

Abstract
Pseudouridine synthases introduce the most common RNA modification and likely use the same catalytic mechanism. Besides a catalytic aspartate residue, the contributions of other residues for catalysis of pseudouridine formation are poorly understood. Here, we have tested the role of a conserved basic residue in the active site for catalysis using the bacterial pseudouridine synthase TruB targeting U55 in tRNAs. Substitution of arginine 181 with lysine results in a 2500-fold reduction of TruB’s catalytic rate without affecting tRNA binding. Furthermore, we analyzed the function of a second-shell aspartate residue (D90) that is conserved in all TruB enzymes and interacts with C56 of tRNA. Site-directed mutagenesis, biochemical and kinetic studies reveal that this residue is not critical for substrate binding but influences catalysis significantly as replacement of D90 with glutamate or asparagine reduces the catalytic rate 30- and 50-fold, respectively. In agreement with molecular dynamics simulations of TruB wild type and TruB D90N, we propose an electrostatic network composed of the catalytic aspartate (D48), R181 and D90 that is important for catalysis by fine-tuning the D48-R181 interaction. Conserved, negatively charged residues similar to D90 are found in a number of pseudouridine synthases, suggesting that this might be a general mechanism.
INTRODUCTION
Pseudouridine synthases are tRNA-uridine uracil mutases (E.C. 5.4.99) that catalyze the most common modification in functional RNA, namely the posttranscriptional conversion of uridines to pseudouridines (1,2). Similar to DNA glycosylases, these enzymes catalyze the breakage of a glycosidic bond, albeit not by hydrolysis. Instead, the uracil base is reoriented and subsequently reattached to the ribose through an unusual C5–C1’ glycosidic bond. Based on this reaction mechanism including breakage of the glycosidic bond and formation of a new C-C bond, pseudouridine synthases are sometimes also considered RNA lyases. Through numerous structural studies, it has become clear that all pseudouridine synthases share a common catalytic domain despite significant heterogeneity in primary structure (3–10). Therefore, it has been suggested that all pseudouridine synthases share the same catalytic mechanism (11).
After more than a decade of research, the details of the catalytic mechanism of pseudouridine formation still remain under debate with three different mechanisms being proposed. The only universally conserved residue in the active site of all pseudouridine synthases is an aspartate residue that is essential for catalysis (12–20). This aspartate may contribute to catalysis either by forming a covalent bond to C1’ of the ribose (13), to C6 of the uracil base (21) or by abstracting a proton from C2’ of the ribose (22). Currently the two mechanisms are favored where the aspartate acts on the ribose rather than the uracil (22). Hardly anything is known about the contribution of other residues to the formation of pseudouridine. Within the active site, there are two additional residues that are highly conserved, a basic and an aromatic residue (2). The basic residue in the active site of pseudouridine synthases is either an arginine or a lysine that forms a salt bridge interaction with the catalytic aspartate. It may be the role of the basic residue to render the aspartate more nucleophilic, to position the aspartate for catalysis or to serve as a source of protons (2). Additionally, an aromatic residue in the active site is found to stack against the target uracil base likely stabilizing the conformation of the base within the active site (23). All pseudouridine synthases use a tyrosine for this role, except for TruD where this residue is instead a phenylalanine (7–9). The tyrosine has also been proposed to act as a general base abstracting a proton from C5 to complete the isomerization process (23).
One of the best characterized pseudouridine synthases that often serves as a model for the other enzymes is the bacterial pseudouridine synthase TruB. This enzyme modifies U55 in the TΨC arm of all elongator tRNAs (24). Although not the first pseudouridine synthase to be identified, TruB was the first to be crystallized in the presence of RNA providing significant insight into the architecture of the active site of pseudouridine synthases (5). Subsequent structures of TruB, including its apo form, revealed an induced-fit mechanism of substrate binding involving the movement of TruB’s thumb loop, insert 1 segment, as well as its C-terminal PUA domain (found in pseudouridine synthases and archaeosine-transglycosidases) (25–27). The RNA also undergoes some conformational changes, as three consecutive bases including the target uridine are flipped out of the T loop into the active site of TruB. In addition, a number of biochemical studies on TruB have been reported including a detailed analysis of its substrate specificity (28), the importance of the catalytic aspartate residue (14,25), the role of the conserved tyrosine residue in the active site (23), investigations on the pH profile (29) and determination of catalysis as the rate-limiting step in TruB’s kinetic mechanism (30). The bacterial TruB enzyme is a homologue of the archaeal and eukaryotic Cbf5 protein (31) that acts as the pseudouridine synthase in H/ACA small (nucleolar) ribonucleoproteins (32).
In this study, we have investigated the function of the basic residue in the active site (R181 in TruB) as well as the role of a nearby second-shell residue (D90 in insert 1 of TruB) using a combination of site-directed mutagenesis, biochemistry, biophysics and computational methods. Our results underline the importance of both residues for catalysis of pseudouridine formation, but not for tRNA interaction. In addition, we identify the electrostatic properties of these residues as their most important characteristic, which—together with molecular dynamic simulations—suggests that the catalytic aspartate, the conserved basic residue and the second-shell D90 residue form an electrostatic interaction network that is critical for catalysis of pseudouridylation.
MATERIALS AND METHODS
Buffers and reagents
TAKEM4 buffer: 50 mM Tris–HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, 1 mM EDTA, 4 mM MgCl2. [C5-3H]-UTP for in vitro transcriptions was purchased from Moravek. T4 RNA ligase was purchased from New England Biolabs. All other enzymes and chemicals were obtained from Fermentas (Fisher Scientific). 2-aminopurine (2AP)-labeled 3′-half tRNA was purchased from Dharmacon (Thermo Fisher Scientific).
Protein expression and purification
The Escherichia coli TruB coding region in the pET28a-EcTruB plasmid (30) was mutated using QuikChange® site-directed mutagenesis (Stratagene) to encode the amino acid substitutions D90E, D90N, D90A, R181K, R181M and R181A. TruB wild type and variants were expressed and purified as previously described (30) using affinity and size-exclusion chromatography. Final protein concentration was determined using A280 (molar extinction coefficient of 20 985 M−1 cm−1) and by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) comparison. A260 and urea-PAGE analysis showed that protein preparations were not detectably contaminated with nucleic acid, and final protein purity was >95% as judged by SDS-PAGE.
[3H]-labeled tRNA preparation
E. coli [3H]-tRNAPhe was prepared as described (30). In brief, template DNA for tRNAPhe was amplified from the pCF0 plasmid (33) and used for in vitro transcription of tRNAPhe including 100 µM [C5-3H]-UTP. The [3H]-tRNA was purified with a Nucleobond PC100 column (Macherey & Nagel). Scintillation counting and absorbance measurements at 260 nm were used to quantify the tRNA concentration and specific activity.
Tritium release assay
[3H]-tRNA was refolded in 1× TAKEM4 buffer by incubation at 65°C for 5 min followed by cooling to room temperature for 10 min. TruB was diluted in 1× TAKEM4 buffer and pre-warmed at 37°C before adding the folded tRNA and incubating at 37°C. Samples were taken from the reaction at various time points and the amount of released tritium corresponding to the amount of pseudouridine formation was determined as described (30). To determine the apparent rate of catalysis (kapp), single-turnover experiments were analyzed by one-exponential fitting:
2AP-tRNA preparation
2AP-tRNA was prepared by annealing two halves of the tRNA and ligating to give full-length tRNA. The following 3′-half was purchased from Dharmacon and contained a 2AP label at position 57 (full-length numbering): 5′-AAAUCCCCGUGUCCUUGGUUCG-2AP-UUCCGAGUCCGCGCACCA-3′. The 5′-half was generated by assembly and amplification of template DNA followed by in vitro transcription. First, the template was assembled by polymerase chain reaction using the 5′-half sense primer (5′-GCGTAATACGACTCACTATAGCGCGGATAGCTCAGTCG-3′) and the 5′-half antisense primer (5′-TCAATCCCCTGCTCTACCGACTGAGCTATCCG-3′); second, the product of this assembly was further amplified using the sense T7 promoter primer (5′-GCGTAATACGACTCACTATAG-3′) and a methylated reverse primer (5′-mUmCAATCCCCTGCTCTACCGAC-3′). The 5′-half tRNA template was then used in 2 ml in vitro transcription reactions using the same procedure as outlined for [3H]-tRNA, but including 3 mM nonradioactive UTP. Following ethanol precipitation, the RNA was purified using the BioRad Model 491 Prep Cell continuous electrophoresis purification system. The RNA (1.44 ml) was loaded on a 5-cm 12% urea-PAGE (28 mm diameter) and a constant voltage of 250 V was applied while eluting with 1× Tris/Borate/EDTA (TBE) buffer at a flow rate of 0.75 ml/min at the bottom of the gel. Peak fractions (A254) were analyzed by 12% urea-PAGE, pooled and ethanol-precipitated in the presence of 0.05 µg/µl glycogen. To generate full-length 2AP-tRNA, 15 µM 5′-half tRNA was annealed in 1.5× excess to 2AP-3′-half tRNA in 1× T4 RNA ligase buffer (New England Biolabs) by heating at 95°C for 3 min, 65°C for 10 min and then slowly cooling from 65 to 20°C over 45 min. T4 RNA ligase (0.02 U/ml) was added to the mixture followed by incubation at 37°C for 4 h. Ligated tRNA was purified by urea-PAGE, ultraviolet-shadowing and electroelution (EluTrap electroelution system, Whatman).
Fluorescence spectroscopy and stopped-flow experiments
2AP-tRNA (0.05–0.2 µM) diluted in 1× TAKEM4 buffer was excited at 325 nm, titrated with TruB and the emission was measured from 340 to 400 nm using a Quanta Master 60 fluorescence spectrometer (Photon Technology International). After the first addition of TruB, the sample was incubated at room temperature for 30 min in the dark to allow for pseudouridine formation to occur. Similarly, TruB was titrated into buffer, and the measured fluorescence intensity was subtracted from the titration with 2AP-tRNA. Relative fluorescence changes at 364 nm were plotted against protein concentration. The data were fitted with a hyperbolic function to determine the KD for TruB binding to 2AP-tRNA:
Pre–steady-state fluorescence experiments were performed using a KinTek SF-2004 stopped-flow apparatus. A final concentration of 0.1 µM 2AP-tRNA was rapidly mixed with 3 µM TruB and excited at 325 nm. Emission was monitored at wavelengths >350 nm. For TruB wild type, the time courses were fitted with a 3-exponential function:
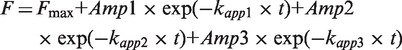 |
where Fmax is the fluorescence end-level; Amp is the respective amplitude of fluorescence change for each phase; t is time; and kapp indicates the apparent rate of the respective phase. Experiments with the TruB variants were best fit with a 2-exponential function:
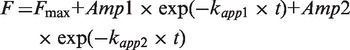 |
TruB molecular dynamics simulations
TruB wild type was simulated in two different conformations as observed in the presence (bound) and absence (apo) of RNA. In crystal structures of E. coli TruB in the absence of RNA, insufficient resolution was obtained to place the thumb-loop structure (residues 124–152) (26). Therefore, to simulate TruB-apo, first a homology model of E. coli TruB-apo was generated using the Swiss-Model server (34,35) and the Thermotoga maritima TruB-apo crystal structure [PDBID: 1ZE1 (27)] as a template such that the ordered thumb-loop could be added to the crystal structure of E. coli TruB-apo (PDBID: 1R3F). To do this, the T. maritima TruB structure was superimposed with the crystal structure of E. coli TruB-apo with respect to the catalytic domain. The coordinates of residues 124–152 corresponding to the thumb-loop of T. maritima TruB were added to the coordinates of E. coli TruB-apo.
To generate a starting structure for TruB-bound, the crystal structure for E. coli TruB bound to a short RNA [PDBID: 1K8W (5)] and the TruB-apo model were used. Since the crystal structure for TruB-bound was missing amino acids at both the N- and C- termini, E. coli TruB-bound was superimposed with TruB-apo with respect to residue 10, and the coordinates of residues 8–9 from TruB-apo were added to the coordinates of TruB-bound. The structures were then superimposed with respect to residues 310–311, and the coordinates of residues 312–314 from TruB-apo were added to the coordinates of TruB-bound. Water molecules within 10 Å of the protein as observed in the crystal structure (PDBID: 1K8W) were also included in the TruB-bound model. This was not done for the TruB-apo model because there were no water molecules identified in the crystal structure. The model for the TruB D90N simulation was generated from the TruB-bound model, modifying D90 to N90 with the Mutator plugin in Visual Molecular Dynamics (VMD) (36). Hydrogen atoms were added to each model using the Psfgen plugin in VMD.
All models were solvated using the solvate package in VMD to create a water box that extended at least 10 Å around the protein in each direction. The models were then minimized in five stages using a 0.5 fs time step, in which the protein molecules were fixed for the first 10 000 steps, followed by fixing the water molecules for the next 10 000 steps, repetition of the first two stages and releasing all constraints for 100 000 steps or until the energy of the system stabilized. The final minimized structures were ionized and neutralized using the autoionize package in VMD. Since wet-lab work with TruB was conducted using TAKEM4 buffer, the structures were ionized and subsequently neutralized by placing 100 mM K+, 100 mM Cl− and 3 mM Mg2+ no closer than 5 Å to each other or to the protein. The models were further minimized for 100 000 steps at 300 K without any atom constraints. The final model for TruB-apo contained 4811 protein atoms, 45 321 water atoms, 37 K+ ions, 30 Cl− ions and 1 Mg2+ ion (50 200 atoms total). The final model for TruB-bound contained 4811 protein atoms, 44 634 water atoms, 37 K+ ions, 30 Cl− ions and 1 Mg2+ ion (49 513 atoms total). The final model for TruB D90N contained 4813 protein atoms, 44 826 water atoms, 36 K+ ions, 30 Cl− ions and 5 Mg2+ ions (49 710 atoms total).
Before starting the simulations, the models were equilibrated at 310 and 360 K for 150 ps with a step size of 0.5 fs at constant pressure (1 atm) and variable volume (NPT ensemble) while invoking periodic boundary conditions. A Nosé-Hoover Langevin piston was used for pressure control, and Langevin dynamics were used to maintain a constant temperature. Production phase simulations were performed at 310 K using the final atomic velocities from the equilibration at 310 K and the atomic coordinates from the equilibration at 360 K. The particle mesh Ewald method was used to calculate full-system electrostatics. Simulations were generated in NAMD2 with CHARMM27 parameters (37,38) for 40 ns using periodic boundary conditions, NPT ensemble conditions with a Nosé-Hoover Langevin piston for pressure control and Langevin dynamics for temperature control. The simulations were analyzed using scripts written in house, evoked in VMD. Data from the trajectories was sampled every 2 ps. Root-mean-square deviation (RMSD) was calculated using the backbone atoms of all residues and all residues excluding the thumb-loop (residues 124–152). Root-mean-square fluctuation (RMSF) was calculated for the backbone atoms of each residue excluding the first 5 ns when the simulation was still stabilizing. Active site interactions were analyzed by measuring the distance between the terminal carbon atom of the carboxylate side chain group of aspartate and the terminal carbon of the guanidinium side chain group of arginine.
RESULTS
To identify and analyze interactions between active site residues of TruB, a multiple sequence alignment of 100 bacterial TruB sequences was used to determine highly conserved residues that were then mapped onto the E. coli TruB structure (Figure 1). As known before, the active site and the RNA-interaction face of the protein are highly conserved including three known active site residues, i.e. catalytic aspartate 48, tyrosine 76 that is thought to help stabilize the conformation of the flipped-out RNA bases after binding (23) and arginine 181 that interacts with D48 (5,25,26,39). Although R181 has been proposed to be a catalytic residue of pseudouridine synthases based on its conservation and location in the active site, its function has not been tested before (to the best of our knowledge). In addition, residues L200 and R202 are universally conserved and located on the β-sheet behind the active site. Notably, the only other invariant residue close to the active site is aspartate 90, a second-shell residue located in insert 1. As reported previously, this D90 residue contacts the strictly required C56 of the RNA substrate (26–28).
Figure 1.

Conserved residues in the active site of the bacterial pseudouridine synthase TruB. (A) Sequence alignment of TruB from different bacteria (E. coli, Serratia marcescens, Yersinia enterocolitica, Vibrio cholera, Haemophilus influenzae), Saccharomyces cerevisiae Pus4 and Pyrococcus furiosus Cbf5. The conserved active site residues D48 and R181 as well as the conserved second-shell residue D90 in insert 1 (E. coli numbering) are indicated by asterisks. (B) TruB sequence conservation mapped onto the E. coli TruB apo structure (PDBID: 1R3F) and represented by a color gradient of red to white to blue, indicating 0–100% conserved, respectively (a colour version of this figures is available online). The conservation of each residue was determined from a multiple sequence alignment of 100 bacterial TruB sequences. Stick models of the conserved residues D48, D90 and R181 are shown in yellow. D48–R181 C-C distance: 4.5 Å, D90–R181 C-C distance: 3.8 Å. (C) Close-up view of TruB active site (cyan), including 22-mer T-loop RNA (dark blue). Stick models show proximity of D48 (green) and R181 (blue) of TruB to U55 of RNA [red, 5-fluoro-6-hydroxy-pseudouracil (5FhΨ55) in this structure], as well as proximity of D90 of TruB (purple) in insert 1 (also purple) to C56 of RNA (yellow). D48–R181 C-C distance: 4.0 Å, D90-R181 C-C distance: 5.7 Å. Image was generated in VMD using PDBID: 1K8W.
Here, we aimed to elucidate the exact role of the highly conserved active site residues R181 and D90 for the function of E. coli TruB. Toward this goal, site-directed mutagenesis was used to substitute D90 (with glutamate, asparagine or alanine) and R181 (with lysine, methionine or alanine). The substitutions were chosen to disrupt the charge and alter the size of the original residue at each position. These TruB variants were over-expressed and purified by affinity and size-exclusion chromatography resulting in >95% purity with no detectable RNA contamination.
tRNA affinity of TruB D90 and R181 variants
First, the ability of the TruB variants to bind tRNA was tested by fluorescence titrations using a novel assay based on tRNAPhe containing a 2AP in position 57; at this position, TruB has been reported to accept any nucleobase (28). 2AP is a fluorescent adenine analog that is quenched when involved in base-stacking interactions and is often used to study RNA folding (40). We hypothesize that 2AP at position 57 will undergo a fluorescence change upon binding to TruB when nucleotides 55–57 of the tRNA are flipped into the active site of the enzyme. The 2AP-tRNA was generated by ligating an in vitro transcribed 5′ half of the tRNA to a chemically synthesized 3′ half containing the 2AP label (for details see ‘Materials and Methods’ section). 2AP-tRNA was titrated with TruB wild type, D90 and R181 variants, and the relative fluorescence change at 364 nm was recorded (Figure 2). A hyperbolic function was fitted to the data yielding the dissociation constant (KD) (Table 1). For TruB wild-type, a KD of 0.7 ± 0.1 µM was recorded, which is in excellent agreement with previous absorbance-based measurements (30). Therefore, we conclude that the 2AP label does not significantly affect tRNA binding and is useful for TruB–tRNA interaction studies. The TruB D90 variants showed no decrease in tRNA affinity compared with the wild type, and the same is true for TruB R181K and R181M. Only TruB R181A displayed a 3-fold increase in the dissociation constant compared with the wild type enzyme (Table 1). During the incubation period before fluorescence measurements, active TruB variants will modify the tRNA, and consequently the affinity for product tRNA is measured. For inactive TruB variants, the affinity for unmodified substrate tRNA is determined. Since TruB binds substrate and product tRNA with similar affinities (14,30), the determined dissociation constants should be comparable even for heterogeneous populations of substrate and product tRNA. Hence, our results show that substitutions of R181 or D90 in the active site of TruB do not strongly affect tRNA binding.
Figure 2.

Binding of 2AP-tRNA by TruB variants. 2AP-tRNA (50–200 nM) was titrated with increasing concentrations of TruB, and the relative fluorescence change of 2AP at 364 nm was recorded. Three representative titrations are shown for TruB wild type (closed circles, dashed line), TruB D90E (closed squares) and TruB R181K (open squares). Fitting a hyperbolic function to each titration curve provided the dissociation constants (KD) summarized in Table 1.
Table 1.
Dissociation constants determined from fluorescence titrations (Figure 2)
| TruB | KD, μM |
|---|---|
| Wild type | 0.7 ± 0.1 |
| D90E | 0.4 ± 0.2 |
| D90N | 0.4 ± 0.1 |
| D90A | 0.4 ± 0.2 |
| R181K | 0.7 ± 0.1 |
| R181M | 0.5 ± 0.2 |
| R181A | 2 ± 1 |
KD values are stated with their standard deviation derived from hyperbolic fitting of the titration curves (Figure 2).
Effects of amino acid substitutions on catalysis of pseudouridine formation
Next, the catalytic activity of each TruB variant was assessed in tritium release assays using [C5-3H]-labeled tRNA. Upon formation of the new C5-C1’ glycosidic bond in pseudouridine, the tritium is released and can be quantified by extraction and scintillation counting (41). Initially, multiple-turnover conditions were used by incubating 20 nM of TruB with 950 nM tRNA. Surprisingly, hardly any pseudouridine formation was detected within 60 min for all D90 and R181 variants, while TruB wild type rapidly converted nearly all substrate into product within the first 5 min (data not shown). When the enzyme concentration was increased to 200 nM, complete conversion of substrate to product was seen for TruB wild type in <1 min, whereas only TruB D90E and D90N showed significant product formation under these conditions (Figure 3A). TruB D90A was able to form pseudouridine, but only reached 30% product formation after 120 min. When incubating for 30 h, pseudouridylation could be detected with TruB R181K, but not TruB R181M and R181A (Figure 3B). In conclusion, substitutions of residue R181 and D90 lead to severe reductions in the ability of TruB to form pseudouridines, and the yields in multiple-turnover reactions are too low to conduct Michaelis–Menten titrations.
Figure 3.

Pseudouridine formation by TruB variants under multiple-turnover conditions. Tritium-labeled tRNAPhe (0.95 μM) was incubated with TruB (0.2 μM) at 37°C, and pseudouridine formation was quantified at selected time points with the help of a tritium release assay. (A) Two-hour time courses with TruB wild type (closed circles), TruB D90E (closed squares), TruB D90N (closed triangles) and TruB D90A (closed diamonds). (B) Thirty-hour time courses with TruB wild type (closed circles) and TruB R181K (open squares) to confirm detectable activity (no enzyme control, open circles).
To detect residual activity of the TruB R181M and TruB R181A variants, as well as to determine the rates for catalysis by all TruB variants, single-turnover experiments were performed, again using 950 nM tritium-labeled tRNA, but an excess of TruB enzyme (3 µM). Under these conditions of high enzyme concentration, product formation was observed for all D90 and R181 TruB variants (Figure 4A and B), albeit at much slower rates than for TruB wild type. In fact, TruB R181M and R181A required incubation with tRNA for up to 30 h before pseudouridylation activity could be reliably detected (Figure 4C). The single-turnover time courses were fit with single-exponential functions to obtain the rate of pseudouridine formation (Table 2). Compared with the single-turnover rate of TruB wild type (30), a drastic reduction in catalytic rate was seen for each variant. The D90 variants were generally more active than the R181 variants, showing a reduction in catalytic rate of 30 -, 50- and 500-fold for substitutions D90E, D90N and D90A, respectively. TruB R181K displayed a 2500-fold reduction in catalytic activity compared with wild type, and TruB R181M and R181A were the most impaired at pseudouridine formation with a reduction in catalytic rate of >20 000-fold. Importantly, the tritium release assay is able to detect pseudouridine formation while the tRNA is still in the active site of TruB (the enzyme is denatured and the tritium released during quenching of the reaction). Also, the single-turnover assays are independent of product release as each enzyme modifies only one substrate tRNA. Therefore, the decrease in rate is due to slow catalysis (or an earlier step in the kinetic mechanism), but cannot be explained by slow product release alone.
Figure 4.

Pseudouridine formation by TruB variants under single-turnover conditions. The percentage of pseudouridine formed was determined during incubations of 0.95 μM titrium-labeled tRNAPhe with an excess of 3 μM TruB at 37°C. (A) Analysis of TruB variants with substitutions of D90: TruB wild type (closed circles), TruB D90E (closed squares), TruB D90N (closed triangles) and TruB D90A (closed diamonds). (B) Effect of substitutions of R181: TruB wild type (closed circles), TruB R181K (open squares), TruB R181M (open triangles) and TruB R181A (open diamonds). (C) Long time courses with TruB R181M (open triangles) and TruB R181A (open diamonds). No enzyme control, open circles. (A–C) A single-exponential equation was fitted to the data to determine the rate of catalysis, kapp (smooth lines, see Table 2).
Table 2.
Apparent rates of pseudouridine formation by TruB variants
| TruB | kapp, s−1 | Fold decrease |
|---|---|---|
| Wild type | 0.5 ± 0.2 | |
| D90E | 0.017 ± 0.003 | 30 |
| D90N | 0.010 ± 0.003 | 50 |
| D90A | 0.0010 ± 0.0002 | 500 |
| R181K | 0.0002 ± 0.0001 | 2500 |
| R181M | < 3 × 10−5 | >20 000 |
| R181A | <3 × 10−5 | >20 000 |
Apparent rates, kapp, were determined by single-exponential fitting of time courses recorded under single-turnover conditions (Figure 4). The fold decrease in catalytic rate is given relative to the rate constant of pseudouridine formation by TruB wild type as previously determined (30). kapp represents average rate from duplicate data ± standard deviation.
Kinetics of tRNA binding to TruB variants
To convert uridine to pseudouridine, a number of steps have to take place including binding of tRNA, flipping of the target uridine and the two subsequent bases into TruB’s active site, breaking of the N1–C1’ glycosidic bond, rotation or flipping of the detached uracil base, formation of a new C5–C1’ glycosidic bond and release of the modified tRNA. To narrow down the step in catalysis in which the TruB variants were impaired, rapid-kinetic stopped-flow analysis was performed using tRNAPhe containing a 2AP in position 57. To observe the interaction of tRNA with TruB wild type and variants in real time, 0.1 µM 2AP-tRNA was rapidly mixed with an excess of TruB (3 µM). When 2AP-tRNA interacted with TruB wild type, a two-phase rapid increase in fluorescence was detected followed by a slower decrease in fluorescence (Figure 5). In contrast, when mixing 2AP-tRNA with TruB D90 or TruB R181 variants, only the biphasic fluorescence increase was observed without a subsequent fluorescence decrease. Fitting of the time courses with three- or two-exponential functions yielded the apparent rates for each fluorescence change. The first rapid fluorescence increase has a relatively small amplitude rendering it difficult to determine precise apparent rates (82 ± 61 s−1 for wild type), but qualitatively there is no difference for kapp1 among the different TruB variants analyzed. The second fluorescence increase has a larger amplitude and takes place with a rate of 4.2 ± 0.5 s−1 for the wild type enzyme. This apparent rate is similar to a previously determined overall rate of tRNA binding to TruB based on absorbance changes (30). As these earlier studies showed that this rate is independent of the TruB concentration, it was concluded that tRNA binding to TruB takes place in at least two steps. With the 2AP reporter group, this second phase has the largest amplitude and likely represents base-flipping into the active site of TruB. As the 2AP label is highly sensitive to its environment, it might be that the first rapid fluorescence increase detected here, which has a comparably small amplitude, represents the initial bimolecular encounter of tRNA and TruB. The D90 and R181 variants display kapp2 values that are only 2-fold lower than observed for TruB wild type (see Table 3 for all kapp2 values), suggesting that the substitutions do not significantly influence the kinetics of tRNA binding and the subsequent base-flipping into TruB's active site. This is in agreement with comparable affinities of these proteins to tRNA (Table 1). Hence, the observed strong impairments in pseudouridylation are not a result of deficiencies in tRNA binding and likely not of base-flipping, but stem directly from effects on the chemical steps in TruB’s mechanism.
Figure 5.

Kinetics of tRNA interaction with TruB observed by 2AP fluorescence. tRNA (0.1 µM) was rapidly mixed in a stopped-flow apparatus with TruB (3 µM) wild type (gray), TruB D90 variants (A) and R181 variants (B). Each time course was fitted with a 3-exponential function for TruB wild type (smooth black line), or a 2-exponential function for TruB D90E and R181K (smooth white lines), D90N and R181M (dashed white lines) and D90A and R181A (dotted white lines). Apparent rates for the strong fluorescence increase (kapp2) derived from the fitting are summarized in Table 3; other apparent rates are described in the results.
Table 3.
Apparent rates of tRNA binding to TruB wild type and variants
| TruB | kapp2, s−1 |
|---|---|
| Wild type | 4.2 ± 0.5 |
| D90E | 1.5 ± 0.7 |
| D90N | 1.9 ± 0.4 |
| D90A | 2.0 ± 0.4 |
| R181K | 1.9 ± 0.5 |
| R181M | 3 ± 1 |
| R181A | 4 ± 1 |
Apparent rates (kapp2) for the strong fluorescent increase were determined by 3-exponential fitting of stopped-flow time courses for TruB wild type and 2-exponential fitting for the TruB variants (Figure 5). kapp2 represents the average apparent rate from at least five time courses ± standard deviation.
The apparent rate of the fluorescence decrease observed uniquely with TruB wild type was 0.26 ± 0.02 s−1 and similar to the previously determined rate of catalysis for TruB [0.5 ± 0.2 s−1 (30)]. Presumably, this fluorescence decrease reflects release of the tRNA, which is rate-limited by catalysis for TruB wild type (30). As the TruB D90 and TruB R181 variants are extremely slow in pseudouridine formation (Figure 4), it seems that the tRNA stays bound to the protein within this observation time (<1 min). Longer stopped-flow time courses with the TruB D90 and TruB R181 variants did not allow reliable determination of the rate of fluorescence decrease as this took place on a similar timescale as photobleaching. In summary, TruB D90 and TruB R181 variants interact with tRNA on a similar timescale as TruB wild type, but do not release tRNA rapidly due to their impairment in catalysis.
Molecular dynamics simulations of TruB
As D90 is not in direct contact with the substrate tRNA, the strong effects of substitutions of this residue on catalysis of pseudouridine formation are not immediately apparent. To shed more light on the intramolecular interactions and functional dynamics in the active site of TruB, we conducted a series of molecular dynamics (MD) simulations of TruB wild type as well as TruB D90N. To account for the different structural states of TruB, TruB wild type was modeled in two functionally different conformations, its apo conformation as observed in crystal structures lacking RNA [PDBID: 1R3F (26)], as well as its conformation when bound to tRNA [PDBID: 1K8W (5)]. To avoid complications of simulating a protein–RNA complex, the RNA was removed from the latter structure (see ‘Materials and Methods’ section for details of model building). In the following, the two simulations of TruB wild type are named ‘TruB-apo’ and ‘TruB-bound’. Furthermore, a model for TruB D90N was generated based on the TruB-bound model by substituting D90 for asparagine, as this conformation is more likely to represent an active conformation. All models were solvated and ionized with the addition of K+, Cl− and Mg2+ ions and simulated for 40 ns at 310 K.
As a first analysis of the MD simulations, the RMSD of the backbone atoms were calculated for all residues of TruB wild type (Figure 6A) and TruB D90N (Figure 6C), as well as for all residues excluding the thumb-loop residues 124–152 (shown only for TruB D90N, Figure 6C). After 5 ns of TruB wild type simulations, the RMSD values stabilized at ∼3.5 Å and at ∼3 Å when the thumb-loop residues were omitted from the calculations (data not shown). Similarly, the RMSD values from the TruB D90N simulations stabilized at ∼3 Å when omitting the highly mobile thumb loop (Figure 6C). Based on these findings, further analyses were performed omitting the first 5 ns of the simulations. Next, RMSF values were calculated for all TruB residues from 5 to 40 ns (Figure 6B and D) to identify the most flexible regions of TruB during the simulations. As expected, insert 1 (residues 85–100), the thumb loop, helix α6 and the C-terminal PUA domain had the highest RMSF values, as these elements have been shown to undergo conformational changes on RNA binding (26). Interestingly, the flexibility of insert 1 differed between the three MD simulations with high RMSF values for the TruB-bound conformation, but 2-fold lower values in the TruB-apo and the TruB D90N simulations—even though the latter one is built on the TruB-bound conformation. As D90 is located in insert 1, these findings suggest that the presence of an asparagine residue at position 90 in some way restricts the motion of this structural element.
Figure 6.

Global fluctuations of TruB during MD simulations. TruB wild type (in the absence of RNA, panels (A and B) was simulated at 310 K in the apo and bound conformation based on available crystal structures with and without RNA, respectively (for details see ‘Materials and Methods’ section). TruB D90N (C and D) was simulated in the bound conformation only. (A) RMSD of the backbone atoms of all residues for TruB wild type simulations (apo conformation, gray; bound conformation, black). (B) RMSF values for simulation of TruB wild type from 5–40 ns, with insert 1 (asterisk) and thumb-loop (double asterisk) residues indicated by dashed boxes (apo conformation, gray; bound conformation, black). (C) TruB D90N simulation. RMSD of the backbone atoms of all residues (black) and for backbone atoms of all residues excluding thumb-loop residues 124–152 (gray). (D) RMSF values for TruB D90N residues from 5 to 40 ns of simulation; insert 1 (asterisk) and thumb loop (double asterisk).
Specifically, changes in the interaction patterns between D48, R181 and D90 were analyzed to determine potential roles of these residues for catalysis and to better understand the impact of the D90N substitution. These three residues may interact through hydrogen bonds and/or electrostatic interactions. To best characterize these interactions in a single measurement, the distance between the carboxylate carbon atom of the aspartate side chains and the guanidinium carbon atom of the arginine side chain was measured for D48–R181 and D90–R181 throughout each simulation (Figure 7). Interestingly, the D48–R181 interaction was not as stable as predicted based on the analysis of TruB crystal structures (5,25–27). In particular in both TruB wild type MD simulations, there were significant fluctuations in the distance between D48 and R181. However, in the TruB D90N simulation, these two residues approached each other closely (<5 Å) for most of the interaction time; this interaction appeared stable from 15 to 40 ns of the simulation. In contrast to the D48–R181 interaction, we observed a short distance (<5.5 Å) between D90 and R181 for the majority of time in all three MD simulations. The TruB-bound simulation was the only one where some fluctuations of the D90–R181 distance were observed in the first 10 ns and the last 5 ns of simulation. For the wild-type simulations, the D90–R181 distance was in general shorter than the D48–R181 distance as evident in the histograms in Figure 7. For the TruB D90N simulation, however, the D48–R181 distance was generally shorter than the N90–R181 distance. This is particularly evident during the last 25 ns of the TruB D90N simulation (Figure 7C). In summary, the MD simulations reveal a stable D90–R181 interaction that was not described based on crystal structures (5,26,27) and a D48–R181 interaction that is less stable with the exception of the TruB D90N simulation.
Figure 7.

Active site residue interactions during MD simulations. Every 2 ps of simulation time, distances were measured between the guanidinium carbon of R181 and the carboxylate carbon of D48 and D90. The D48–R181 interaction is depicted in gray while the D90–R181 interaction is shown in black. Both time-resolved changes (left) as well as histograms of C-C distances (right) are reported. (A) TruB wild type in the apo conformation. (B) TruB wild type in the bound conformation. (C) TruB D90N in the bound conformation.
DISCUSSION
In the present study, we have analyzed by biochemical, biophysical and computational means the detailed role of residues R181 and D90 for the function of the pseudouridine synthase TruB. Our data suggest that the active site residue R181 and in particular its charge is critical for catalysis of efficient pseudouridine formation without influencing substrate binding. Interestingly, residue D90 is also important for catalysis although it is not in direct contact with the target uridine and is located in the second-shell surrounding the active site. Again, the charge of this residue seems to be its most important characteristic, and substitutions of this residue do not affect the interaction of TruB with substrate tRNA. MD simulations of TruB wild type and TruB D90N suggest that the three residues, D48, R181 and D90, form an electrostatic interaction network.
Residue R181 of TruB is a catalytic residue
Although the conserved basic residue in the active site, R181 in TruB, has been discussed in a number of publications describing pseudouridine synthases (5,25), no biochemical studies testing the exact role of this residue have been reported. Here, we unambiguously demonstrate the importance of R181 in bacterial TruB for catalysis of pseudouridine formation. Even a conservative substitution of this residue with lysine leads to a 2500-fold reduction in the rate of pseudouridylation while substitutions with methionine or alanine almost abolish activity. This is particularly remarkable considering that lysine is found in the corresponding position in enzymes from the RsuA, TruD and Pus10 families (3,8,42). A basic residue in this position is evidently essential for catalyzing pseudouridine formation, and therefore this residue can be classified as a catalytic residue. For TruB specifically, the lysine substitution might result in significantly less activity compared with TruB wild type (with arginine in position 181) either because lysine has fewer potential hydrogen-bond donors or because it is shorter. In this respect, it is noteworthy that we observed in the MD simulations that R181 often interacted simultaneously with both D48 and D90 (Figure 8), which might be facilitated by the guanidinium group forming multiple interactions. Also, visual inspection of substituting R181 with lysine in the crystal structure of TruB in the bound conformation revealed that lysine should still be long enough to effectively interact with either D48 or D90. As expected for a catalytic residue and similar to the catalytic aspartate (14,30), R181 does not contribute significantly to substrate binding by TruB, presumably since the majority of contacts between TruB and tRNA are formed outside the active site. In conclusion, a basic residue in position 181 is essential for catalysis of pseudouridine formation, and in TruB an arginine might be strictly required as it is able to simultaneously interact with D48 and other residues such as D90.
Figure 8.

Selected conformation of TruB active site residues during MD simulations. Representative active site conformations where R181 (blue) was found in close proximity to both D48 (green) and D90 within insert 1 (purple) are shown. RNA from the TruB-RNA crystal structure (PDBID 1ZL3) was modeled into the simulation frames (red/yellow/light purple). (A) Snapshot at 9.1 ns of the TruB-bound simulation. D48–R181 C-C distance: 3.7 Å, D90–R181 C-C distance: 3.9 Å. (B) Snapshot at 15.6 ns of the TruB D90N simulations. D48–R181 C-C distance: 3.6 Å, N90-R181 C-C distance: 4.6 Å. In neither frame do the interactions between the active site residues clash with the crystal structure position of the RNA substrate.
Residue D90 in insert 1 is critical for pseudouridine formation by TruB
We initially predicted the importance of residue D90 in insert 1 of TruB based on its remarkably high conservation and proximity to the active site. Since D90 has been seen in crystal structures to interact with C56 in the tRNA (26,27), it was expected that D90 substitutions would have a greater effect on tRNA binding than on catalysis. However, our results show that all TruB D90 variants had a similar affinity for tRNA as TruB wild type, indicating that D90 is not critical for substrate binding and that the hydrogen bond to C56 is not a major binding determinant. As D90 and C56 are located on the surface of the TruB–tRNA complex, we suspect that this hydrogen bond can be replaced by interactions with the surrounding water. Moreover, C56 can also be recognized by residues in the thumb loop (27), and this interaction may also compensate for substitutions of D90. Surprisingly though, all three substitutions of D90 severely impaired pseudouridine formation, with even the most active variant (TruB D90E) displaying a 30-fold decrease in catalytic rate. As the effect of replacing aspartate 90 with asparagine was even larger than substituting with glutamate, it seems that the negative charge of this residue is most important for TruB’s activity. This highlights a role of this residue for catalysis rather than tRNA binding as the hydrogen bond of residue 90 to C56 of the tRNA should be retained in the TruB D90N variant. The critical importance of the negative charge at position 90 suggests that electrostatic interactions of this residue contribute to pseudouridine formation. Both the TruB apo crystal structure (26) as well as our MD simulations reveal that R181 is a potential electrostatic interaction partner of D90 and the only positively charged group in the vicinity of D90. The potential role of the electrostatic interactions of D90 for catalysis of pseudouridine formation will be discussed in more detail below.
Dynamics of active site interactions in TruB
Here, we also report the first MD simulations of TruB in two different conformations (i.e. the apo conformation and the RNA-bound conformation) although the RNA was omitted from the simulations for technical reasons. It is noteworthy that our data confirm the presence of several flexible regions in TruB; namely, insert 1, the thumb loop, helix 6 and the PUA domain as evident in the RMSF values (Figure 6B and D). The flexible nature of these structural elements likely contributes to the relatively high RMSD values (∼3 Å) observed throughout our simulations (Figure 6A and C) (43,44). Since the RMSDs (excluding the thumb loop for TruB D90N) do not display any significant drifts after the first 5 ns of simulation, we consider the simulations to be stable allowing us to analyze interactions in TruB’s active site.
The MD simulations of TruB revealed that R181 was capable of forming a salt bridge with D90 (Figure 8) in addition to the previously described interaction with D48 (5,25,26,39). Interestingly, such an interaction between R181 and D90 is also visible in the apo TruB crystal structure (Figure 1A), while in all other crystal structures of TruB, R181 is in a bent orientation pointing away from D90 (Figure 1B). This difference might be explained by the absence of RNA in the MD simulations, as the RNA may influence the orientation of nearby residues, in particular of R181. However, superimposing the RNA from crystal structures onto representative frames from the MD simulations reveals that a close network of interactions between D48, R181 and D90 is compatible with RNA binding and does not result in steric clashes (Figure 8). It is therefore feasible that these three residues adopt multiple conformations before and during the interaction with tRNA and that one of these conformations is a close network between D48, R181 and D90 as observed in the MD simulations.
The MD simulations presented here strongly support the suggestion that the proximity of the negatively charged D90 residue influences the nearby R181—independent of the exact orientation of these residues. This is particularly evident in the simulations of TruB D90N where this interaction is reduced to the formation of a hydrogen bond instead of an electrostatic interaction. In this case, the interaction between R181 and D48 seems to become even stronger as the two residues are in close proximity without major fluctuations for 25 ns of the simulation. Therefore, changing D90 to asparagine apparently stabilized the interaction between R181 and D48, but did not drastically alter the interaction between R181 and N90. The only other notable difference in the TruB D90N simulations, compared with simulations of TruB wild type in the bound conformation, was the reduced mobility of insert 1 containing the D90N substitutions (Figure 6D). The exact molecular nature of this stabilization of insert 1 is unclear. These observations offer two explanations for the 50-fold reduced catalytic activity of TruB D90N compared with wild type. It might be that flexibility of insert 1 is important for catalysis; however, this is difficult to reconcile as movements of this structural element are typically associated with RNA binding or release. Alternatively, and maybe more likely, catalysis may be impaired in the TruB D90N variant because of the strengthened interaction between the catalytic aspartate 48 and arginine 181.
Model for the role of R181 and D90 in pseudouridine formation by TruB
Based on the findings from biochemical and computational studies, we propose that a finely balanced interaction network of residues D48, R181 and D90 is required for efficient catalysis of pseudouridylation by TruB. As evident from the TruB R181M and R181A variants, catalysis is almost completely abolished if D48 is not able to interact with R181. On the other hand, if the interaction between R181 and D48 is too stable, as was seen in the TruB D90N MD simulation, the rate of catalysis is also reduced. We propose that R181 must interact with D48, but the strength of the interaction needs to be modulated by additional contacts such as to D90. For example, the contact between R181 and D90 may be important for disrupting the interaction of R181 with D48 to make the catalytic aspartate 48 residue available for nucleophilic attack of the target uridine. The D90 residue could thereby sequester the positively charged R181 residue while D48 is participating in the chemical reaction. This would mitigate a highly disfavored uncompensated positive charge on R181 in the protein’s interior. Previously, it was hypothesized that the thumb loop could partially fulfill this role (25), but it seems likely that the negatively charged D90 residue contributes to interacting with R181 while D48 is engaged in the chemical reaction.
Lastly, we asked whether the contribution of an electrostatic network to catalysis could be a common principle in pseudouridine synthases. The catalytic role of a basic residue for pseudouridine formation seems to be conserved in all pseudouridine synthases, as they all contain either an arginine or a lysine in the corresponding position. In contrast, D90 is 100% conserved in insert 1 in all bacterial TruBs, but insert 1 is unique to TruB. Therefore, we searched for similarly conserved, negatively charged residues in other pseudouridine synthases that are in the spatial vicinity of the conserved basic residue. We identified an aspartate in position 178 of E. coli RluA that is 100% conserved in all aligned RluA sequences and in close proximity to RluA's catalytic arginine (Figure 9A). This residue is located in a unique insert of the RluA protein compared with other pseudouridine synthases. Also, in RluF, we found a highly conserved aspartate residue, D109, found in 80% of the sequences, that could interact with the catalytic arginine 190 (Figure 9B). Lastly, there is a negatively charged residue at position 243 in 73 of 100 analyzed TruA sequences that could potentially interact with the conserved arginine 205 on changing its orientation (Figure 9C). However, we could not find such a conserved negatively charged residue in Cbf5 or in Nop10 in H/ACA small (nucleolar) ribonucleoproteins. Also, the nearest phosphate in the H/ACA RNA is 13 Å away from the active site arginine such that it is unlikely to fulfill the same role as D90 in TruB. Furthermore, there seems to be no conserved acidic residue in TruD, which is the most divergent pseudouridine synthase. Interestingly, the conserved basic residue (K21) in TruD is located in a different structural element than in other pseudouridine synthases. This further supports the suggestion that residues in nonequivalent positions, such as the different second-shell acidic residues identified here, can fulfill the same functional role (7–9). In conclusion, it seems highly likely that the proposed network of the catalytic aspartate with a basic residue and another acidic residue plays a role in catalysis in several pseudouridine synthases in addition to TruB.
Figure 9.

Conserved negatively charged residues in the active site of bacterial pseudouridine synthases. Sequence conservation mapped onto crystal structures of E. coli pseudouridine synthases (A) RluA (PDBID: 2I82), (B) RluF (PDBID: 3DH3) and (C) TruA (PDBID: 2NQP), each bound to RNA. The conservation of each residue was determined from a multiple sequence alignment of 100 bacterial sequences of the protein and is represented by a colour gradient of red to white to blue, indicating 0–100% conserved, respectively (a colour version of this figures is available online). Stick models are shown in yellow of conserved residues for the catalytic aspartate and arginine (D48 and R181 in TruB) as well as a highly conserved negatively charged residue in close proximity. (A) The alignment of RluA showed D178 to be 100% conserved, which has a C-C distance to R165 of 4.0 Å. (B) D109 in RluF is 77% conserved (16% P, 4% E) with a C-C distance to R190 of 3.6 Å. (C) E243 in TruA was found to be 62% conserved (11% D, 9% G) and has a C-C distance to R205 of 7.9 Å in accordance with its orientation pointing away from R205.
In summary, we have proven the catalytic role of the conserved basic residue in the active site of pseudouridine synthases. While in the different pseudouridine synthases this residue can be either an arginine or a lysine interacting with the catalytic aspartate residue, the exact type of residue is highly conserved for each enzyme family. Substitutions of an arginine for lysine result in a 2500-fold reduced activity in the case of TruB, corresponding to an increase of activation energy from 77.8 kJ mol−1 to 97.5 kJ mol−1 for this reaction. This discovery indicates that the interactions in the active sites of pseudouridine synthases are finely balanced to optimize activity. For TruB, we have identified D90 in insert 1 as a critical residue for efficient pseudouridine formation. Rather than contributing significantly to substrate binding, D90 seems to form an electrostatic interaction network with D48 and R181 thereby facilitating catalysis. Similar conserved, negatively charged second-shell residues have been identified in RluA, RluF and TruA. These findings shed new light onto the complexity of pseudouridine formation. The challenge remains to identify the exact catalytic mechanism of pseudouridine synthases; a comprehensive description thereof will have to include the contribution of the catalytic basic residue and the second-shell acidic residue identified here.
FUNDING
Natural Science and Research Council of Canada [NSERC Discovery Grant 341996-2009 to U.K. and Discovery Grant 326979-2011 to H.-J. W.]; the Canada Foundation for Innovation (CFI Leaders Opportunity Fund, Project Number 13152, U.K.); the University of Lethbridge [University of Lethbridge Research Fund No. 13724, U.K.]; an NSERC CGS-M and an Alberta Innovates—Technology Futures Graduate Student Scholarship (to J.F.). Supercomputer time was provided by the WestGrid/Compute Canada Resource Allocation Committee. Funding for open access charge: The publication charges will be covered by research grants to Ute Kothe (NSERC Discovery Grant) and Hans-Joachim Wieden (AITF Strategic Chair).
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Adrian Ferré-D’Amaré, Juli Feigon, Marc Roussel and Steven Mosimann for critical discussion of this research. Furthermore, we are grateful to Laura Keffer-Wilkes for initial nitrocellulose filtration assays and preparation of tritium-labeled tRNA. Lastly, we appreciate the advice by Daniel Lafontaine on tRNA ligation.
REFERENCES
- 1.Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- 2.Hamma T, Ferré-D'Amaré AR. Pseudouridine synthases. Chem. Biol. 2006;13:1125–1135. doi: 10.1016/j.chembiol.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 3.McCleverty CJ, Hornsby M, Spraggon G, Kreusch A. Crystal structure of human Pus10, a novel pseudouridine synthase. J. Mol. Biol. 2007;373:1243–1254. doi: 10.1016/j.jmb.2007.08.053. [DOI] [PubMed] [Google Scholar]
- 4.Hoang C, Chen J, Vizthum CA, Kandel JM, Hamilton CS, Mueller EG, Ferré-D'Amaré AR. Crystal structure of pseudouridine synthase RluA: indirect sequence readout through protein-induced RNA structure. Mol. Cell. 2006;24:535–545. doi: 10.1016/j.molcel.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 5.Hoang C, Ferré-D'Amaré AR. Cocrystal structure of a tRNA Psi55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell. 2001;107:929–939. doi: 10.1016/s0092-8674(01)00618-3. [DOI] [PubMed] [Google Scholar]
- 6.Hur S, Stroud RM. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Mol. Cell. 2007;26:189–203. doi: 10.1016/j.molcel.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ericsson UB, Nordlund P, Hallberg BM. X-ray structure of tRNA pseudouridine synthase TruD reveals an inserted domain with a novel fold. FEBS Lett. 2004;565:59–64. doi: 10.1016/j.febslet.2004.03.085. [DOI] [PubMed] [Google Scholar]
- 8.Hoang C, Ferré-D'Amaré AR. Crystal structure of the highly divergent pseudouridine synthase TruD reveals a circular permutation of a conserved fold. RNA. 2004;10:1026–1033. doi: 10.1261/rna.7240504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaya Y, Del Campo M, Ofengand J, Malhotra A. Crystal structure of TruD, a novel pseudouridine synthase with a new protein fold. J. Biol. Chem. 2004;279:18107–18110. doi: 10.1074/jbc.C400072200. [DOI] [PubMed] [Google Scholar]
- 10.Alian A, DeGiovanni A, Griner SL, Finer-Moore JS, Stroud RM. Crystal structure of an RluF-RNA complex: a base-pair rearrangement is the key to selectivity of RluF for U2604 of the ribosome. J. Mol. Biol. 2009;388:785–800. doi: 10.1016/j.jmb.2009.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald MK, Miracco EJ, Chen J, Xie Y, Mueller EG. The handling of the mechanistic probe 5-fluorouridine by the pseudouridine synthase TruA and its consistency with the handling of the same probe by the pseudouridine synthases TruB and RluA. Biochemistry. 2010;50:426–436. doi: 10.1021/bi101737z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raychaudhuri S, Niu L, Conrad J, Lane BG, Ofengand J. Functional effect of deletion and mutation of the Escherichia coli ribosomal RNA and tRNA pseudouridine synthase RluA. J. Biol. Chem. 1999;274:18880–18886. doi: 10.1074/jbc.274.27.18880. [DOI] [PubMed] [Google Scholar]
- 13.Huang L, Pookanjanatavip M, Gu X, Santi DV. A conserved aspartate of tRNA pseudouridine synthase is essential for activity and a probable nucleophilic catalyst. Biochemistry. 1998;37:344–351. doi: 10.1021/bi971874+. [DOI] [PubMed] [Google Scholar]
- 14.Ramamurthy V, Swann SL, Paulson JL, Spedaliere CJ, Mueller EG. Critical aspartic acid residues in pseudouridine synthases. J. Biol. Chem. 1999;274:22225–22230. doi: 10.1074/jbc.274.32.22225. [DOI] [PubMed] [Google Scholar]
- 15.Del Campo M, Kaya Y, Ofengand J. Identification and site of action of the remaining four putative pseudouridine synthases in Escherichia coli. RNA. 2001;7:1603–1615. [PMC free article] [PubMed] [Google Scholar]
- 16.Conrad J, Niu L, Rudd K, Lane BG, Ofengand J. 16S ribosomal RNA pseudouridine synthase RsuA of Escherichia coli: deletion, mutation of the conserved Asp102 residue, and sequence comparison among all other pseudouridine synthases. RNA. 1999;5:751–763. doi: 10.1017/s1355838299990167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaya Y, Ofengand J. A novel unanticipated type of pseudouridine synthase with homologs in bacteria, archaea, and eukarya. RNA. 2003;9:711–721. doi: 10.1261/rna.5230603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zebarjadian Y, King T, Fournier MJ, Clarke L, Carbon J. Point mutations in yeast CBF5 can abolish in vivo pseudouridylation of rRNA. Mol. Cell. Biol. 1999;19:7461–7472. doi: 10.1128/mcb.19.11.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan CM, Huang RH. Enzymatic characterization and mutational studies of TruD-the fifth family of pseudouridine synthases. Arch. Biochem. Biophys. 2009;489:15–19. doi: 10.1016/j.abb.2009.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Behm-Ansmant I, Urban A, Ma X, Yu YT, Motorin Y, Branlant C. The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite-multisubstrate RNA:Psi-synthase also acting on tRNAs. RNA. 2003;9:1371–1382. doi: 10.1261/rna.5520403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kammen HO, Marvel CC, Hardy L, Penhoet EE. Purification, structure, and properties of Escherichia coli tRNA pseudouridine synthase I. J. Biol. Chem. 1988;263:2255–2263. [PubMed] [Google Scholar]
- 22.Miracco EJ, Mueller EG. The products of 5-fluorouridine by the action of the pseudouridine synthase TruB disfavor one mechanism and suggest another. J. Am. Chem. Soc. 2011;133:11826–11829. doi: 10.1021/ja201179f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phannachet K, Elias Y, Huang RH. Dissecting the roles of a strictly conserved tyrosine in substrate recognition and catalysis by pseudouridine 55 synthase. Biochemistry. 2005;44:15488–15494. doi: 10.1021/bi050961w. [DOI] [PubMed] [Google Scholar]
- 24.Nurse K, Wrzesinski J, Bakin A, Lane BG, Ofengand J. Purification, cloning, and properties of the tRNA psi 55 synthase from Escherichia coli. RNA. 1995;1:102–112. [PMC free article] [PubMed] [Google Scholar]
- 25.Hoang C, Hamilton CS, Mueller EG, Ferré-D'Amaré AR. Precursor complex structure of pseudouridine synthase TruB suggests coupling of active site perturbations to an RNA-sequestering peripheral protein domain. Protein Sci. 2005;14:2201–2206. doi: 10.1110/ps.051493605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan H, Agarwalla S, Moustakas DT, Finer-Moore J, Stroud RM. Structure of tRNA pseudouridine synthase TruB and its RNA complex: RNA recognition through a combination of rigid docking and induced fit. Proc. Natl Acad. Sci. USA. 2003;100:12648–12653. doi: 10.1073/pnas.2135585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phannachet K, Huang RH. Conformational change of pseudouridine 55 synthase upon its association with RNA substrate. Nucleic Acids Res. 2004;32:1422–1429. doi: 10.1093/nar/gkh287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu X, Yu M, Ivanetich KM, Santi DV. Molecular recognition of tRNA by tRNA pseudouridine 55 synthase. Biochemistry. 1998;37:339–343. doi: 10.1021/bi971590p. [DOI] [PubMed] [Google Scholar]
- 29.Hamilton CS, Spedaliere CJ, Ginter JM, Johnston MV, Mueller EG. The roles of the essential Asp-48 and highly conserved His-43 elucidated by the pH dependence of the pseudouridine synthase TruB. Arch. Biochem. Biophys. 2005;433:322–334. doi: 10.1016/j.abb.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Wright JR, Keffer-Wilkes LC, Dobing SR, Kothe U. Pre-steady-state kinetic analysis of the three Escherichia coli pseudouridine synthases TruB, TruA, and RluA reveals uniformly slow catalysis. RNA. 2011;17:2074–2084. doi: 10.1261/rna.2905811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koonin EV. Pseudouridine synthases: four families of enzymes containing a putative uridine-binding motif also conserved in dUTPases and dCTP deaminases. Nucleic Acids Res. 1996;24:2411–2415. doi: 10.1093/nar/24.12.2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meier UT. How a single protein complex accommodates many different H/ACA RNAs. Trends Biochem. Sci. 2006;31:311–315. doi: 10.1016/j.tibs.2006.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampson JR, DiRenzo AB, Behlen LS, Uhlenbeck OC. Nucleotides in yeast tRNAPhe required for the specific recognition by its cognate synthetase. Science. 1989;243:1363–1366. doi: 10.1126/science.2646717. [DOI] [PubMed] [Google Scholar]
- 34.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 35.Kiefer F, Arnold K, Kunzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37:D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 37.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaudhuri BN, Chan S, Perry LJ, Yeates TO. Crystal structure of the apo forms of psi 55 tRNA pseudouridine synthase from Mycobacterium tuberculosis: a hinge at the base of the catalytic cleft. J. Biol. Chem. 2004;279:24585–24591. doi: 10.1074/jbc.M401045200. [DOI] [PubMed] [Google Scholar]
- 40.Souliere MF, Haller A, Rieder R, Micura R. A powerful approach for the selection of 2-aminopurine substitution sites to investigate RNA folding. J. Am. Chem. Soc. 2011;133:16161–16167. doi: 10.1021/ja2063583. [DOI] [PubMed] [Google Scholar]
- 41.Cortese R, Kammen HO, Spengler SJ, Ames BN. Biosynthesis of pseudouridine in transfer ribonucleic acid. J. Biol. Chem. 1974;249:1103–1108. [PubMed] [Google Scholar]
- 42.Sivaraman J, Sauve V, Larocque R, Stura EA, Schrag JD, Cygler M, Matte A. Structure of the 16S rRNA pseudouridine synthase RsuA bound to uracil and UMP. Nat. Struct. Biol. 2002;9:353–358. doi: 10.1038/nsb788. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Hassan SA, Mehler EL. Long dynamics simulations of proteins using atomistic force fields and a continuum representation of solvent effects: calculation of structural and dynamic properties. Proteins. 2005;60:464–484. doi: 10.1002/prot.20470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wieden HJ, Mercier E, Gray J, Steed B, Yawney D. A combined molecular dynamics and rapid kinetics approach to identify conserved three-dimensional communication networks in elongation factor Tu. Biophys. J. 2010;99:3735–3743. doi: 10.1016/j.bpj.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]