

Abstract
Although engineered nucleases can efficiently cleave intracellular DNA at desired target sites, major concerns remain on potential ‘off-target’ cleavage that may occur throughout the genome. We developed an online tool: predicted report of genome-wide nuclease off-target sites (PROGNOS) that effectively identifies off-target sites. The initial bioinformatics algorithms in PROGNOS were validated by predicting 44 of 65 previously confirmed off-target sites, and by uncovering a new off-target site for the extensively studied zinc finger nucleases (ZFNs) targeting C-C chemokine receptor type 5. Using PROGNOS, we rapidly interrogated 128 potential off-target sites for newly designed transcription activator-like effector nucleases containing either Asn-Asn (NN) or Asn-Lys (NK) repeat variable di-residues (RVDs) and 3- and 4-finger ZFNs, and validated 13 bona fide off-target sites for these nucleases by DNA sequencing. The PROGNOS algorithms were further refined by incorporating additional features of nuclease–DNA interactions and the newly confirmed off-target sites into the training set, which increased the percentage of bona fide off-target sites found within the top PROGNOS rankings. By identifying potential off-target sites in silico, PROGNOS allows the selection of more specific target sites and aids the identification of bona fide off-target sites, significantly facilitating the design of engineered nucleases for genome editing applications.
INTRODUCTION
The efficiency of genome editing in cells is greatly increased by specific DNA cleavage with zinc finger nucleases (ZFNs) or transcription activator-like (TAL) effector nucleases (TALENs), which have been used to create new model organisms (1–6), correct disease-causing mutations (7) and genetically engineer stem cells (8). However, both ZFNs (6,9–11) and TALENs (5,8) have off-target cleavage that can lead to genomic instability, chromosomal rearrangement and disruption of the function of other genes. It is vitally important to identify the locations and frequency of off-target cleavage to reduce these adverse events, and ensure the specificity and safety of nuclease-based genome editing. Although the emerging systems utilizing clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR associated (Cas) proteins are highly active at their intended target sites, recent publications indicate that they likely have much greater levels of off-target cleavage than ZFNs or TALENs (12–14).
Experimental identification of ZFN and TALEN off-target sites is a daunting task because of the size of the genome and the large number of potential cleavage sites to assay. Previous attempts to identify new off-target sites based entirely on bioinformatics search methods have all failed to locate any off-target cleavage sites (1–4,7,15), which has led to the belief that identifying off-target activity based on sequence homology alone would not be fruitful (10). In contrast, efforts using experimental methods to characterize the specificity of nucleases have successfully identified several off-target cleavage sites for ZFNs (6,9–11,16) and TALENs (5,8). While most of these characterization methods incorporate a bioinformatics component to search through the genome, the final decision of what sites to investigate is dictated by the experimental data; for example, Perez et al. applied a classifier based on their characterization of the nucleases to narrow the full list of 136 genomic sites with two or fewer mismatches in each ZFN down to the top 15 sites they chose to interrogate (16). However, these experimental characterization methods, including SELEX (5,8,16), bacterial one-hybrid (6), in vitro cleavage (9) or IDLV trapping (10), can be very time consuming, costly and technically challenging (Supplementary Note 2). This has severely limited the number of laboratories undertaking these experiments and the number of nucleases characterized for off-target effects. There is a clear unmet need for a rapid and scalable online method that can predict nuclease off-target sites with reasonable accuracy without requiring the user to have specialized computational skills, especially for application of nucleases in disease treatment.
MATERIALS AND METHODS
Major features of PROGNOS ranking algorithms
All PROGNOS algorithms only require the DNA target sequence as input; prior construction and experimental characterization of the specific nucleases are not necessary. Based on the differences between the sequence of a potential off-target site in the genome and the intended target sequence, each algorithm generates a score that is used to rank potential off-target sites. If two (or more) potential off-target sites have equal scores, they are further ranked by the type of genomic region annotated for each site with the following order: Exon > Promoter > Intron > Intergenic. A final ranking by chromosomal location is employed as a tie-breaker to ensure consistency in the ranking order. Full descriptions and formulae of each PROGNOS algorithm are provided in Supplementary Method M1.
The average 5′-base and RVD-nucleotide frequencies for engineered TALEs were calculated by compiling previously published SELEX results of nine engineered TALEs (5,8,17) and calculating frequency matrices (Supplementary Table S16).
PROGNOS Homology, RVDs and Conserved G’s Algorithms
The ‘Homology’, ‘RVDs’ and ‘Conserved G’s’ algorithms in PROGNOS all apply the ‘energy compensation’ model of dimeric nuclease cleavage (9) to account for the interactions between the two half-sites, but the scores for each half-site are calculated in different ways. The Homology algorithm can be applied to both ZFNs and TALENs and is based largely on the number of mismatches relative to the intended target sequence. The RVDs algorithm is designed for use with TALENs and utilizes the RVD-nucleotide binding frequencies of natural TAL Effectors (18); alternate ‘5T’ and ‘5TC’ versions require either a thymidine or a pyrimidine to be in the 5′ position of each half-site. The Conserved G’s algorithm is designed for use with ZFNs and applies a weighting factor to the Homology algorithm that biases the rankings towards sites where intended guanosine contacts are maintained. More details for these algorithms can be found in Supplementary Method M1.
PROGNOS Algorithms ‘ZFN v2.0’ and ‘TALEN v2.0’
The weightings of the parameters for the refined PROGNOS algorithms ‘ZFN v2.0’ and ‘TALEN v2.0’ were developed by training the algorithms to maximize recovery of previously confirmed off-target sites, as well as the novel off-target sites found using the initial algorithms developed in this study. For each algorithm, ∼105 randomly assigned parameter sets (within a constrained range) were analyzed for their performance using the Perl off-target-ranking script. The top performing parameter sets were further optimized by running further analyses allowing each parameter to vary slightly from the original value.
ZFN v2.0 Algorithm
The ZFN v2.0 algorithm was constructed based on the binding of individual zinc finger subunits rather than treating all mismatches equally. Specifically, the scoring algorithm in ZFN v2.0 for each finger is based on: (i) an initial score of 100 is given as a starting point, (ii) if there is at least one mismatch, a ‘First_Penalty’ is subtracted, (iii) if there are additional mismatches, an ‘Additional_Penalty’ is subtracted for each additional mismatch, (iv) if a guanosine is the intended base at positions 2 or 3 and it matches the target sequence, a ‘G_Bonus’ is added, (v) if a guanosine is the intended base at position 1 and it matches the target sequence, a double ‘G_Bonus’ is added, (vi) if the resulting score is <0, it is set to zero. We further introduced parameters to model polarity effects by weighting the impact of each of the 2nd–4th nucleotide triplets away from the FokI domain. The score for each zinc finger subunit is multiplied by the corresponding polarity parameter and all scores for the half-site are summed together. The sum is then divided by the score of a subunit that has a perfect match to the intended target sequence of that half-site. To allow for compensation between the two ZFN dimers, the score for each half-site is raised to the power of ‘Dimer_Exponent’ before being summed together, divided by two, and multiplied by 100 to generate a score from 0 to 100 (100 being a perfect match). More details for the construction of the ZFN v2.0 algorithm can be found in Supplementary Method M1.
TALEN v2.0 Algorithm
In constructing the TALEN v2.0 algorithm, a score for each RVD-nucleotide interaction is calculated using the same formula as in TALE-NT (18) (as in the original RVDs algorithm) except that the RVD-nucleotide frequencies used were derived from engineered TAL domains instead of naturally occurring TAL Effectors. If no RVDs are specified by the user in the PROGNOS online input form, RVDs are assumed to follow the standard code based on the intended target sequence: NI→A, HD→C, NN→G, NG→T. Based on the finding that the presence of the ‘strong’ RVDs NN and HD are key to TAL binding (19), we hypothesized that these RVDs may impart excess binding energy that could compensate for local effects of adjacent RVD-nucleotide mismatches. Accordingly, we developed two parameters, ‘Single_Strong’ and ‘Double_Strong’ that were applied to the score of RVDs that were flanked on one or both sides by NNs or HDs correctly bound to their respective intended bases (guanosines or cytidines). If these criteria are met, a fraction (defined by the parameter) of the difference between the mismatched RVD binding to its intended base and the base at the potential off-target site is subtracted from the score for that RVD-nucleotide interaction. Since a polarity effect exists in TAL–DNA binding where mismatches further from the N-terminus have a less disruptive effect (20), the scores for the 14th RVD and any RVDs further towards the C-terminus are all multiplied by the ‘Polarity’ parameter.
The scores of all positions in each half-site are summed together to create the ‘Off_Target’ score for that half-site and the full score for the potential off-target sites is computed using the ‘Dimer_Exponent’ parameter and the score for a complete match between the RVDs and their intended target bases to yield a score from 0 to 100 (a perfect match). More details of the TALEN v2.0 algorithm can be found in Supplementary Method M1.
Nuclease construction
Four novel TALEN pairs and two novel ZFN pairs were designed to target sequences near the A to T mutation that causes sickle-cell anemia in the human beta-globin gene. TALENs were assembled using the Golden Gate method (21) and cloned into a mammalian expression destination vector containing the wild-type FokI domain (available through AddGene #40788). ZFNs were rationally designed to target overlapping sites. As these ZFNs target the same site, the activity and specificity of the 3-finger (3F) and 4-finger (4F) ZFNs can be directly compared. ZFN1-4F contains an additional finger added to ZFN1-3F, extending the target site from 9 to 12 bp. ZFN2-4F shares two proximal fingers with ZFN2-3F, and uses a long linker between fingers two and three, extending the target site from 9 to 13 bp (Supplementary Figure S5). The coding sequences for the ZFNs were ordered (IDT) and cloned into a wild-type FokI expression vector (Supplementary Data D2 and D3). The PROGNOS search settings that were used for investigation of the novel nucleases are available in Supplementary Table S14.
Cellular transfection of nucleases
HEK-293T cells were cultured under standard conditions (37°C, 5% CO2) in Dulbecco’s Modified Eagle’s Medium (Sigma Aldrich), supplemented with 10% FBS. Plates were coated with 0.1% gelatin. Passaging was performed with 0.25% Trypsin-EDTA. For TALENs, 2 × 105 cells/well were seeded in 6-well plates 24 h prior to transfection with FuGene HD (Promega). Along with 80 ng of an eGFP plasmid, 3.3 µg of each nuclease plasmid were transfected with 19.8 µl of FuGene reagent. Media was changed 24 and 48 h after transfection. Seventy-two hours after transfection, cells were trypsinized and the genomic DNA extracted using the DNeasy Kit (Qiagen). A small fraction of the cells were analyzed with the Accuri C6 flow cytometer to determine transfection efficiency by GFP fluorescence. For ZFNs, 8 × 104 cells/well were seeded in 24-well plates and 100 ng of each ZFN was transfected using 3.4 µl of FuGene HD along with 10 ng of eGFP and 340 ng of a Mock vector containing FokI but no DNA-binding domain. Seventy-two hours after transfection, cells were harvested and the genomic DNA extracted using 100 µl of QuickExtract (EpiCentre). Mock transfections were performed similarly to the TALEN transfections, except that 6.6 µg of the mock FokI vector was transfected instead of TALEN plasmid.
PCR amplification of regions of interest
The primers designed by PROGNOS (ordered from Eurofins-MWG-Operon, Supplementary Table S18) were used in a high-throughput manner to amplify genomic regions of interest in a single-plate PCR reaction. Each 25 µl reaction contained 0.5 units of AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen) in AccuPrime Buffer 2 along with 150 ng of genomic DNA or 0.5 µl of QuickExtract, 0.2 µM of each primer and 5% DMSO vol/vol. Touchdown PCR reactions were found to yield the highest rate of specific amplification. Following an initial 2-minute denaturing at 94°C, 15 cycles of touchdown were performed by lowering the annealing temperature 0.5°C per cycle from 63.5°C to 56°C (94°C for 30 s, anneal for 30 s, extend at 68°C for 90 s). After the touchdown, an additional 29 cycles of amplification were performed with the annealing temperature at 56°C before a final extension at 68°C for 10 min. Reactions were purified using MagBind EZ-Pure (Omega), quantified using a Take3 Plate and SynergyH4 Reader (Biotek) and normalized to 10 ng/µl.
High-throughput sequencing
Amplicons from each transfection were pooled in roughly equimolar ratios and SMRT sequenced using the C2/C2 Chemistry and Consensus Sequencing options, according to the manufacturer’s protocol (Pacific Biosciences). Sequencing reads were aligned and processed using a pipeline of custom Perl scripts, BLAST and Needle (Supplementary Method M4).
Statistical analysis
P-values for off-target cleavage in Table 1 and Supplementary Tables S6–S10 were calculated exactly as previously described (9). Briefly, the t-statistic was calculated based on the fraction of mutated reads in the nuclease-treated sample compared to the fraction of mutated reads in the mock-treated sample and the number of sequencing reads was given as the degrees of freedom. In a similar manner, 90% confidence intervals were calculated by determining the upper and lower bounds of the fractions of mutated sequences that would yield P-values of 0.05.
Table 1.
SMRT Sequencing confirms on- and off-target activity at sites ranked by PROGNOS
 |
We interrogated 138 highly ranked genomic loci for the novel TALENs and ZFNs using SMRT, and observed off-target activity in 13 cases, nine of which were outside the globin gene family. The ‘match type’ indicates the orientation of the left (L) and right (R) nucleases at the site and the length of the spacer sequence. In sequences, lower-case red letters indicate mutations compared to the target site. Site sequences are listed as 5′—(+) half-site—spacer—(–) half-site—3′. Therefore, the (–) half-site for TALENs and the (+) half-site for ZFNs are listed in the reverse anti-sense orientation compared to the DNA sequence that the nuclease binds. Rankings by the initial PROGNOS algorithms Homology (column H), RVDs for NK (RK), RVDs for NN (RN) and Conserved G’s (C) are displayed as well as the rankings by the refined ‘TALEN v2.0’ algorithm for NK (TK) and for NN (TN) and the ‘ZFN v2.0’ algorithm (Z). 293T modification frequency is the percentage of observed sequences showing evidence of non-homologous end-joining repair. For the CCR5 ZFNs, 15 off-target sites ranked by PROGNOS that had not been previously investigated were interrogated using SMRT, validating a novel off-target site. Additionally, six known highly active off-target sites were sequenced as positive controls. The PROGNOS rankings for the site near KDM2A are listed as ‘N/A’ because the site was not found by PRGONOS due to the high number of mismatches. *P < 0.05 in cells expressing active nuclease compared to cells expressing empty vector. **P < 0.05 for the difference in activity between NK and NN at that site.
Source code for PROGNOS search algorithm
PROGNOS exhaustively searches for matches by moving the query mask iteratively across the entire genomic sequence, base by base. PROGNOS was implemented in Strawberry Perl 5.12 on a Windows machine (Supplementary Method M3). Source code and user manual are available at http://baolab.bme.gatech.edu/bao/Research/BioinformaticTools/prognos.html or http://bit.ly/PROGNOS. The probabilistic estimate of the number of expected off-target sites in a genome with a given level of homology is described in Supplementary Figure S1 and Supplementary Method M5. Details of the online server implementation are available in Supplementary Methods M6. The current list of genomes available on the online server is available in Supplementary Table S15.
RESULTS
Construction of initial bioinformatics ranking algorithms
The initial PROGNOS algorithms codified several established factors influencing nuclease specificity, including sequence homology, zinc fingers’ preference for binding guanine residues (6) and RVD-nucleotide binding frequencies of natural TAL effectors (22). To improve upon simple ‘mismatch counting’, we incorporated the recently proposed ‘energy compensation’ model of dimeric nuclease interactions (9). Using these factors, three different algorithms were initially developed. The ‘Homology’ algorithm, which could be used for both ZFNs and TALENs, generates a score based primarily on sequence divergence from the intended target site, including the number of mismatches in the left and right nuclease half-sites, and the maximum number of mismatches allowed per half-site. The ‘Conserved G’s’ algorithm (for ZFNs only) ranks potential ZFN off-target sites by counting the number of guanine bases and adding a weighting factor to the homology score accordingly. The ‘RVDs’ algorithm (for TALENs only) weighs mismatches based on RVD nucleotide preferences observed in natural TAL effectors and then applies the energy compensation model. Since all three of the TALEN off-target sites discovered previously using experiment-based off-target prediction methods contained a pyrimidine at the 5′ position, a ‘5TC’ version of the ‘Homology’ and ‘RVDs’ algorithms was also applied to TALEN rankings that required a thymidine or cytidine in the preceding 5′ position of each half-site. For any given potential off-target site, these algorithms generate a score that allows ranking of all potential off-target sites in a genome for a specific nuclease target site. Search parameters, such as target sites, maximum mismatches per half-site and allowed spacer lengths are entered as inputs using the online interface (Figure 1A and Supplementary Note 4) and ranked lists of potential cleavage sites in the selected genome are given as PROGNOS outputs for further analysis. Although two online tools—ZFN Site (23) and TALE-NT (18)—exist to help search genomes for cleavage sites with homology to intended nuclease on-target sites, neither automatically ranks the potential off-target sites, nor has led to a report of any new experimentally verified off-target cleavage sites. In a direct comparison, we found that TALE-NT was only able to predict two of the seven bona fide TALEN off-target sites in unrelated gene families—three sites from previous work (5,8) and four from this work—while PROGNOS could predict six (Supplementary Note 3). Recently, a new tool for identifying TALEN off-target sites, TALENoffer, was published (24). Although it performs better than TALE-NT and does provide a rank-order for the potential off-target sites, it is outperformed by the refined TALEN v2.0 algorithm (Supplementary Note 3).
Figure 1.

PROGNOS search interface and comparison to previous prediction methods. (A) The PROGNOS online interface allows users to enter the target site of their nuclease pair and specify search parameters and primer design considerations. (B) A comparison of PROGNOS predictions to previously reported methods identifying off-target sites for different ZFNs (6,9). The Homology and Conserved G’s algorithms were used to determine what percentage of the sites with previously identified off-target activity fell within the top fractions of PROGNOS rankings. The ‘1X’ top fraction corresponds to searching the same number of top PROGNOS sites as were investigated in the original paper and ‘3X’ corresponds to searching three times as many PROGNOS sites as were investigated in the original manuscript. (C) A comparison of the PROGNOS search algorithms to previously reported methods identifying off-target sites for TALENs (5,8). The top PROGNOS rankings using the Homology-5TC and RVD-5TC algorithms were searched to determine what percentage of off-target sites found to have activity fell within the top fractions of PROGNOS rankings. (D) Venn diagram displaying the 13 known off-target sites identified for the heterodimeric CCR5 ZFNs during development and testing of the original PROGNOS algorithms (9,10). The sites ranked at the top of the PROGNOS Homology and Conserved G’s in silico algorithms [allowing 3X the number of sites searched by Pattanayak et al. (9)] are compared to the 12 sites identified previously and one site uncovered in this study.
Validation of PROGNOS algorithms with previously confirmed off-target sites
To validate the initial PROGNOS ranking algorithms, we compared PROGNOS predictions with the off-target sites of ZFN and TALEN pairs identified by others using experimental characterization methods. If the same number of sites (1X) were interrogated as in the original studies, but the sites were chosen by taking the top-ranked PROGNOS predictions, (33 ± 21)% (mean ± SD) of the off-target sites previously found in studies of ZFNs targeting CCR5 (9), VEGF (9) and kdrl (6) could be located. Since off-target searches using the in silico PROGNOS predictions can be scaled up readily, we tripled (3X) the number of sites interrogated from PROGNOS top-ranked lists, and found that PROGNOS could identify (65 ± 24)% of the off-target sites previously confirmed experimentally (Figure 1B and Supplementary Tables S1–S3). Excluding sites in highly homologous gene pairs such as CCR5/CCR2, only three bona fide TALEN off-target sites had previously been experimentally identified to date (5,8) (Supplementary Note 5), making a rigorous analysis of the predictive power of PROGNOS for ranking TALEN off-target sites more difficult (25). Nevertheless, we found that the ‘Homology-5TC’ and ‘RVD-5TC’ algorithms in PROGNOS could predict several off-target sites confirmed previously for TALEN pairs targeting the AAVS1 (8) and IgM (5) loci (Figure 1C and Supplementary Tables S4–S5). Since no single off-target analysis method has yet been able to provide a comprehensive list of all off-target sites of a nuclease (Figure 1D) (9,10), the comparison of PROGNOS predictions with previously published results may underestimate the power of PROGNOS. Specifically, these comparisons are limited by the small number of off-target sites experimentally validated previously, and do not reflect the ability of PROGNOS to predict new off-target sites.
Validation of novel CCR5 ZFN off-target site predicted by PROGNOS
To date, the only nuclease pair to have its off-target sites experimentally interrogated using two independent methods is a ZFN-pair targeting CCR5 [analyzed using in vitro cleavage (9) and IDLV (10)]. These two studies located a total of 12 hetero-dimeric bona fide off-target sites, verified by sequencing the resulting mutations. A comparison between PROGNOS predictions using the ‘Homology’ and ‘Conserved G’s’ algorithms and those 12 sites identified experimentally shows that PROGNOS [analyzing the top 3X number of sites interrogated by Pattanayak et al. (9)] was able to predict 10 out of the 12 off-target sites (Figure 1D and Supplementary Table S1). Additionally, through investigating 16 potential off-target sites predicted by PROGNOS, but not identified by any other existing methods (9,10,16), a novel CCR5 ZFN off-target site was experimentally validated (Table 1 and Supplementary Tables S10–S11).
PROGNOS search output
PROGNOS provides ranked lists of potential nuclease cleavage sites that can be used to guide experimental evaluation of ZFN and TALEN off-target activities (Figure 2A). Specifically, for each pair of ZFNs or TALENs, the user-friendly online interface of PROGNOS (http://bit.ly/PROGNOS (13 December 2013, date last accessed) or http://baolab.bme.gatech.edu/bao/Research/BioinformaticTools/prognos.html) allows entry of the nuclease search parameters (the guidelines for de novo investigation of nucleases are given in Supplementary Note 4) and returns lists of the top-ranked off-target sites according to the PROGNOS algorithms, as well as a full list of un-ranked potential off-target sites meeting the search parameters (Figure 2B). While the top-ranked sites provide a list of likely locations in a genome where off-target cleavage may occur, neither the PROGNOS rankings nor any published method can yet directly correlate the ranking with the precise level of observed off-target mutagenesis at a given site (Supplementary Figure S3). Furthermore, to aid experimental analysis, PROGNOS also provides PCR primer sequences that can be used to amplify the potential nuclease cleavage sites in a high-throughput manner (Supplementary Method M2), a unique feature not present in other online search tools. Automated design of PCR primers significantly facilitates the analysis of off-target sites, since an initial experimental study of off-target cleavage by a single pair of nucleases typically requires at least 40 primers (1,8), and an in-depth investigation of nuclease off-target effects may require >250 primers (6,9). Although tools such as Primer3 (26) can assist in primer design, they require a large amount of effort to generate primers optimal for off-target analysis due to specific requirements of where the nuclease site must be positioned within the amplicon. Although PCR amplification is an essential step in examining a potential off-target site, in previous investigations the success rates of amplifying off-target loci varied from 31% (1) to 95% (8). In contrast, the primers automatically designed by PROGNOS had a robust 95% success rate across the 116 potential off-target loci interrogated in this study (Figure 2C and Supplementary Methods M1). PROGNOS also provides the sequences, the sizes of expected cleavage products of the amplicons, and site of expected cleavage. This information is used when testing for nuclease-induced mutations—typically short insertions and deletions (indels) resulting from error-prone resolution of the DNA double-strand break through the non-homologous end-joining (NHEJ) repair pathway—using methods such as the Surveyor Nuclease assay, high-throughput sequencing or Sanger sequencing of TOPO-cloned fragments (Figure 2D).
Figure 2.

Using PROGNOS to identify nuclease off-target sites. (A) Outline of the procedure to identify nuclease off-target activity. (B) Sample outputs of the PROGNOS online software showing all sites found and what types of genomic regions they are located in as well as rankings of the top potential off-target sites. The rankings include the closest gene, the number of mismatches, the size of PCR product from the automatically designed primers, and other helpful information. (C) Comparison of the success of the automatically designed PROGNOS primers used in high-throughput full-plate PCR of off-target sites to primers designed in other off-target publications. (D) Sequencing reads of an off-target location for the 3F ZFN pair that show evidence of NHEJ. In the wild-type (WT) sequence, the ZFN binding sites are highlighted in yellow and mismatches to the intended target sequence are lowercase red. In the sequencing reads, inserted bases are lowercase and highlighted in blue. The size of the indel is displayed to the right of the sequence, along with the number of times that mutation was observed.
Determination of NHEJ-mediated indels using high-throughput SMRT sequencing
To experimentally measure nuclease activity at on-target and potential off-target sites identified by PROGNOS, we used single molecule real-time (SMRT) sequencing of the PCR amplicons. The consensus sequencing mode of the SMRT platform provides highly accurate long length reads (27) that allowed determination of nuclease activity and specificity with reasonable sensitivity, and at a lower cost per run than other deep sequencing platforms (other advantages of SMRT sequencing for smaller laboratories are described in Supplementary Note 7). The good agreement between SMRT sequencing results and Sanger sequencing of TOPO-cloned samples further confirmed the accuracy of the SMRT-based analysis of nuclease cleavage (Figure 3A). Further, the high quality of the SMRT consensus sequence reads allowed us to achieve a much better signal to noise ratio for the mutation analysis than other sequencing methods (1). We found that only three sequencing reads from mock treated control cells (∼0.003% of the total) contained indels flagged by the analysis and all three were from the same genomic site, which in retrospect should have been excluded from sequencing analysis due to several long adjacent homopolymer stretches known to be error-prone during the sequencing process (Supplementary Tables S6–S9 and Supplementary Data D1).
Figure 3.

Using SMRT Sequencing to analyze nuclease activity. (A) SMRT sequencing produced very similar results to standard TOPO sequencing over a range of mutation rates from ∼20% to ∼76%. Error bars are 90% confidence intervals. S2/S5 NK and S2/S5 NN are the TALENs targeting beta-globin compared in this study. S116/S120 and J7/J8 are NK-TALENs targeting beta-globin and CDH1, respectively (30). (B) Comparison of the range and frequency of different sizes of indels observed in cells treated with TALENs or ZFNs. The observed frequencies of the different sizes are normalized to the frequency of the most common indel size for each nuclease type.
Although the spectrums of indels induced by ZFNs (6) or TALENs (1,28) have been investigated previously, the long SMRT read lengths provided a more comprehensive analysis (Figure 3B). We found that ZFNs induced predominately 3-, 4- and 5-bp insertions or deletions, with just a small number of large deletions. In contrast, TALENs induced indels over a much broader range, centered at 5–20 bp deletions, possibly due to the flexibility of the +63 C-terminal TAL domain (29).
Prediction and validation of off-target sites for novel nucleases
To demonstrate the application of PROGNOS in analyzing newly designed nucleases, we investigated the off-target cleavage of four pairs of TALENs and two pairs of ZFNs (Table 1). TALENs containing the Asn-Asn (NN) RVD have been shown to be less specific than corresponding TALENs containing the Asn-Lys (NK) RVD (29); however the difference in off-target activity of NN-TALENs and NK-TALENs has not been demonstrated in a genome-wide context. For ZFNs, although both 3F and 4F ZFNs have been shown to have off-target cleavage (6,9,10), there has been no direct comparison of off-target cleavage induced by 3F- and 4F-ZFNs that target the same DNA sequence. We expressed the TALENs and ZFNs in HEK-293T cells, and analyzed the PROGNOS top-ranked off-target sites (Table 1 and Supplementary Tables S6–S9). We found that TALENs exclusively using the NN RVD to target all of the guanosine nucleotides in the target sequence imparted higher activity level than TALENs exclusively using the NK RVD at corresponding positions, in agreement with previous reports (1,29). However, the NN-TALENs tested in this study had higher off-target cleavage activity than the corresponding NK-TALENs. For the first time, off-target cleavage by NK-TALENs was uncovered, as well as bona fide TALEN off-target sites with substantial (>5%) sequence divergence from the intended target that lacked a 5′ pyrimidine and a site with a spacer >24 bp (Table 1). For ZFNs, we found that the 4F-ZFNs had higher on-target activity [consistent with previous reports that additional fingers increased activity (31)] and much lower off-target activity compared with the corresponding 3F-ZFNs targeting the same DNA site. Specifically, all six of the off-target sites found for the 3F-ZFNs had equal or greater activity than the off-target site of the 4F-ZFNs (a single site with 0.2% activity), with three sites having activity >1% (Table 1).
Refinement of PROGNOS ranking algorithms
Although the set of initial PROGNOS algorithms (two for ZFNs and four for TALENs) performed well in locating bona fide off-target sites for newly designed nucleases based solely on in silico prediction, a user would still need to choose a specific algorithm or use all the available algorithms without knowing a priori which one would be most predictive for their nuclease. Using the expanded set of bona fide off-target sites including those found in this study (Table 1) as well as new insights into TALEN-DNA binding (19,20), we refined the PROGNOS algorithms so that they are more sensitive, efficient and user friendly compared with the initial algorithms. Although the ‘Homology’, ‘Conserved G’s’ and ‘RVDs’ algorithms (including the ‘5TC’ version for TALENs) all located bona fide off-target sites, no algorithm was consistently superior across all ZFNs or all TALENs studied (Figure 1B and C and Table 1). In developing the refined algorithms, we were able to unify the different algorithms for each type of nuclease into a single algorithm (ZFN v2.0 for ZFNs, TALEN v2.0 for TALENs). Compared with the original PROGNOS algorithms, ZFN v2.0 and TALEN v2.0 predicted a larger total number of bona fide off-target sites within the top 16 rankings (representing the minimum recommended size of a small-scale off-target analysis), located higher mean percentages of known off-target sites per nuclease across all nucleases tested (within the top 3X rankings for previously investigated nucleases and within the same number of sites as in the PROGNOS-based investigations, Supplementary Table S17), and had lower standard deviations of the mean percentages, demonstrating that the refined algorithms performed more consistently across all nucleases tested.
In developing the refined and unified ZFN algorithm, we added factors weighing a model of the binding energy of each zinc finger subunit (9) and polarity effects reflecting the distance of a mismatch from the FokI domain and allowed more flexible models of the previous concepts of energy compensation between the two half-sites of a nuclease pair and a stronger affinity for guanosine residues (Supplementary Method M1). This new ‘ZFN v2.0’ algorithm outperforms the initial ‘Homology’ and ‘Conserved G’s’ algorithms for ZFNs in terms of both identifying a larger set of bona fide off-target sites for the nucleases tested and having a superior true discovery rate in the Top 16 rankings (Figure 4A). The Top 16 ranked sites were chosen as a cutoff (instead of the Top 24, as recommended in Supplementary Note 4) because by necessity nearly all of the novel off-target sites found were within the Top 24 rankings of one of the original algorithms since that was their initial criteria for being selected for investigation. Therefore, a stricter cutoff was required in order to observe differential performances between the algorithms for these new sites.
Figure 4.

Improved performance of the refined PROGNOS algorithms. (A) The performance of the two initial ZFN algorithms and the refined ‘ZFN v2.0’ algorithm are compared for their ability to predict off-target sites for all the ZFNs in the training and validation sets. Percentages of off-target sites located were calculated according to 3X limits for previous studies and within the number of sites interrogated for PROGNOS-based studies (typically the top 24 ranked sites). Error bars represent SD. (B) The expanded landscape of 38 total heterodimeric off-target sites for the CCR5 ZFNs found by four different experiment-based prediction methods and the refined ‘ZFN v2.0’ PROGNOS algorithm. The PROGNOS sites are drawn from the top rankings spanning 3X the number of predictions by the Bayesian abstraction of the in vitro cleavage profile. (**) Note that only six of the sites found using ChIP-Seq were provided by Sander et al. (11), so the full degree of overlap of all ChIP-Seq sites with sites found by other methods is unclear. (C) The performance of the four original TALEN algorithms and the refined ‘TALEN v2.0’ algorithm are compared for their ability to predict off-target sites for all TALENs in the training and validation sets.
Recently, Sander et al. (11) used Bayesian machine learning to re-analyze the original results of the in vitro cleavage experiments for CCR5 and VEGF ZFNs (9) and subsequently developed two separate classifiers that ranked all sequences in the human genome for their potential as off-target sites of either the CCR5 or VEGF ZFNs, respectively. Their work validated 25 new bona fide off-target sites for the CCR5 ZFNs and 26 new sites for the VEGF ZFNs, but did not locate—among any of the 15 882 possible off-target sites predicted for the CCR5 ZFNs by their classifier system—the novel off-target site for the CCR5 ZFNs predicted by the PROGNOS algorithms near CSNK1G3 that was validated in this study. Although the analysis by Sander et al. combined machine learning and in vitro cleavage experiments, it was unable to locate all the known off-target sites for the CCR5 ZFNs. Details of the comparison to the Sander et al. analysis (11) can be found in Supplementary Note 6.
Since the 51 new sites found by Sander et al. (11) were not part of the training set for the ‘ZFN v2.0’ algorithm, this provided an opportunity to test the new algorithm for its ability to locate additional off-target sites. By extending the standard PROGNOS search limit recommendations (Supplementary Note 4) for the CCR5 ZFNs to allow for a larger number of possible off-target sites (3X the number of possible off-target sites considered by Sander et al.), we found that the refined ZFN algorithm successfully identified more than half (13 of 25 = 52%) of the new off-target sites for those ZFNs (Figure 4B and Supplementary Note 5). For the VEGF ZFNs, the standard PROGNOS search provided enough potential off-target sites to make an appropriate 3X comparison to Sander et al. (11), and the refined algorithm again located more than half (18 of 26 = 69%) of the new off-target sites for those ZFNs (Supplementary Note 5). Three additional pairs of ZFNs (a 3F pair, a 4F pair and a 5F CompoZr pair from Sigma-Aldrich) which had previously been investigated using the Homology and Conserved G’s PROGNOS algorithms (Mussolino,C. et al. and Abarrategui-Pontes,C. et al., manuscripts in preparation) were also re-analyzed using the refined algorithm and all six of the previously located bona fide off-target sites were highly ranked by ZFN v2.0 (Supplementary Table S17). Taken together, these results provide significant evidence that the refined ZFN algorithm was not over trained to existing sites during its development and is able to robustly predict additional bona fide off-target sites. An analysis of each of the components of the ZFN v2.0 algorithm showed that while all play a part in the improved performance, some parameters are more critical to the algorithm than others (Supplementary Figure S7).
In developing the refined and unified TALEN algorithm, we added new parameters based on compensatory effects of strong RVDs (NN and HD) (19) on adjacent mismatches and polarity effects indicating that mismatches further from the N-terminus are less disruptive (20). These new considerations were combined with a model of dimeric nuclease interactions, as well as RVD-nucleotide association frequencies. To improve upon the RVD-nucleotide association frequencies derived from natural TAL effectors (18), as were used in the initial ‘RVDs’ algorithm and the TALE-NT online tool (18), we calculated association frequencies based on SELEX data from engineered TAL domains (5,8,17) (Supplementary Figure S6 and Table S16). Importantly, this generated an association frequency for the 5′ ‘Position 0’ in the TALEN-binding site that allowed us to use this parameter to unify the ‘5TC’ and unrestricted versions of the ‘RVDs’ algorithm. Further, we found that while the nucleotide frequencies for the RVDs NI, HD, NK and NG did not appreciably vary between engineered TALEs and natural TALEs, the results for NN were substantially different. Although the NN RVD is still the least specific of all the standard RVDs, in engineered TALEs it showed a stronger preference for its intended base (guanosine) and a reduced preference for adenosines and cytidines compared with that of naturally occurring TALEs (Supplementary Table S16). We found that the new unified ‘TALEN v2.0’ algorithm outperforms the four initial algorithms for TALENs in terms of both finding a larger number of bona fide off-target sites in the Top 16 rankings and locating a higher mean percentage of known off-target sites per nuclease across all nucleases tested (Figure 4C). The refined TALEN algorithm was additionally able to predict several bona fide TALEN off-target sites not in its training set that were found using the initial PROGNOS algorithms (Supplementary Table S17, Mussolino,C. et al., manuscript in preparation), demonstrating that the refined algorithm was not over trained during development and retains robust predictive capabilities. An analysis of each of the components of the TALEN v2.0 algorithm showed that while all play a part in the improved performance, some parameters are more critical to the algorithm than others (Supplementary Figure S8).
Sensitivity and specificity of PROGNOS search algorithms
When applying the initial PROGNOS algorithms to identify off-target sites for newly constructed NN-TALENs and 3F and 4F ZFNs, we obtained a very manageable average false positive ratio—defined as the number of interrogated sites with no detectable activity compared to the number with detectable activity—of only ∼11:1, which is less than 2-fold greater than current experimental prediction methods (Figure 5A and Supplementary Table S12). When interrogating three additional pairs of NN-TALENs with the initial algorithms, we observed a similarly low false positive ratio of 11:1 (Mussolino,C. et al., in preparation). For NK-TALENs, the false positive ratio was higher (∼21:1); however, since no previously published method has identified any off-target sites for NK-TALENs, we were not able to make a meaningful comparison of the false positive ratio with experiment-based prediction methods. As the new ‘ZFN v2.0’ and ‘TALEN v2.0’ algorithms have a higher true discovery rate among the top 16 rankings, we would expect that their false positive ratios would be even lower than the initial algorithms when used as the basis for investigations of novel nucleases.
Figure 5.
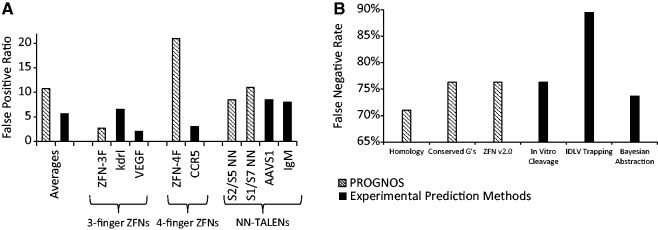
Sensitivity and specificity analysis of PROGNOS algorithms. (A) Average false positive ratios are shown for the PROGNOS investigation of novel nucleases using the initial algorithms, and for previous experimental prediction methods. Ratios are also shown for individual nucleases in the three different categories of nuclease that have been investigated previously by experimental prediction methods. (B) The false negative rates of the different PROGNOS algorithms and previous experimental prediction methods are shown. These were determined by each method’s ability to identify the 38 known hetero-dimeric off-target sites of the CCR5 ZFNs in their top ranking predictions.
As mentioned above, to date only a single nuclease pair (the heterodimeric sites of the CCR5 ZFNs) has had its off-target cleavage investigated by independent experimental prediction methods (9–11), and it is therefore the only pair for which a false negative rate analysis can be conducted. Defining the false negative rate as the percentage of all known off-target sites that are not predicted by the particular method within a top portion of the rankings, the PROGNOS algorithms had false negative rates equal or superior to the IDLV and in vitro cleavage experimental prediction methods (Figure 5B and Supplementary Table S13). An ROC-like analysis of the different predictive methods for the CCR5 ZFNs using the false discovery and true positive rates also demonstrates that the PROGNOS algorithms perform comparably to experimental based prediction methods (Supplementary Figure S4).
DISCUSSION
Engineered nucleases can readily be designed and optimized to target specific endogenous sequences in a genome. However, to reach their potential for generating model research systems and treating human diseases, the specificity of engineered nucleases must be better understood. However, the analysis of the location and frequency of TALEN and ZFN off-target effects has been beyond the reach of most laboratories due to the limitations of the existing methods. We created PROGNOS, an online search tool solely based on bioinformatics and the current understanding of nuclease–DNA interactions, which allows users to predict potential nuclease off-target sites by following a simple set of instructions (Supplementary Note 4), and to evaluate the sites using standard molecular biology techniques if so desired (Supplementary Figure S2). The novel bioinformatics ranking algorithms in PROGNOS predict many of the off-target sites of the CCR5 ZFNs that were identified previously using experimental methods and also identified a novel off-target site that was missed in those studies. However, there are several very active (>5% mutation rate) off-target sites for these ZFNs that PROGNOS did not rank highly, suggesting that there are still unknown factors influencing ZFN off-target activity that are not accounted for in our current models. Future unbiased genome-wide analyses of off-target activity [such as the IDLV method (10)] will be critical to build a larger database of sites with low sequence homology from which further insight into the factors affecting off-target activity can be gained. Nevertheless, PROGNOS is able to successfully predict many off-target sites and overcomes the drawbacks of the current experiment-based prediction methods that limit the number of nucleases tested, as evidenced by the fact that no bona fide off-target sites for new ZFNs or TALENs have been reported over the last 2 years (5,8) (see Supplementary Note 5). The improved performance of the refined ‘ZFN v2.0’ and ‘TALEN v2.0’ algorithms over the initial algorithms highlights a key advantage of bioinformatics-based predictions: as more bona fide off-target sites are discovered, increasingly better predictive models can be incorporated.
PROGNOS allowed interrogation and comparison of the off-target activities of several novel nucleases targeting the beta-globin gene. We directly compared 3F versus 4F ZFNs that targeted the same site, and compared NK-TALENs versus NN-TALENs that shared target sites. We found that these NN-TALENs and 3F ZFNs had more off-target activity than the corresponding NK-TALENs and 4F ZFNs, respectively. While NN-TALENs generally have high on-target cleavage, this may be accompanied by decreased specificity leading to high off-target activity. To confirm the conclusion that the 4F-ZFNs targeting this site are more specific than the 3F versions, we interrogated several of the validated 3F-ZFN off-target sites in cells expressing 4F-ZFNs and found no statistically significant off-target activity (Supplementary Table S9). Our comparison of the specificity of NN-TALENs versus NK-TALENs is somewhat limited by the fact that the NN-TALENs had higher on-target activity than the corresponding NK-TALENs, but the dramatic difference in off-target activity at HBD for the S2/S5 NN- and NK-TALENs (Table 1) strongly supports the notion that NK-TALENs have improved specificity over NN-TALENs. The nature of the new off-target sites and their implications are discussed further in Supplementary Note 1.
In summary, PROGNOS provides a user-friendly, web-based tool for rapid identification of potential nuclease off-target cleavage sites that can be evaluated using standard molecular biology techniques. The bioinformatics-based ranking algorithms in PROGNOS identify most nuclease off-target cleavage sites found by existing experimental methods. PROGNOS has relatively low false positive ratios and comparable false negative rates to experiment-based predictions, making it a robust method that can be readily implemented by most laboratories. Screening potential target sites using PROGNOS can facilitate the selection of superior nuclease target sites that minimize the number of likely genomic off-target sites. PROGNOS allows nuclease off-target analysis to become a routine component of nuclease design and testing, facilitating the discovery of new off-target sites for ZFNs and TALENs, which expand the off-target database and may improve future versions of the PROGNOS algorithms. These capabilities give PROGNOS the potential to help expand and expedite the application of engineered nucleases for a wide range of biological and medical applications.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [32–40].
ACKNOWLEDGEMENTS
We thank Mike Tschannen at the Medical College of Wisconsin Sequencing Core for his help and Dr Ayal Hendel at Stanford University for his suggestions on SMRT sequencing. We thank Dr Claudio Mussolino and Dr Toni Cathoman at the University of Freiburg for the genomic DNA from HEK-293T cells transfected with the CCR5 ZFNs.
FUNDING
National Institutes of Health (NIH Nanomedicine Development Center Award; grant number PN2EY018244 to G.B.); National Science Foundation Graduate Research Fellowship (DGE-1148903 to E.J.F.). Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1.Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B. Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotech. 2011;29:699–700. doi: 10.1038/nbt.1939. [DOI] [PubMed] [Google Scholar]
- 2.Lei Y, Guo X, Liu Y, Cao Y, Deng Y, Chen X, Cheng CHK, Dawid IB, Chen Y, Zhao H. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs) PNAS. 2012;109:17484–17489. doi: 10.1073/pnas.1215421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zschemisch N-H, Glage S, Wedekind D, Weinstein EJ, Cui X, Dorsch M, Hedrich H-J. Zinc-finger nuclease mediated disruption of Rag1 in the LEW/Ztm rat. BMC Immunol. 2012;13:60. doi: 10.1186/1471-2172-13-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watanabe T, Ochiai H, Sakuma T, Horch HW, Hamaguchi N, Nakamura T, Bando T, Ohuchi H, Yamamoto T, Noji S, et al. Non-transgenic genome modifications in a hemimetabolous insect using zinc-finger and TAL effector nucleases. Nat. Commun. 2012;3:1017. doi: 10.1038/ncomms2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tesson L, Usal C, Ménoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, et al. Knockout rats generated by embryo microinjection of TALENs. Nat. Biotechnol. 2011;29:695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 6.Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 2011;39:381–392. doi: 10.1093/nar/gkq787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LFZ, et al. In Situ Genetic Correction of the Sickle Cell Anemia Mutation in Human Induced Pluripotent Stem Cells Using Engineered Zinc Finger Nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat. Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotech. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 11.Sander JD, Ramirez CL, Linder SJ, Pattanayak V, Shoresh N, Ku M, Foden JA, Reyon D, Bernstein BE, Liu DR, et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013;41:e181. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;21:9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding Q, Lee Y-K, Schaefer EAK, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, et al. A TALEN Genome-Editing System for Generating Human Stem Cell-Based Disease Models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee Y-L, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotech. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nat. biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 18.Doyle EL, Booher NJ, Standage DS, Voytas DF, Brendel VP, VanDyk JK, Bogdanove AJ. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL Effector Design and Target Prediction. Nucleic Acids Res. 2012;40:W117–W122. doi: 10.1093/nar/gks608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Streubel J, Blücher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat. Biotechnol. 2012;30:593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 20.Meckler JF, Bhakta MS, Kim M-S, Ovadia R, Habrian CH, Zykovich A, Yu A, Lockwood SH, Morbitzer R, Elsäesser J, et al. Quantitative analysis of TALE–DNA interactions suggests polarity effects. Nucleic Acids Res. 2013;41:4118–4128. doi: 10.1093/nar/gkt085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 23.Cradick TJ, Ambrosini G, Iseli C, Bucher P, McCaffrey AP. ZFN-site searches genomes for zinc finger nuclease target sites and off-target sites. BMC Bioinform. 2011;12:152. doi: 10.1186/1471-2105-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grau J, Boch J, Posch S. TALENoffer: genome-wide TALEN off-target prediction. Bioinformatics. 2013;29:2931–2932. doi: 10.1093/bioinformatics/btt501. [DOI] [PubMed] [Google Scholar]
- 25.Mussolino C, Morbitzer R, Lütge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Research. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. In: Misener S, Krawetz SA, editors. Bioinformatics Methods and Protocols: Methods in Molecular Biology. Totowa, NJ: Humana Press; 1999. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 27.Travers KJ, Chin C-S, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y, Kweon J, Kim J-S. TALENs and ZFNs are associated with different mutation signatures. Nat. Methods. 2013;10:185. doi: 10.1038/nmeth.2364. [DOI] [PubMed] [Google Scholar]
- 29.Christian ML, Demorest ZL, Starker CG, Osborn MJ, Nyquist MD, Zhang Y, Carlson DF, Bradley P, Bogdanove AJ, Voytas DF. Targeting G with TAL effectors: a comparison of activities of TALENs constructed with NN and NK repeat variable di-residues. PLoS ONE. 2012;7:e45383. doi: 10.1371/journal.pone.0045383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y, Fine EJ, Zheng Z, Antico C, Voit R, Porteus M, Cradick T, Bao G. SAPTA: a new design tool for improving TALE nuclease activity. Nucleic Acids Res. 2013:e47. doi: 10.1093/nar/gkt1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, et al. Highly active zinc finger nucleases by extended modular assembly. Genome Res. 2013;23:530–538. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 33.Bogdanove AJ, Voytas DF. TAL Effectors: Customizable Proteins for DNA Targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 34.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler AD. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, et al. Ensembl 2012. Nucleic Acids Res. 2011;40:D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray). Science (New York, N.Y.) 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 37.Jeong H, Barbe V, Lee CH, Vallenet D, Yu DS, Choi S-H, Couloux A, Lee S-W, Yoon SH, Cattolico L, et al. Genome Sequences of Escherichia coli B strains REL606 and BL21(DE3) J. Mol. Biol. 2009;394:644–652. doi: 10.1016/j.jmb.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 38.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 39.Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, Miller JC, Urnov FD, Gregory PD, Holmes MC. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat. Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- 40.Osborn MJ, Starker CG, McElroy AN, Webber BR, Riddle MJ, Xia L, Defeo AP, Gabriel R, Schmidt M, Von Kalle C, et al. TALEN-based Gene Correction for Epidermolysis Bullosa. Mol. Ther. J. Am. Soc. Gene Ther. 2013;21:1151–1159. doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.