic site exhibited a strong inhibition of the relative amplification rate compared to the unmodified template. Interestingly, also 5-hydroxymethylcytosine (5hmC), which plays an important role in epigenetic regulation and stem-cell development (20–22), but not 5-methylcytosine (5mC), led to complete inhibition of rtPCR amplification. In contrast, other tested base modifications including 8-oxo-deoxyadenosine and 8-oxo-deoxyguanosine had only a minor influence on the amplification rate (Figure 3C, Supplementary Figure S2). Thus, LORD-Q is able to quantify DNA-strand breaks, thymine dimers, abasic and 5hmC sites in an accurate and highly sensitive (≥0.3 detectable lesions/10 kb) manner.
The LORD-Q method is suitable for a wide range of scientific applications
Finally, we wished to provide some examples for potential applications of the LORD-Q assay in biomedical research. Dysfunctional DNA repair and genomic instability are tightly associated with age-related disorders such as Xeroderma pigmentosum, Ataxia telangiectasia, Fanconi’s anemia, Parkinson’s disease and cancer (23–27). Hence, besides the identification and characterization of the genotoxic potential of chemical substances, a quantitative determination of cell-type-specific susceptibility to DNA-damage and -repair velocity remains of great interest. We therefore employed LORD-Q to monitor DNA vulnerability and repair processes upon genotoxic treatment. Exemplarily, hiPS cells, which have been reported to exhibit high DNA-repair capacities (28,29) and dermal fibroblasts of identical genetic background were analyzed for their vulnerability towards genotoxic treatment. We generated hiPS cells by transducing dermal fibroblasts with retroviruses carrying genes coding for hOCT4, hSOX2, hKLF4 and hcMYC (OSKM) reprogramming factors. Subsequently, cells were characterized for the expression of pluripotency markers and their capacity to differentiate into the three germ layers (Supplementary Figure S3).
It has been proposed that stem cells minimize DNA damage in order to prevent spreading of DNA damage-associated mutations during development (30), whereas differentiated fibroblasts can exit the cell cycle and enter a quiescent or senescent state if DNA repair fails. Indeed, compared to hiPS cells, fibroblasts revealed a significantly higher initial DNA damage after both UV irradiation (mtDNA, P = 0.004; nDNA, P = 0.04) and bleomycin exposure (mtDNA, P = 0.004; nDNA, P = 0.0005) (Figure 4A). In order to examine DNA-repair processes, we compared the detected initial DNA-damage rate to the lesion frequencies remaining 2 h after genotoxic treatment. For comparison of hiPS cells and fibroblasts, bleomycin and UV radiation were each applied at different doses that roughly induced a similar quantity of DNA damage in both cell types. LORD-Q analyses revealed that hiPS cells were able to repair detectable DNA lesions within the first 2 h after genotoxic treatments more efficiently than isogenic fibroblasts (Figure 4B), which is consistent with the finding that hiPS cells possess a high DNA-repair capacity (29) and rapidly upregulate DNA repair enzymes following DNA damage (30).
Figure 4.

Exemplary applications of the LORD-Q method. (A) Comparative analysis of genotoxic vulnerability. Human primary fibroblasts (hFib) and corresponding isogenic hiPS cells were treated in serum-free medium with 10 µM bleomycin (20 min) or 10 mJ/cm2 UVC radiation before mitochondrial and genomic lesion rates were determined by LORD-Q analysis (n = 3, mean ± SD). (B) Investigation of mtDNA repair. Human primary fibroblasts and corresponding isogenic hiPS cells were treated with low and high doses of bleomycin (see Materials and methods section for details) or UV radiation in serum-free medium, and then harvested either directly after treatment (indicated ‘initial’) or after 2 h of recovery under cell culture conditions (n = 3, mean ± SD). (C) Comparison of DNA damage in different genes. QRT-PCR expression analysis of Oct4 and Col1a1 in fibroblasts and corresponding hiPS cells (left, n = 3, mean ± SD). Corresponding qPCR analysis of histone H3 acetylation in ChIP-enriched Oct4 and Col1a1 loci (middle). Fibroblasts and corresponding hiPS cell clones were stimulated with 10 µM bleomycin (20 min) and the DNA lesion rates in both Oct4 and Col1a1 gene loci were determined (right, n = 6, mean ± SD). The ratio of detected lesions per 10 kb in both genes is shown. *P < 0.05, **P < 0.01 and ***P < 0.001.
Besides cell-type-specific differences in sensitivity to DNA damage, DNA vulnerability might correlate with the transcriptional activity of the respective DNA locus. To investigate whether differences in the vulnerability of distinct DNA regions could be monitored by LORD-Q, we evaluated DNA damage in two nDNA loci. In hiPS cells, the gene encoding the stem cell renewal factor OCT4 (gene ID: POU5F1) but not the collagen 1 locus (COL1A1) is intensely transcribed, whereas fibroblasts reveal an opposite expression pattern (Figure 4C, left panel). The corresponding chromatin status was confirmed by ChIP analysis of histone H3K9/14 acetylation, which marks transcriptionally active DNA regions (Figure 4C, middle panel). LORD-Q analysis revealed that bleomycin-treated hiPS cells accumulated more DNA lesions in the OCT4 promoter than in the COL1A1 promoter, whereas fibroblasts exhibited a significantly (P = 1E – 6) inverted DNA damage pattern (Figure 4C, right panel).
These examples demonstrate that LORD-Q can successfully be applied for the analysis of DNA-repair processes. In addition, the method enables the identification of locus- and/or cell-type dependent DNA-damage incidence and represents a versatile tool for the surveillance of hot spots of DNA damage, e.g. in tumor-associated genes.
DISCUSSION
Various severe diseases including neurological disorders or cancer are tightly connected to the accumulation of nuclear or mitochondrial mutations and subsequent cellular dysfunction (27–31). Mutations mainly evolve during replication processes (i.e. by non-complementary nucleotide insertion) or by a failure of proper DNA-damage repair. For this reason, DNA-damage-analysis methods are needed to elucidate the mechanisms underlying malignancy and neurodegeneration.
The main principle of DNA-damage quantification employing PCR technologies relies on the inhibition of polymerase-driven DNA duplication of template sequences harboring certain types of DNA lesions. The LORD-Q method described in this study represents a substantial advance over previous methods in terms of sensitivity, amplicon length, lesion specificity and potential applications. Previously described qPCR-based assays require labor-intensive establishment of quantitative PCR and normalization procedures using ‘50% template’ controls (5–7). In addition, as the range of the linear phase during amplification is strongly influenced by the extent of DNA damage, the required PCR cycle number has to be individually established in a time-consuming manner for each DNA sample. Therefore, those PCR methods are hardly applicable for high-throughput analyses.
Powerful methods of DNA-damage detection must be automation-compatible and suitable for standard microtiter formats, e.g. for genotoxicity screenings of chemical compound libraries. Moreover, methods for DNA-damage detection require sufficient sensitivity to reliably determine DNA-damage rates at sublethal and physiological conditions. Real-time PCR-based DNA-damage quantification methods possess most of these properties. However, previous rtPCR approaches, including a semi-long-run PCR method developed in our laboratory (8), lack sensitivity and accuracy, thereby failing to enable reliable and reproducible damage quantification in nDNA templates. Instead, those methods are restricted to the analysis of damage incidence in mtDNA templates that exhibit a high copy number, but cannot be employed for genome-wide DNA-damage analysis or the quantification of DNA lesions in tumor-associated nuclear genes.
By using a novel rapid high-fidelity DNA polymerase, a second-generation fluorescent DNA dye, by screening of a large number of candidate primer pairs and by optimizing the rtPCR parameters, we were able to considerably increase the sensitivity of DNA-damage detection and to establish a protocol for the quantification of DNA lesions in mitochondrial and nuclear probes of up to 4-kb length. The improved sensitivity of long DNA-damage probes becomes evident in Table 1, which displays the correlation between probe length and detectable ΔCp value.
Table 1.
Correlation between probe length and expected ΔCp for commonly used rtPCR methods applied for a hypothetical sample carrying one detectable lesion per 10 kb DNA
| Method | Probe length (bp) | Calculated ΔCp |
|---|---|---|
| Classical rtPCR (short template) | 50 | 0.01 |
| Classical rtPCR (long template) | 300 | 0.04 |
| Semi-long-run rtPCR | 974 | 0.14 |
| LORD-Q | 3723 | 0.54 |
Using standard DNA templates with a defined number of dsDNA breaks we validated the analytical power and accuracy of LORD-Q, which demonstrated a tight correlation of calculated and experimentally detected DNA lesions. Moreover, by comparing LORD-Q with a previously established qPCR technique and the comet assay, we show that LORD-Q is significantly more sensitive than other methods. Consistent with this result and as confirmed by ROC analyses, we demonstrate that even sublethal DNA-damage rates caused by extremely low concentrations of genotoxic agents can reliably be quantified. LORD-Q could reproducibly distinguish DNA damage in untreated controls and cells treated with 10 nM bleomycin. This concentration of bleomycin is ∼20-fold less than the serum levels that are usually achieved during chemotherapy of cancer patients (32). Moreover, it is 100-fold lower than the LC50 of hiPS cells and 10 000-fold lower than the LC50 of Jurkat cells determined 6 h after bleomycin treatment (data not shown). In particular, human iPS cells exhibit a high susceptibilty to programmed cell death after treatment with DNA-damaging agents (33), thereby further illustrating the sensitivity of the assay.
We show that LORD-Q allows the separate assessment of nDNA and mtDNA damage. Lesions or mutations of mtDNA have been implicated in a wide range of diseases and are causally involved in the side effect of several drugs (34). Owing to their role in energy metabolism, redox homeostasis and apoptosis, mitochondria are also considered as an attractive target for cancer therapy. In this context, we found that the short-term incubation of cells with bleomycin, a reactive oxygen species-inducing genotoxic agent, primarily induced mtDNA damage, whereas the topoisomerase II inhibitor etoposide preferentially targeted the nuclear genome. In line with our findings, it was shown that bleomycin induces mitochondrial rather than nDNA damage and that depletion of mtDNA confers drug resistance (35,36). In contrast, inhibition of topoisomerase II, which is restricted to the nucleus, triggers mainly nDNA strand breaks (37). Thus, LORD-Q might be useful to assess pathological processes or drug actions that differentially affect the mitochondrial or nuclear genome.
Using various synthetic oligonucleotides that carry a single base or nucleotide modification as rtPCR templates, we further characterized the lesion specificity of the LORD-Q assay. Besides DNA strand breaks, we thereby identified that thymine dimers, abasic sites and 5hmC strongly impaired the duplication of the template sequence. Thymine dimers, which are implicated in melanoma development (38), completely abrogated template amplification, making LORD-Q a potential tool for UV-induced DNA-damage analysis. Similar to thymine dimers, 5hmC inhibited the polymerase-driven template amplification. The conversion of 5mC to 5hmC has been identified as an essential step for the erasure of CpG methylation during totipotency reprogramming of germ cells (39). In pluripotent cells, distinct DNA regions exhibit up to ∼30% 5-hydroxymethylated cytosines (39,40). In this context, LORD-Q might serve as a tool for the facilitated and site-specific monitoring of 5hmC frequency.
LORD-Q is further suitable to detect cell-type-specific DNA-repair capacities or DNA-damage patterns. Exemplarily, we show that hiPS cells acquired less detectable DNA lesions during equal genotoxic treatment and were able to repair DNA lesions more efficiently than isogenic fibroblasts. Moreover, a comparative analysis of hiPS cells and differentiated fibroblasts demonstrated that two differentially transcribed genes (OCT4 and COL1A1) differed in their susceptibility to DNA damage in the two cell types. A potential link of the DNA vulnerability of certain gene with the chromatin status and active gene transcription certainly requires further investigation. Our finding of a distinct susceptibility of differentially expressed genes could, nevertheless, imply that the sequence-specific quantification of DNA damage might allow to monitor disease-associated genes or to identify hot spots of DNA damage that could be relevant in tumorigenesis or neurodegeneration.
In summary, LORD-Q exhibits substantial advances over existing PCR-based methods and the comet assay. The LORD-Q method represents a highly sensitive and accurate technique, which enables the sequence-specific quantification of specified DNA lesions not only in abundant mitochondrial genomes, but also at defined chromosomal loci. The method is high-throughput compatible and robust. Moreover, LORD-Q is suitable for the identification of DNA damage hot spots, the characterization of genotoxic susceptibility of distinct cell types and for monitoring of DNA-repair capacities. It is therefore a simple, precise, labor- and cost-effective method for a wide range of applications in the field of biomedical sciences.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [SFB 685, SFB 773, GRK1302]; Deutsche Krebshilfe; Innovation Grant of the University of Tübingen. Funding for open access charges: University of Tübingen.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Reinhard Gilniorz for technical assistance. O.R. conceived of the project. S.L. and O.R. were responsible for the experimental work. S.L., D.G.H., B.M., P.N.M., M.S.B., P.S. and O.R. designed experiments and analyzed data. S.L., F.E., K.S.O. and O.R. wrote the manuscript. All authors discussed the data and commented on the manuscript. S.L., O.R. and K.S.O. have filed a patent application on the method described in this manuscript.
REFERENCES
- 1.Wagener S, Volker T, De Spirt S, Ernst H, Stahl W. 3,3'-Dihydroxyisorenieratene and isorenieratene prevent UV-induced DNA damage in human skin fibroblasts. Free Rad. Biol. Med. 2012;53:457–463. doi: 10.1016/j.freeradbiomed.2012.05.022. [DOI] [PubMed] [Google Scholar]
- 2.Balajee AS, May A, Bohr VA. DNA repair of pyrimidine dimers and 6-4 photoproducts in the ribosomal DNA. Nucleic Acids Res. 1999;27:2511–2520. doi: 10.1093/nar/27.12.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seo J, Kim SC, Lee HS, Kim JK, Shon HJ, Salleh NL, Desai KV, Lee JH, Kang ES, Kim JS, et al. Genome-wide profiles of H2AX and gamma-H2AX differentiate endogenous and exogenous DNA damage hotspots in human cells. Nucleic Acids Res. 2012;40:5965–5974. doi: 10.1093/nar/gks287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 6.Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 2012;920:111–132. doi: 10.1007/978-1-61779-998-3_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter SE, Jung D, Di Giulio RT, Meyer JN. The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods. 2010;51:444–451. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rothfuss O, Gasser T, Patenge N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 2010;38:e24. doi: 10.1093/nar/gkp1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gudnason H, Dufva M, Bang DD, Wolff A. Comparison of multiple DNA dyes for real-time PCR: effects of dye concentration and sequence composition on DNA amplification and melting temperature. Nucleic Acids Res. 2007;35:e127. doi: 10.1093/nar/gkm671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards JG. Quantification of mitochondrial DNA (mtDNA) damage and error rates by real-time QPCR. Mitochondrion. 2009;9:31–35. doi: 10.1016/j.mito.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 12.Mourier T, Hansen AJ, Willerslev E, Arctander P. The Human Genome Project reveals a continuous transfer of large mitochondrial fragments to the nucleus. Mol. Biol. Evol. 2001;18:1833–1837. doi: 10.1093/oxfordjournals.molbev.a003971. [DOI] [PubMed] [Google Scholar]
- 13.Greve B, Bolling T, Amler S, Rossler U, Gomolka M, Mayer C, Popanda O, Dreffke K, Rickinger A, Fritz E, et al. Evaluation of different biomarkers to predict individual radiosensitivity in an inter-laboratory comparison—lessons for future studies. PLoS One. 2012;7:e47185. doi: 10.1371/journal.pone.0047185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmezer P, Rajaee-Behbahani N, Risch A, Thiel S, Rittgen W, Drings P, Dienemann H, Kayser KW, Schulz V, Bartsch H. Rapid screening assay for mutagen sensitivity and DNA repair capacity in human peripheral blood lymphocytes. Mutagenesis. 2001;16:25–30. doi: 10.1093/mutage/16.1.25. [DOI] [PubMed] [Google Scholar]
- 15.Park IH, Daley GQ. Human iPS cell derivation/reprogramming. Curr. Protoc. Stem Cell Biol. 2009 doi: 10.1002/9780470151808.sc04a01s8. Chapter 4, Unit 4A.1, doi:10.1002/9780470151808.sc04a01s8. [DOI] [PubMed] [Google Scholar]
- 16.Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, et al. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum. Mol. Genet. 2009;18:3832–3850. doi: 10.1093/hmg/ddp327. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Horakova H, Polakovicova I, Shaik GM, Eitler J, Bugajev V, Draberova L, Draber P. 1,2-propanediol-trehalose mixture as a potent quantitative real-time PCR enhancer. BMC Biotechnol. 2011;11:41. doi: 10.1186/1472-6750-11-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oliver R, Bjoertomt O, Greenwood R, Rothwell J. ‘Noisy patients'–can signal detection theory help? Nat. Clin. Pract. Neurol. 2008;4:306–316. doi: 10.1038/ncpneuro0794. [DOI] [PubMed] [Google Scholar]
- 20.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M, McLoughlin EM, Brudno Y, Mahapatra S, Kapranov P, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S. Quantitative Sequencing of 5-Methylcytosine and 5-Hydroxymethylcytosine at Single-Base Resolution. Science. 2012;336:215–228. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 22.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C, Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broyde S, Patel DJ. DNA repair: How to accurately bypass damage. Nature. 2010;465:1023–1024. doi: 10.1038/4651023a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu. Rev. Pathol. 2012;7:303–321. doi: 10.1146/annurev-pathol-011811-132509. [DOI] [PubMed] [Google Scholar]
- 25.Zhou W, Otto EA, Cluckey A, Airik R, Hurd TW, Chaki M, Diaz K, Lach FP, Bennett GR, Gee HY, et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012;44:910–915. doi: 10.1038/ng.2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devine MJ, Plun-Favreau H, Wood NW. Parkinson's disease and cancer: two wars, one front. Nat. Rev. Cancer. 2011;11:812–823. doi: 10.1038/nrc3150. [DOI] [PubMed] [Google Scholar]
- 27.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 28.Luo LZ, Gopalakrishna-Pillai S, Nay SL, Park SW, Bates SE, Zeng X, Iverson LE, O’Connor TR. DNA repair in human pluripotent stem cells is distinct from that in non-pluripotent human cells. PLoS One. 2012;7:e30541. doi: 10.1371/journal.pone.0030541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maynard S, Swistowska AM, Lee JW, Liu Y, Liu ST, Da Cruz AB, Rao M, de Souza-Pinto NC, Zeng X, Bohr VA. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells. 2008;26:2266–2274. doi: 10.1634/stemcells.2007-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Momcilovic O, Knobloch L, Fornsaglio J, Varum S, Easley C, Schatten G. DNA damage responses in human induced pluripotent stem cells and embryonic stem cells. PLoS One. 2010;5:e13410. doi: 10.1371/journal.pone.0013410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mabeta P, Dippenaar N, Shelver G. A validated HPLC method for the simultaneous determination of bleomycin A2 and B2 in human plasma. Int. J. Pharm. Biomed. Res. 2012;3:191–194. [Google Scholar]
- 33.Dumitru R, Gama V, Fagan BM, Bower JJ, Swahari V, Pevny LH, Deshmukh M. Human embryonic stem cells have constitutively active Bax at the Golgi and are primed to undergo rapid apoptosis. Mol. Cell. 2012;46:573–583. doi: 10.1016/j.molcel.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 35.Lim LO, Neims AH. Mitochondrial DNA damage by bleomycin. Biochem. Pharmacol. 1987;36:2769–2774. doi: 10.1016/0006-2952(87)90263-2. [DOI] [PubMed] [Google Scholar]
- 36.Brar SS, Meyer JN, Bortner CD, Van Houten B, Martin WJ., 2nd Mitochondrial DNA-depleted A549 cells are resistant to bleomycin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012;303:L413–L424. doi: 10.1152/ajplung.00343.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Champoux JJ. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 2001;70:369–413. doi: 10.1146/annurev.biochem.70.1.369. [DOI] [PubMed] [Google Scholar]
- 38.Rass K, Reichrath J. UV damage and DNA repair in malignant melanoma and nonmelanoma skin cancer. Adv. Exp. Med. Biol. 2008;624:162–178. doi: 10.1007/978-0-387-77574-6_13. [DOI] [PubMed] [Google Scholar]
- 39.Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA. Germline DNA Demethylation Dynamics and Imprint Erasure Through 5-Hydroxymethylcytosine. Science. 2012;339:448–452. doi: 10.1126/science.1229277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hackett JA, Reddington JP, Nestor CE, Dunican DS, Branco MR, Reichmann J, Reik W, Surani MA, Adams IR, Meehan RR. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development. 2012;139:3623–3632. doi: 10.1242/dev.081661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.