


Abstract
Dicer is a specialized nuclease that produces RNA molecules of specific lengths for use in gene silencing pathways. Dicer relies on the correct measurement of RNA target duplexes to generate products of specific lengths. It is thought that Dicer uses its multidomain architecture to calibrate RNA product length. However, this measurement model is derived from structural information from a protozoan Dicer, and does not account for the helicase domain present in higher organisms. The Caenorhabditis elegans Dicer-related helicase 3 (DRH-3) is an ortholog of the Dicer and RIG-I family of double-strand RNA activated ATPases essential for secondary siRNA production. We find that DRH-3 specifies 22 bp RNAs by dimerization of the helicase domain, a process mediated by ATPase activity and the N-terminal domain. This mechanism for RNA length discrimination by a Dicer family protein suggests an alternative model for RNA length measurement by Dicer, with implications for recognition of siRNA and miRNA targets.
INTRODUCTION
RNA interference (RNAi) is a conserved mechanism that uses small RNAs to guide sequence specific regulation of gene expression, chromatin structure and immunity (1). Small RNAs, such as micro-RNAs (miRNAs), small interfering RNAs (siRNAs) and piwi-interacting RNAs (piRNAs), are defined by their short length and their association with members of the Argonaute family of proteins (2). Once bound to an Argonaute protein, small RNAs are delivered to their regulatory target, where they typically act to silence gene expression. Primary siRNAs are typically 20–22 nucleotides in length and are often derived from long double-stranded RNA (dsRNA) (3). In Caenorhabditis elegans, primary siRNAs activate the production of secondary siRNAs, which amplify the silencing signal (4,5). To initiate secondary siRNA synthesis, a primary siRNA binds to RDE-3, an Argonaute protein, which guides it to a target mRNA (6). There, a RNA-dependent RNA polymerase (RdRP) is recruited to synthesize siRNAs that are 22 nucleotides in length (7,8). Because the secondary siRNAs are transcribed and not produced by dicing, they contain 5′ triphosphates and are exclusively antisense to their target mRNA. These secondary siRNAs are known as 22G-RNAs, due to their length and propensity for a 5′ guanosine residue (9).
The C. elegans Dicer-related helicase 3 (DRH-3) is an ortholog of the Dicer- and retinoic acid-inducible gene I (RIG-I) like family of proteins, and it is essential for RdRP-dependent secondary siRNA production, as evidenced by the depletion of 22G-RNA when DRH-3 is knocked down or mutated (9). However, in worms where DRH-3 contains mutations in the helicase domain, 22G-RNAs are not uniformly depleted. Based on the target mRNA polarity, there is a 3′ end enrichment of 22G-RNA, and a noted absence of upstream 22G-RNA (9). The enrichment of terminal 22G-RNA suggests that secondary siRNA biogenesis is initiated at the 3′ end of the target mRNA. The mutant data also indicate that DRH-3 activity promotes propagation of 22G-RNAs by the RdRP along the mRNA template in the 3′ to the 5′ direction. Because the DRH-3 mutations are in the helicase domain (residues 834 and 840), they likely disrupt RNA binding and/or ATP hydrolysis. Thus, a mechanistic analysis of target RNA recognition by DRH-3, and of its closely linked ATPase activity, is essential for understanding DRH-3 function.
Phylogenetically related proteins, such as the RIG-I-like receptors and Dicer, discriminate among RNA ligands by sensing their relative length. Melanoma differentiation-associated gene-5 (MDA-5), a RIG-I-like receptor, assembles by forming cooperative filaments along RNA duplexes (dsRNA), thereby selecting specifically for long dsRNA targets (10). This contrasts with the behavior of RIG-I, which binds dsRNA as a monomer and prefers duplexes that contain a 5′ triphosphate (11–14). DRH-3 lacks apparent affinity for 5′ triphosphorylated dsRNA, despite the sequence and structural similarity of its C-terminal domain to the RIG-I C-terminal domain (15). Thus, although DRH-3, MDA-5 and RIG-I share similar domain organizations, the functions of these domains may be different. The related Dicer proteins also have length-discrimination mechanisms for recognizing RNA targets. Based on the Giardia intestinalis Dicer structure (16), it has been proposed that Dicer proteins use their PAZ domains to measure RNA and subsequently cleave products of a discrete length. However, generalization from this system is problematic, as the G. intestinalis Dicer lacks the N-terminal helicase domain that typifies other Dicer proteins. A mechanism for length-dependent RNA recognition by DRH-3 has yet to be specifically examined. The biologically relevant RNA ligand for DRH-3 is expected to be ∼22 base pairs (bp) and the protein has been proposed to function as an oligomer (15), although the determinants for RNA binding and ATPase activation remain undefined.
Using a combination of direct RNA binding assays, steady state kinetics and transient kinetic analysis, we show that DRH-3 forms a cooperative dimer on duplex RNAs of biologically relevant lengths (∼22 bp). The ATP hydrolysis activity of DRH-3 is similarly length-dependent, and it appears to mediate dimer assembly. In addition, studies of a deletion mutant show that the N-terminal domain (NTD) plays a key role in RNA length recognition by DRH-3. Collectively, these studies provide a mechanism for 22G-RNA recognition by DRH-3, whereby functional dimerization of the helicase domains provides the molecular ruler for measuring dsRNAs of appropriate lengths. Because DRH-3 and Dicer both contain the same type of phylogenetically conserved, specialized helicase domain, DRH-3 may provide generalizable insights into mechanisms of RNA length discrimination by metazoan Dicer proteins.
MATERIALS AND METHODS
Protein expression and purification
Full length DRH-3 was expressed and purified as previously described (15). The N-terminal deletion variant of DRH-3 (ΔN-DRH3) containing residues 300–1120 was cloned into the pET-SUMO expression vector and transformed into Rosetta II cells (Novagen). Cells were grown at 37°C to an Abs600 of 0.6 and induced overnight with 0.3 mM IPTG at 16°C. The ΔN-DRH3 construct was purified using essentially the same protocol as previously described for full length DRH3 (15). After concentration, ΔN-DRH3 was quantified by absorbance (ε280 = 79.19 mM−1 cm−1), flash frozen and stored at −80°C.
Full-length RIG-I was purified as previously described (11). Human MDA-5 was cloned into the pET-SUMO vector and transformed into Rosetta II cells (Novagen). Cells were grown at 37°C to an Abs600 of 0.6, and MDA-5 expression was induced with 0.5 mM IPTG at 16°C overnight. Like RIG-I, MDA-5 was purified using nickel affinity chromatography, followed by cationic exchange and size exclusion chromatography using Heparin Sepharose and HiLoad Superdex 200 columns (GE Healthcare). After concentration, MDA-5 was quantified by absorbance (ε280 = 92.71 mM−1 cm−1), flash frozen and stored at −80°C.
RNA preparation
Oligoribonucleotides were synthesized on an automated MerMade synthesizer (BioAutomation) using standard phosphoramidite chemistry. The oligonucleotides were deprotected and gel purified as previously described (17). Fluorophore-labeled RNA used in stopped-flow fluorescence spectroscopy was generated using amino-modified RNA, which is synthesized using a 3′ amino modifier with a C3 linker (Glen Research). Cy3 Mono NHS ester (GE Healthcare) was conjugated to the modified oligonucleotides as per the manufacturer’s instructions. Cy3-labeled and unlabeled RNA were separated on a 20% denaturing polyacrylamide gel, and purified by gel extraction. Duplex RNA substrates were hybridized by mixing complementary sequences in equal molar ratio, and rapid heating to 95°C followed by slow cooling to 25°C in annealing buffer (10 mM MOPS, pH 6.5, 1 mM EDTA, 150 mM NaCl). For electrophoretic mobility shift assays, single-stranded RNA was 5′-end labeled with [γ-32P] ATP (Perkin Elmer) using polynucleotide kinase (New England Biolabs). Labeled RNA was then annealed to its complement as described above. The duplex was then passed over a G25 column to remove free [γ-32P] ATP and gel purified from a 15% native polyacrylamide gel.
Triphosphorylated RNA was prepared by in vitro transcription by T7 RNA polymerase using DNA oligomers containing 3′ 2′-O-methyl modifications as templates. After a 4 h transcription at 37°C, the resulting transcripts were purified by gel extraction from a 20% denaturing polyacrylamide gel. The purified RNA was then run on a 20% polyacrylamide denaturing gel alongside 5′OH synthesized RNA of the same sequence to confirm purity and triphosphorylation state. The 5′ triphosphorylated RNA was annealed to its complementary strand as described above. All RNA sequences are listed in Supplementary Table S1.
Electrophoretic mobility shift assay
Electrophoretic mobility shift assays (EMSAs) were performed to determine the apparent dissociation constants of each protein construct for various length RNA duplexes. Reactions (20 μl) containing 1 nM radiolabeled RNA duplex and varying concentrations of protein were incubated at room temperature in RNA binding buffer (25 mM HEPES pH 7.4, 100 mM NaCl, 2 mM DTT, 10 mM MgCl2, 10% glycerol). Incubation times were varied to ensure the system was equilibrated. A portion of the reactions were loaded onto a precast 6% native polyacrylamide gel (Invitrogen) and run in 0.5× TBE at 100V for 1–1.25 h at 4°C. The gels were then dried and exposed to phosphorimaging screens for up to 48 h. The gels were imaged using a Storm 820 PhosphorImager (GE Healthcare) and analyzed using ImageQuant software (GE Healthcare). The fraction bound RNA was quantified for each protein concentration and plotted versus protein concentration. The data were fit to an equation describing cooperative binding (Equation 1) using GraFit 5 (18),
| (1) |
where L is the protein (ligand) concentration, n is the cooperativity coefficient, Cap is the capacity or maximal fraction bound, K is the microscopic dissociation constant and y is the fraction of occupied sites (or fraction bound RNA).
Malachite green ATP hydrolysis assay
To determine the steady state kinetic parameters (kcat and KM) of ATP hydrolysis for DRH-3, an established malachite green assay was used. DRH-3 (or ΔN-DRH3) was first preincubated with a 3-fold molar excess of RNA duplex for 30 min at room temperature in ATP hydrolysis buffer (25 mM HEPES pH 7.4, 100 mM NaCl, 2 mM DTT). The reactions were then initiated by the addition of a 1:1.5 ATP:MgCl2 complex. For initial velocity measurements, aliquots of the reaction were quenched at 6 time points between 15 s and 10 min. The reactions were quenched by the addition of 5× quench buffer (250 mM EDTA) for a final concentration of 50 mM EDTA (≥50-fold excess over ATP). Malachite green reagent was added (9:1 malachite green: reaction volume) and allowed to age for 30 min at room temperature. The Abs650 was then measured using a Synergy 2 plate reader (BioTek). The steady state kinetic parameters, kcat and KM, were obtained by determining the initial velocity (v0) as a function of ATP concentration, and fitting the data to the Michaelis–Menten equation (Equation 2) using nonlinear regression using GraFit 5 (18),
| (2) |
where kobs = v0/[Etot], and Etot is the total protein concentration.
Dissociation rate constant determination
To obtain RNA dissociation rate constants (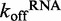 ) for DRH-3, stopped-flow fluorescence spectroscopy was used. Stopped-flow experiments were performed in ATP hydrolysis buffer (described above) at 24°C using an Applied Photophysics SX20 stopped-flow instrument (Applied Photophysics Ltd.) supplied with a 150 W xenon arc lamp. For detection, the Cy3-labeled duplex RNA was excited at 515 nm and the fluorescence emission was monitored at ≥570 nm using a 570 bandpass filter (Newport Corporation). Briefly, a slight excess (20%) of DRH-3 was pre-incubated with duplex RNA at room temperature for 30 min to 1 h to form a protein–RNA complex. The protein–Cy3 RNA complex was then rapidly mixed with a 50-fold excess of unlabeled duplex RNA for a specified period of time in which 2000 points were collected. For experiments with nucleotide analog, adenosine 5′-diphosphate (ADP) (Sigma-Aldrich) or adenosine 5′-[γ-thio]triphosphate (ATPγS) (Sigma-Aldrich) and MgCl2 were included with the trap RNA and rapidly mixed with the protein–Cy3 RNA complex. The average fluorescence measurements for each condition were then used in data analysis.
) for DRH-3, stopped-flow fluorescence spectroscopy was used. Stopped-flow experiments were performed in ATP hydrolysis buffer (described above) at 24°C using an Applied Photophysics SX20 stopped-flow instrument (Applied Photophysics Ltd.) supplied with a 150 W xenon arc lamp. For detection, the Cy3-labeled duplex RNA was excited at 515 nm and the fluorescence emission was monitored at ≥570 nm using a 570 bandpass filter (Newport Corporation). Briefly, a slight excess (20%) of DRH-3 was pre-incubated with duplex RNA at room temperature for 30 min to 1 h to form a protein–RNA complex. The protein–Cy3 RNA complex was then rapidly mixed with a 50-fold excess of unlabeled duplex RNA for a specified period of time in which 2000 points were collected. For experiments with nucleotide analog, adenosine 5′-diphosphate (ADP) (Sigma-Aldrich) or adenosine 5′-[γ-thio]triphosphate (ATPγS) (Sigma-Aldrich) and MgCl2 were included with the trap RNA and rapidly mixed with the protein–Cy3 RNA complex. The average fluorescence measurements for each condition were then used in data analysis.
Data was fit using nonlinear regression to a single (Equation 3) or double exponential equation (Equation 4) using GraFit 5 (18),
| (3) |
| (4) |
where A is the amplitude, k is the rate constant, t is the reaction time (s) and the offset is the fluorescence value (V) of free Cy3–RNA.
RESULTS
DRH-3 binds preferentially to RNA duplexes containing at least 22 base pairs
To characterize the length dependence of DRH-3 affinity for duplex RNA, binding titrations were conducted with RNA duplexes of increasing length. Complex formation was analyzed by EMSA, and the resultant data were fit to Equation (1). Quantitation of the data revealed relatively high affinity of DRH-3 for RNA duplexes of 22 bp and longer, with apparent dissociation constants (Kd) of 230 ± 7, 218 ± 7 and 384 ± 19 nM for lengths of 22, 25 and 34 bp (ds22, ds25 and ds34), respectively (Figure 1A). By comparison, the binding affinity for a 12 bp RNA duplex (ds12) was approximately 4-fold weaker (Kd of 839 ± 53 nM, Figure 1A and Table 1). The binding affinity for an 18 bp RNA duplex was also weak, with a Kd value of 679 ± 135 nM (Figure 1A and Table 1). The tighter affinity of DRH-3 for ds22 and ds25 suggests that optimal DRH-3 recognition occurs with RNA duplexes of ∼22 bp, i.e. 22G-RNA. Structural studies on the closely related RIG-I and MDA-5 proteins indicate that the monomeric form of these proteins occupy a footprint of 10–12 bp on duplex RNA. If DRH-3 has a similar structure, which would be expected given its domain organization and phylogenetic similarity, it is likely that two DRH-3 load upon a 22 bp duplex.
Figure 1.

Equilibrium binding experiments. (A) DRH-3 binding curves for ds12 (black circle), ds18 (blank square), ds22 (gray down triangle), ds25 (black square) and ds34 (gray up triangle) RNA. DRH-3 (0.002 to 8 μM) was added incrementally to a 1 nM 32P-labeled duplex RNA solution, and allowed to incubate for 30 min and up to 2 h (see ‘Materials and Methods’ section). For ds12 (black circle), Kd = 839 ± 53 nM, cooperativity coefficient = 1.2 ± 0.07; ds18 (blank square), Kd = 679 ± 135 nM, cooperativity coefficient = 1.2 ± 0.2; ds22 (gray down triangle), Kd = 230 ± 7 nM, cooperativity coefficient = 2.2 ± 0.1; ds25 (black square), Kd = 218 ± 7 nM, cooperativity coefficient = 1.9 ± 0.1; and ds34 (gray up triangle), Kd = 384 ± 19 nM, cooperativity coefficient = 1.9 ± 0.1. Plotted values are mean ± SD (n = 3). (B) Representative EMSA for DRH-3 binding to a 25 base-paired RNA duplex. Lanes 1–11 contain 1 nM labeled RNA and increasing DRH-3 concentrations (0.003–3 μM), and lane 12 contains only 1 nM labeled RNA. Dimer formation is clearly demonstrated by the second, slower-mobility band that evolves at higher protein concentrations. (C) Comparative binding curves for DRH-3, RIG-I and MDA-5. Each protein was added incrementally (0.001–3 μM) to a 1 nM solution of 32P-labeled ds25 and allowed to incubate for 30 min in the case of DRH-3 or 1 h in the case of MDA-5 and RIG-I. In the RIG-I binding reactions, it was necessary replace the 10% glycerol with 0.1 mg/mL BSA (14). Values for RIG-I (gray up triangle), Kd = 38 ± 6 nM, cooperativity coefficient = 0.8 ± 0.07; DRH-3 (black circle), Kd = 218 ± 7 nM, cooperativity coefficient = 1.9 ± 0.1; MDA-5 (blank square), Kd = 123 ± 4 nM, cooperativity coefficient = 2.6 ± 0.2. Plotted values are mean ± SD (n = 3).
Table 1.
RNA binding affinities
| Protein | RNA | Kd (nM) | Cooperativity coefficient |
|---|---|---|---|
| DRH-3 | ds12 | 839 ± 53 | 1.2 ± 0.07 |
| ds18 | 679 ± 135 | 1.2 ± 0.2 | |
| ds22 | 230 ± 7 | 2.2 ± 0.1 | |
| ds25 | 218 ± 7 | 1.9 ± 0.1 | |
| ds34 | 384 ± 19 | 1.9 ± 0.1 | |
| ΔN-DRH3 | ds25 | 376 ± 5 | 1.9 ± 0.03 |
| dsPPP25 | 120 ± 6 | 2.1 ± 0.2 | |
| ds12 | 522 ± 67 | 1.3 ± 0.2 | |
| dsPPP12 | 174 ± 12 | 1.4 ± 0.1 | |
| RIG-I | ds25 | 38 ± 6 | 0.8 ± 0.07 |
| MDA-5 | ds25 | 123 ± 4 | 2.6 ± 0.2 |
DRH-3 binds cooperatively to RNA, unlike the related RIG-I protein
To assess the cooperativity of DRH-3 binding to RNA duplexes, cooperativity coefficients were determined from EMSA experiments on DRH-3 binding to duplexes of increasing length (Figure 1A). For a duplex that is likely to represent an approximately monomeric site size (12 bp), DRH-3 binding did not display cooperativity (n = 1.2), and the mobility was consistent with migration of a 1:1 complex. Additionally, DRH-3 did not display cooperativity for a slightly longer duplex (n = 1.2 for ds18). In that case, the mobility was again consistent with that of a 1:1 complex. By contrast, distinctly cooperative behavior was observed for DRH-3 binding to RNA duplexes that were 22, 25 and 34 bp in length (n = 2.2 for ds22, n = 1.9 for ds25 and ds34; Figure 1A and Table 1). These values indicate that DRH-3 binds at least as a cooperative dimer to duplexes ≥22 bp, which is supported by the formation of a single, slow-migrating complex in EMSA experiments at high concentrations of DRH-3 (Figure 1B).
To compare the behavior of DRH-3 to that of related proteins, DRH-3, MDA-5 and RIG-I were titrated with the 25 bp RNA duplex in side-by-side experiments that were conducted as described above (Figure 1C). The curves show strongly sigmoidal behavior only for DRH-3 and MDA-5 (Figure 1C), immediately suggesting cooperative binding of both proteins to ds25. To quantitate affinity and cooperativity, the data were fit to Equation (1), which resulted in binding constants of 218 ± 7 nM and 123 ± 4 nM and cooperativity coefficients of 1.9 and 2.6 for DRH-3 and MDA-5, respectively. The data for MDA-5 are consistent with published results (10). In contrast, quantitative analysis of RIG-I binding to ds25 revealed a hyperbolic curve, which is indicative of noncooperative binding (Figure 1C), and which is consistent with the measured RIG-I cooperativity coefficient of 0.8 (Figure 1C and Table 1).
It is important to note that, while RIG-I binding is not cooperative, one ultimately observes the loading of two RIG-I molecules onto the 25 bp duplex (data not shown), as reported previously (11). Thus, while RIG-I molecules do not bind cooperatively, at high protein concentrations they will ultimately occupy all available binding sites on a duplex RNA molecule. Given the published structures of RIG-I bound to RNA, a 25 bp duplex will contain two binding sites for non-interacting molecules of RIG-I. Taken together, these data demonstrate that DRH-3, RIG-I and MDA-5 each displays slightly different behavior, despite similarities in their structures. While RIG-I does not increase its affinity or cooperativity on longer duplexes, DRH-3 behaves like an obligate dimer, and MDA-5 displays even greater cooperativity, as reported previously (10). In addition, these data show that the DRH-3 dimer is specifically optimized to bind an RNA duplex that is 22 bp in length.
DRH-3 is a more efficient ATPase when bound to longer RNA
To establish whether cooperative dimerization influences DRH-3 function, RNA-stimulated ATPase activity of DRH-3 was examined as a function of RNA duplex length. Using a malachite green ATP hydrolysis assay, steady state kinetic parameters were determined for DRH-3 activation by RNA duplexes that ranged in length from 12 to 50 bp. Prior to initiation of this experiment, it was essential to determine the optimal RNA concentrations for each steady state ATPase experiment by varying the RNA concentration while keeping DRH-3 and ATP concentrations constant (data not shown).
The ATPase activity of DRH-3 was robust when bound to ds22, with a kcat of 5.7 ± 0.8 s−1 (Figure 2). When bound to successively longer duplexes, the steady state rate constant did not increase significantly (Figure 2). In contrast, ATP hydrolysis was 4-fold slower when DRH-3 was bound to ds12, resulting in a kcat of 1.4 ± 0.1 s−1 (Figure 2). ATP hydrolysis was also slower when DRH-3 was bound to ds16 and ds18, with kcat values of 1.2 ± 0.2 s−1 and 2.3 ± 0.2 s−1 for ds16 and ds18, respectively (Figure 2). Based on this data, ds22 is the minimal ligand for stimulating for maximal ATP hydrolysis activity. Given that the binding experiments implicate a cooperative DRH-3 dimer on the 22 bp RNA duplex but not shorter duplexes, the ATPase experiments show that this cooperativity is functionally important for stimulating enzymatic function of DRH-3. This is in striking contrast to RIG-I, which does not show cooperativity in RNA binding (Figure 1C) or RNA-stimulated ATPase function (14,19).
Figure 2.
Steady state ATP hydrolysis of DRH-3 with various RNA duplexes. Steady state rate constants for DRH-3 bound to ds12, ds16, ds18, ds22, ds25, ds34 and ds50 are plotted as a function of substrate (rATP) concentration. The data were fitted to the Michaelis–Menten equation, resulting in the following values: ds12 (black circle), kcat = 1.36 ± 0.13 s − 1, KM = 28 μM; ds16 (blank circle), kcat = 1.22 ± 0.18 s−1, KM = 49 μM; ds18 (blank square), kcat = 2.31 ± 0.20 s−1, KM = 72 μM; ds22 (gray down triangle), kcat = 5.69 ± 0.79 s−1, KM = 77 μM; ds25 (black square), kcat = 4.49 ± 0.32 s−1, KM = 54 μM; ds34 (gray up triangle), kcat = 4.51 ± 0.34 s−1, KM = 40 μM; ds50 (gray diamond suit), kcat = 3.98 ± 0.37 s−1, KM = 61 μM. Plotted values are mean ± SD (n = 3). The concentrations of DRH-3 and RNA used were as follows: 400 nM DRH-3 and 800 nM ds12, 400 nM DRH-3 and 800 nM ds16, 200 nM DRH-3 and 600 nM ds18, 100 nM DRH-3 and 300 nM ds22, 100 nM DRH3 and 300 nM ds25, 100 nM DRH-3 and 300 nM ds34, and 100 nM DRH-3 and 300 nM ds50. ATP concentration was varied from 7.8 to 500 μM.
The N-terminal domain contributes to RNA length specification by DRH-3
Studies of RNA binding and ATP hydrolysis reveal striking differences between DRH-3 and RIG-I ligand specificities, as indicated by the preference of DRH-3 for longer RNA duplexes and lack of apparent 5′ triphosphate specificity by DRH-3. These differences, particularly given the similar domain organizations of DRH-3 and RIG-I, suggest that the individual protein domains function differently in these two proteins. Previous RIG-I studies have shown that the N-terminal CARD signaling domains do not improve RNA binding by RIG-I. In fact, removal of RIG-I CARDs slightly increases the affinity for 5′ triphosphorylated RNA duplexes (14). Because the NTD of DRH-3 is not known to be an RNA binding domain or a CARD domain, we thought it was important to test whether it plays a role in RNA recognition and enzymatic function. To this end, we removed the NTD and tested its influence on RNA binding by DRH-3.
We analyzed binding of an N-terminal deletion variant of DRH-3 (ΔN-DRH3) to a 25 bp RNA duplex with and without a 5′ triphosphate (dsPPP25 and ds25) using EMSA. Data quantitation revealed a binding affinity for dsPPP25 that was 3-fold tighter than for ds25, with Kd values of 120 ± 6 nM and 376 ± 5 nM respectively (Figure 3A and Table 1). Thus, removal of the NTD increases the affinity of DRH-3 for a 5′ triphosphate. Surprisingly, NTD removal also weakened overall affinity for a blunt (5′OH) ds25 compared with full-length protein. We next measured the affinity of ΔN-DRH3 for ds12 and dsPPP12. Given that removal of the NTD increased DRH-3 binding affinity for a 5′ triphosphorylated ds25, we hypothesized that ΔN-DRH3 would demonstrate a 5′ triphosphate preference on this shorter RNA, as well. As expected, ΔN-DRH3 bound more tightly to dsPPP12 than ds12, with Kd values of 174 ± 12 nM and 522 ± 67 nM respectively (Figure 3B and Table 1). Importantly, the Kd values were similar for ΔN-DRH3 binding to both ds12 and ds25. In contrast, full length DRH-3 displayed a clear length preference, binding ds25 3.5-fold more tightly than ds12 (Figure 1A and Table 1). Removal of the N-terminus therefore appears to abolish length-dependent RNA binding by DRH-3. Additionally, truncation of the NTD causes DRH-3 to behave more like RIG-I with respect to 5′ triphosphate specificity: similar to the CARD domains in RIG-I, the inhibitory behavior demonstrated by the NTD may be a mechanism to reduce nonspecific binding to RNA. However, NTD removal also eliminated RNA length discrimination by DRH-3. Either directly or indirectly, the NTD facilitates RNA length discrimination, enabling, DRH-3 dimerization to serve as a molecular ‘ruler’ for specifying the sizes of secondary siRNAs.
Figure 3.

RNA binding and ATPase activity of ΔN-DRH-3. (A) ΔN-DRH3 binding curves for ds25 (black circle) and dsPPP25 (blank square). ΔN-DRH3 was added incrementally (0.002 to 2 μM) to a 1 nM 32P-labeled ds25 or dsPPP25 solution and allowed to incubate for 30 min. Values for ds25 (black circle), Kd = 376.2 ± 5 nM, cooperativity coefficient = 1.9 ± 0.03; dsPPP25 (blank square), Kd = 119.6 ± 6 nM, cooperativity coefficient = 2.1 ± 0.2. Plotted values are mean ± SD (n = 3). (B) ΔN-DRH3 binding curves for ds12 (black circle) and dsPPP12 (blank square). The ΔN-DRH3 was added incrementally (0.002–2 μM) to a 1 nM 32P-labeled ds12 or dsPPP12 solution and allowed to incubate for 30 min. Values for ds12 (black circle), Kd = 522.2 ± 67 nM, cooperativity coefficient = 1.3 ± 0.2; dsPPP12 (blank square), Kd = 174.4 ± 12 nM, cooperativity coefficient = 1.4 ± 0.1. Plotted values are mean ± SD (n = 3). (C) Saturation curves for the steady state ATP hydrolysis of ΔN-DRH3. Steady state rate constants for ΔN-DRH3 bound to ds12 and ds25 are plotted as a function of substrate (rATP) concentration. The data were fitted to the Michaelis–Menten equation, resulting in values of ds12 (blank square), kcat = 2.03 ± 0.15 s−1, KM = 59 μM; ds25 (black circle), kcat = 3.09 ± 0.23 s−1, KM = 64 μM. Plotted values are mean ± SD (n = 3). The concentration of DRH-3 in these experiments was 400 nM and the concentration of RNA used was 1200 nM for both ds12 and ds25. ATP concentration was varied from 7.8 to 500 μM. (D) Schematic comparison of the domain architecture of full length DRH-3 and ΔN-DRH3. Full length DRH-3 is composed of 1119 amino acids and contains the NTD, the two helicase domains (Hel1 and Hel2) and the C-terminal domain (CTD). In contrast, ΔN-DRH3 is composed of 819 amino acids (residues 300–1119), and is lacking the NTD.
Steady state ATP hydrolysis for ΔN-DRH3 is not dependent on RNA length
Deletion of the NTD of DRH-3 abolished the apparent RNA length preference in binding experiments, as shown by the equivalent Kd values for ds12 and ds25. However, it was also important to determine whether the truncation influenced the RNA stimulated ATPase activity of ΔN-DRH3. Using the malachite green assay, we obtained steady state kinetic parameters for ΔN-DRH3 when activated by ds12 and ds25. The ATP hydrolysis rate for ΔN-DRH3 when bound to ds25 was similar to the rate for ΔN-DRH3 when bound to ds12, with kcat values of 3.1 ± 0.2 and 2.0 ± 0.1 s−1 respectively (Figure 3C). In the case of full length DRH-3, both Kd and kcat values were significantly lower for ds12 relative to ds25. These experiments show that, unlike the wild type protein, ΔN-DRH3 has lost the ability to discriminate the length of RNA ligands and that this is reflected in both binding and enzymatic activity. This supports the hypothesis that the NTD contributes to the role of DRH-3 as a molecular ‘ruler’ and that the NTD is important for the recognition of longer, double-stranded RNA by DRH-3.
The off-rate of DRH-3 is dependent on RNA duplex length
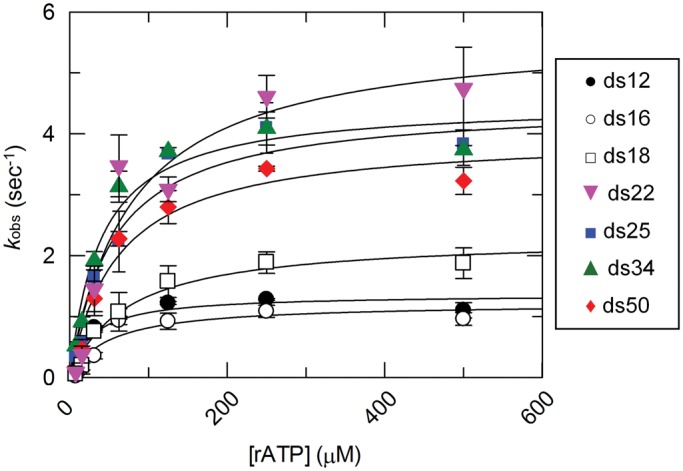
Because ATP hydrolysis is tightly coupled to RNA binding, we asked if ATP binding and/or hydrolysis affects the off-rate of DRH-3 from RNA. To assess the role of ATP hydrolysis in RNA dissociation, we first determined the RNA dissociation rate constants (koffRNA) for DRH-3 using ds12 and ds25 in the absence of nucleotide. The 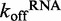 values were determined using stopped-flow fluorescence spectroscopy, where a pre-bound DRH-3:Cy3-labeled dsRNA complex (ds12 or ds25) was rapidly mixed with an excess of unlabeled dsRNA (a pulse-chase experiment). The concentrations of protein and RNA used in these measurements were 10-fold higher than the Kd to ensure fully saturated RNA. In the case of ds25, where Kd = 218 ± 7 nM, we used 2.4 μM DRH-3 and 2 μM ds25-Cy3. These concentrations ensure that DRH-3 dimers, as opposed to monomers, are bound to ds25. In addition, excess unlabeled dsRNA acts to effectively trap free DRH-3 in order to prevent rebinding of the Cy3-labeled RNA. The release of DRH-3 from Cy3-RNA results in a decrease of fluorescence intensity, which allows for real-time measurement of DRH-3 dissociation. Subsequent quantitation of reductions in Cy3 fluorescence intensity were used to determine the dissociation rate constant (see ‘Materials and Methods’ section and Figure 4), as in other studies (20,21).
values were determined using stopped-flow fluorescence spectroscopy, where a pre-bound DRH-3:Cy3-labeled dsRNA complex (ds12 or ds25) was rapidly mixed with an excess of unlabeled dsRNA (a pulse-chase experiment). The concentrations of protein and RNA used in these measurements were 10-fold higher than the Kd to ensure fully saturated RNA. In the case of ds25, where Kd = 218 ± 7 nM, we used 2.4 μM DRH-3 and 2 μM ds25-Cy3. These concentrations ensure that DRH-3 dimers, as opposed to monomers, are bound to ds25. In addition, excess unlabeled dsRNA acts to effectively trap free DRH-3 in order to prevent rebinding of the Cy3-labeled RNA. The release of DRH-3 from Cy3-RNA results in a decrease of fluorescence intensity, which allows for real-time measurement of DRH-3 dissociation. Subsequent quantitation of reductions in Cy3 fluorescence intensity were used to determine the dissociation rate constant (see ‘Materials and Methods’ section and Figure 4), as in other studies (20,21).
Figure 4.

Determination of DRH-3–dsRNA dissociation rate constants. Using fluorescence stopped-flow spectroscopy, 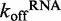 was measured for DRH-3. (A) A comparison of
was measured for DRH-3. (A) A comparison of 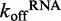 for ds25 and ds12. An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is displaced by 100 μM ds25 and observed for 120 s (20 s shown, red trace). An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12 and observed for 100 s (20 s shown, blue trace). Both traces were fit to a double exponential equation, from which the following parameters were determined. For ds25: k1 = 0.0705 ± 0.0009 s−1, Amp. = 0.0626 ± 0.0009, k2 = 0.4985 ± 0.0022 s−1, Amp. = 0.4437 ± 0.0010. For ds12: k1 = 0.1901 ± 0.0016 s−1, Amp. = 0.2255 ± 0.0021, k2 = 1.5391 ± 0.0114 s−1, Amp. = 0.7378 ± 0.0028 ( ± SEM). (B) Effect of ATPγS and ADP on the
for ds25 and ds12. An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is displaced by 100 μM ds25 and observed for 120 s (20 s shown, red trace). An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12 and observed for 100 s (20 s shown, blue trace). Both traces were fit to a double exponential equation, from which the following parameters were determined. For ds25: k1 = 0.0705 ± 0.0009 s−1, Amp. = 0.0626 ± 0.0009, k2 = 0.4985 ± 0.0022 s−1, Amp. = 0.4437 ± 0.0010. For ds12: k1 = 0.1901 ± 0.0016 s−1, Amp. = 0.2255 ± 0.0021, k2 = 1.5391 ± 0.0114 s−1, Amp. = 0.7378 ± 0.0028 ( ± SEM). (B) Effect of ATPγS and ADP on the 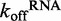 for ds25. An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is displaced by 100 μM ds25 in the presence of 2 mM ATPγS and 3 mM MgCl2 and observed for 120 s (100 s shown, purple trace). An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is rapidly mixed with 200 μM ds12, 2 mM ADP and 3 mM MgCl2 and observed for 100 s (green trace). Both traces were fit to a single exponential equation, resulting the parameters of
for ds25. An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is displaced by 100 μM ds25 in the presence of 2 mM ATPγS and 3 mM MgCl2 and observed for 120 s (100 s shown, purple trace). An average of three traces where 2.4 μM DRH-3 and 2 μM ds25Cy3 is rapidly mixed with 200 μM ds12, 2 mM ADP and 3 mM MgCl2 and observed for 100 s (green trace). Both traces were fit to a single exponential equation, resulting the parameters of 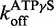 = 0.0630 ± 0.0001 s−1,
= 0.0630 ± 0.0001 s−1,
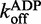 = 0.2726 ± 0.0009 s−1 ( ± SEM). (C) Effect of ATPγS and ADP on the
= 0.2726 ± 0.0009 s−1 ( ± SEM). (C) Effect of ATPγS and ADP on the 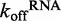 for ds12. An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12, 2 mM ATPγS and 3 mM MgCl2, and observed for 100 s (60 s shown, purple trace). An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12, 2 mM ATPγS and 3 mM MgCl2, and observed for 60 s (green trace). Both traces were fit to a double exponential equation, resulting in parameters of
for ds12. An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12, 2 mM ATPγS and 3 mM MgCl2, and observed for 100 s (60 s shown, purple trace). An average of three traces where 6 μM DRH-3 and 5 μM ds12Cy3 is rapidly mixed with 300 μM ds12, 2 mM ATPγS and 3 mM MgCl2, and observed for 60 s (green trace). Both traces were fit to a double exponential equation, resulting in parameters of 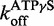 = 0.2093 ± 0.0008 s−1, Amp. = 0.6600 ± 0.0032, k2 = 0.0707 ± 0.0008 s−1, Amp. = 0.1535 ± 0.0033,
= 0.2093 ± 0.0008 s−1, Amp. = 0.6600 ± 0.0032, k2 = 0.0707 ± 0.0008 s−1, Amp. = 0.1535 ± 0.0033, 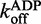 = 0.7846 ± 0.004 s−1, Amp. = 0.7720 ± 0.0026, k2 = 0.1724 ± 0.0021 s−1, Amp. = 0.1670 ± 0.0028 (± SEM).
= 0.7846 ± 0.004 s−1, Amp. = 0.7720 ± 0.0026, k2 = 0.1724 ± 0.0021 s−1, Amp. = 0.1670 ± 0.0028 (± SEM).
The experimental results reveal that DRH-3 dissociates faster from ds12 than ds25, with 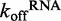 values of 1.54 ± 0.01 and 0.500 ± 0.002 s−1 for ds12 and ds25, respectively. Corresponding closely to the equilibrium binding data, there was a 3-fold increase in
values of 1.54 ± 0.01 and 0.500 ± 0.002 s−1 for ds12 and ds25, respectively. Corresponding closely to the equilibrium binding data, there was a 3-fold increase in 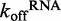 for ds12 as compared with ds25. Additionally, the observed time courses were biphasic and best fit to a double exponential equation (Figure 4A), with minimal contribution from the slower rate constant (i.e. the amplitude corresponding to the slow rate comprised about 10% of the total change in fluorescence). The small population could reflect a fraction of molecules in an alternative conformation or in an alternative oligomeric state. Overall, the inverse correlation between dissociation rate constant and RNA duplex length is consistent with the equilibrium binding data and with the notion that DRH-3 senses the length of RNA duplex targets. These experiments also establish a basal
for ds12 as compared with ds25. Additionally, the observed time courses were biphasic and best fit to a double exponential equation (Figure 4A), with minimal contribution from the slower rate constant (i.e. the amplitude corresponding to the slow rate comprised about 10% of the total change in fluorescence). The small population could reflect a fraction of molecules in an alternative conformation or in an alternative oligomeric state. Overall, the inverse correlation between dissociation rate constant and RNA duplex length is consistent with the equilibrium binding data and with the notion that DRH-3 senses the length of RNA duplex targets. These experiments also establish a basal 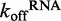 value for DRH-3, which is imperative for developing a full kinetic framework for ligand binding and ATP hydrolysis activity of DRH-3.
value for DRH-3, which is imperative for developing a full kinetic framework for ligand binding and ATP hydrolysis activity of DRH-3.
ATP hydrolysis affects RNA binding, and may aid in DRH-3 function
ATP hydrolysis has been suggested to trigger RNA dissociation by many helicases and motor proteins, including Mss116, DbpA and MDA-5 (10,22–24). Having obtained basal 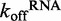 values for DRH-3 dissociation from ds12 and ds25, we set out to determine the effect of ATP binding and hydrolysis on these parameters. Using stopped-flow fluorescence spectroscopy, we rapidly mixed ATPγS (an ATP analog) or ADP and excess trap RNA with pre-formed DRH-3:Cy3RNA complex, resulting in
values for DRH-3 dissociation from ds12 and ds25, we set out to determine the effect of ATP binding and hydrolysis on these parameters. Using stopped-flow fluorescence spectroscopy, we rapidly mixed ATPγS (an ATP analog) or ADP and excess trap RNA with pre-formed DRH-3:Cy3RNA complex, resulting in 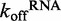 values in the presence of substrate (ATPγS) and product (ADP) nucleotide. In the presence of ATPγS,
values in the presence of substrate (ATPγS) and product (ADP) nucleotide. In the presence of ATPγS, 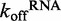 was 8-fold slower for both ds25 and ds12, 0.063 ± 0.001 s−1 and 0.2093 ± 0.0008 s−1, respectively (Figure 4B and C). In contrast,
was 8-fold slower for both ds25 and ds12, 0.063 ± 0.001 s−1 and 0.2093 ± 0.0008 s−1, respectively (Figure 4B and C). In contrast, 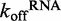 was minimally affected by ADP binding (Figure 4B and C). Taken together, these data demonstrate that ATP binding increases the lifetime of DRH-3 on RNA, while ATP hydrolysis products stimulate DRH-3 dissociation from RNA.
was minimally affected by ADP binding (Figure 4B and C). Taken together, these data demonstrate that ATP binding increases the lifetime of DRH-3 on RNA, while ATP hydrolysis products stimulate DRH-3 dissociation from RNA.
The koff differences suggest a mechanism whereby ATP binding initially promotes DRH-3 assembly on RNA while hydrolysis stimulates enzymatic turnover. Because the effects of nucleotide identity on koff values follow the same trends for both ds12 and ds25, it seems that ATP binding does not aid in RNA length specification, but rather in dimer assembly. For example, when one molecule of DRH-3 is bound to a 22G-RNA, ATP binding would increase its lifetime on RNA. This would allow another DRH-3 molecule to bind, effectively allowing the resultant dimer to ‘recognize’ its RNA target. The increased lifetime of DRH-3 on dsRNA provided by ATP binding would permit subsequent ATP hydrolysis, a process that is favored when DRH-3 is dimerized. Hydrolysis, and therefore ADP binding, would subsequently mediate dimer disassembly on dsRNA.
DISCUSSION
Here we show that DRH-3 forms functional dimers on double-stranded RNA molecules of a specific and biologically relevant length. Through a combination of direct RNA binding studies, ATPase kinetics and mutational analysis, we show that DRH-3 is specifically tuned to recognize RNA duplexes that are 22 bp in length, which is significant because this length is twice the expected site size of the DRH-3 helicase domain and it is the length of secondary siRNA molecules. Remarkably, even the enzymatic activity of DRH-3 has evolved a strict dependence on cooperative, functional dimerization on RNA, as the ATPase active-site is only fully engaged and maximally active upon dimerization of protein units. Thus, the specificity of DRH-3 for 22 bp RNA is controlled at multiple levels, by cooperative dimer binding (Kd) and by cooperative engagement of catalytic activity (kcat).
The fact that DRH-3 forms functional dimers is significant because it provides insight into the role of the Dicer helicase domain in higher organisms. The typical metazoan Dicer recognizes and operates upon RNA duplexes ranging in size from short pre-miRNA to long dsRNA, generating products 21–25 nucleotides in length, depending on their role and species of origin. The DRH-3 helicase domain, like that of metazoan Dicers, is approximately the same size as RIG-I, and likely to share a similar monomeric footprint of ∼10–12 bp. Indeed, a homology model of human Dicer indicates that its helicase domain shares many features with the RIG-I protein (25,26). If two Dicer helicase domains bind the RNA in tandem, the site size is therefore expected to be 21–25 bp.
It has been suggested that the PAZ domain of Dicer proteins, along with the 5’ pocket motif in higher eukaryotes (27), recognizes the length of dsRNA helices and positions them for cleavage by the RNase III catalytic site (16,25). A structure of the G. intestinalis Dicer shows the PAZ domain and RNase III sites separated by a distance that is equivalent to the length of the diced product (typically 21–25 nucleotides), hence providing a means for RNA length measurement (16,25). However, the G. intestinalis Dicer that has been studied structurally is highly unusual in that it lacks the conserved helicase domain present in other Dicer proteins, particularly those of higher organisms. In most cases, Dicer proteins contain a dsRNA binding motif that is structurally homologous to the helicase domain found within the innate immune sensor RIG-I. This conserved and highly specialized helicase domain, which is distinct from the helicase domains of DEAD-box proteins and SF2 helicases (15,28,29), binds to dsRNA with higher affinity than a PAZ domain. For example, the isolated RIG-I helicase domain binds to dsRNA with an affinity of 1 μM (14), while the Argonaute2 PAZ domain binds with an affinity of 10 μM (30). Therefore, most Dicer proteins may be capable of interacting with dsRNA through their helicase domains, in a manner that has been visualized crystallographically and biochemically for RIG-I-like receptors (11,19,31). However, while RIG-I binds as a monomer, preferring short duplexes with a single binding site (Figure 1C) (14), our data show that a Dicer-like protein binds as cooperative dimers to duplexes twice as long as the typical RIG-I site (∼22 bp instead of ∼12 bp) (Figure 5). Hence, a dimerized helicase domain, like that of DRH-3, is potentially capable of RNA length discrimination, particularly during the processing of long dsRNA into endo-siRNA, which contains long stretches of duplex RNA to which the helicase domain can bind.
Figure 5.
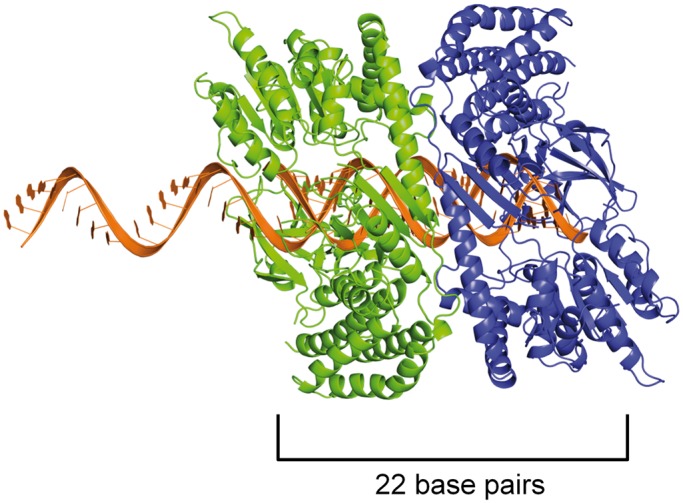
Model of a helicase domain dimer on an mRNA/22G-RNA duplex. As a proxy for the closely related DRH-3 protein, we used the crystal structure of the RIG-I helicase domain (11) and modeled it bound to a 22-bp duplex. Because 22G-RNA is complementary to a longer mRNA target, we imagine that the formation of the DRH-3 dimer occurs immediately after synthesis, when the mRNA and 22G-RNA are in duplex form. The top strand is thus extended beyond the duplex in the 5′ direction, as 22G-RNA is complementary to upstream mRNA target sequences that have yet to be synthesized. We show one protein monomer (in blue) interacting with the 5′ G of the 22G-RNA strand via the C-terminal domain, as is presented in the RIG-I structure, and because we show here that ΔN-DRH3 can recognize 5′-triphosphates. When a second protein molecule (in green) is bound adjacent to the first, the footprint is roughly 22 bp in size, thus creating a means for measurement by the dimer. The protein monomers are oriented such that the Hel2 domains are in close proximity, which is supported by the observation that the DRH-3 dimer is necessary for efficient ATP hydrolysis.
While a Dicer helicase domain may aid in long dsRNA recognition, we are not suggesting that it can replace the PAZ domain for RNA measurement in all cases. In fact, when the PAZ domain was mutated, human Dicer activity on dsRNA with 3′ overhangs was inhibited (32). Additionally, human Dicer does not show a requirement for the helicase domain in long dsRNA processing in vitro. The helicase domain of human Dicer has even been shown to have an inhibitory effect on dsRNA processing (33). Drosophila Dicer-1, which is responsible for miRNA production in the fly, also relies on its PAZ domain for product length measurement (34). Tsutsumi et al. suggest that while the 3′ overhang is recognized by the PAZ domain of Dicer-1, the loop of the pre-miRNA is recognized by the helicase domain. Based on these and other studies, there is not yet experimental evidence to suggest that the helicase domain functions in the measurement of dsRNA segments by human Dicer or Drosophila Dicer-1. By contrast, studies of Schizosaccharomyces pombe Dicer (Dcr1), which lacks a PAZ domain, does require a functional helicase domain to generate siRNA, indicating that the helicase domain plays an essential role in RNA binding and measurement (35). Therefore, the helicase domain may function differently in Dicers of different organisms. If S. pombe Dcr1 and other Dicers use their helicase domains to measure long dsRNA like DRH-3, it will be necessary to re-examine existing models for RNA target recognition and length determination by this family of proteins.
The helicase dimerization model for length determination is also consistent with studies on Drosophila Dicer-2, which shares a domain architecture with human Dicer. Dicer-2 requires its helicase domain and ATP hydrolysis for processing blunt end dsRNA and long dsRNA, but not for short pre-miRNA (36), as suggested by the model above. Similarly, C. elegans DCR-1 also requires an ATPase-competent helicase domain for the production of endo-siRNA, which results from the cleavage of long RNA ligands (37,38). Significantly, the PAZ domain of Dicer recognizes blunt ended dsRNA poorly (16), suggesting that it depends on other domains, such as its helicase domain, for efficient binding to endo-siRNA precursors. These results, along with the low affinity observed for the Argonaute2 PAZ binding to dsRNA (30), are consistent with the fact that PAZ domains are OB folds, which are not typical motifs for binding double-stranded nucleic acid. The OB fold is usually a single-stranded DNA binding motif, and it is abundant in proteins such as RPA, BRCA2 and the telomere binding protein POT1 (39–42). Given that endo-siRNA precursors have blunt ends and long stretches of duplexed RNA, these RNAs are unlikely targets for the PAZ domain of Dicer proteins. By contrast, pre-miRNAs, which often contain single strand overhangs, are likely preferred substrates for PAZ domain-mediated measurement and recognition by Dicer. Significantly, numerous studies reveal that Dicers use their PAZ domain for 3′ overhang recognition when processing miRNA. Human Dicer and Drosophila Dicer-1, which contain helicase domains divergent from the RIG-I helicase domain (34), rely on the PAZ domain and not the helicase domain for length specification, as discussed previously. But, this does not preclude use of the helicase domain by other Dicer proteins when processing endo-siRNA, particularly by Dicers that have helicase domains more similar to RIG-I (i.e. Drosophila Dicer-2 and C. elegans DCR-1). Collectively, these data suggest that certain metazoan Dicer proteins may rely on the RNA binding stoichiometry and enzymatic activity of their helicase domains for recognition of specific RNA substrates, such as long and blunt end dsRNA.
As in the case of DRH-3, ATP hydrolysis may act to position Dicer dimers along the dsRNA. ATP hydrolysis-mediated loading of Dicer along the RNA would result in cleavage every 21–25 nucleotides, which is supported by processive cleavage of long RNA by Dicer-2 in the presence of ATP (36). Thus, functional dimerization may provide tight control over the specificity of certain Dicer proteins, and the dimerized Dicer helicase domain may be a molecular ‘ruler' for controlling duplex RNA product length during the processing of long dsRNA substrates.
It is biologically significant that physical dimerization of DRH-3 is not only required for strong binding to RNA and for site-size definition, but for efficient, coupled ATP hydrolysis (Figure 2). It is suggested that ATP hydrolysis is important for 22G-RNA synthesis in vivo and that ATP hydrolysis is likely to facilitate dsRNA length discrimination (9). This is supported by genetic studies demonstrating a loss of 22G-RNA upon mutation of the DRH-3 helicase domain (9). Importantly, the reduction in 22G-RNA is not total, but rather the 3′ terminal 22G-RNAs accumulate while 22G-RNAs corresponding to 5′ mRNA sequences are depleted (9). This suggests that DRH-3 uses both its binding and ATP hydrolysis activities to propagate the sequential synthesis of 22G-RNA.
To elucidate the precise role of ATP binding and hydrolysis in DRH-3 function, we employed a combination of steady state and transient kinetics. Specifically, we determined the basal dissociation rate constants of DRH-3 from RNA in the absence and presence of various nucleotide analogs, measuring the RNA dissociation rate constants of both ds25 and ds12. In addition, we measured the kcat for ATP hydrolysis when DRH-3 was bound to RNAs of varying length. These studies show that ATP hydrolysis plays a key role in the function of DRH-3 by enhancing RNA length discrimination beyond that achieved through cooperative binding alone. Intriguingly, DRH-3 utilizes differences in RNA dissociation and ATP hydrolysis rate constants as a kinetic proofreading mechanism for sensing the length of different target duplexes. For example, DRH-3 dissociates from ds25 with a 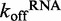 of 0.500 ± 0.002 s−1 (Figure 4A), while the kcat for ATP hydrolysis when DRH-3 is bound to ds25 is 4.5 ± 0.3 s−1 (Figure 2). DRH-3 is thus likely to remain bound to ds25 and hydrolyze ATP, hence promoting a forward commitment to catalysis. Because DRH-3 is not rapidly dissociating from RNA, the coupled ATP hydrolysis reaction is favored. By contrast, DRH-3 dissociates from ds12 with a
of 0.500 ± 0.002 s−1 (Figure 4A), while the kcat for ATP hydrolysis when DRH-3 is bound to ds25 is 4.5 ± 0.3 s−1 (Figure 2). DRH-3 is thus likely to remain bound to ds25 and hydrolyze ATP, hence promoting a forward commitment to catalysis. Because DRH-3 is not rapidly dissociating from RNA, the coupled ATP hydrolysis reaction is favored. By contrast, DRH-3 dissociates from ds12 with a 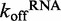 of 1.54 ± 0.01 s−1 (Figure 4A), while the kcat for ATP hydrolysis is 1.4 ± 0.1 s−1 (Figure 2). Thus for short RNA duplexes, DRH-3 is more likely to dissociate than bind and hydrolyze ATP. The transient and steady state kinetic data indicate that DRH-3 uses the disparity (or lack thereof) between RNA dissociation and ATP hydrolysis rate constants to facilitate dsRNA binding specificity and to ensure a forward commitment to catalysis. This kinetic regulation of RNA length discrimination is likely to greatly facilitate the role of DRH-3 in 22G-RNA synthesis. When DRH-3 is favorably bound to an mRNA/22G-RNA duplex, multiple rounds of ATP hydrolysis are probable.
of 1.54 ± 0.01 s−1 (Figure 4A), while the kcat for ATP hydrolysis is 1.4 ± 0.1 s−1 (Figure 2). Thus for short RNA duplexes, DRH-3 is more likely to dissociate than bind and hydrolyze ATP. The transient and steady state kinetic data indicate that DRH-3 uses the disparity (or lack thereof) between RNA dissociation and ATP hydrolysis rate constants to facilitate dsRNA binding specificity and to ensure a forward commitment to catalysis. This kinetic regulation of RNA length discrimination is likely to greatly facilitate the role of DRH-3 in 22G-RNA synthesis. When DRH-3 is favorably bound to an mRNA/22G-RNA duplex, multiple rounds of ATP hydrolysis are probable.
When considering these results, together with those obtained from genetic studies, we propose that DRH-3 cooperatively dimerizes upon binding to the mRNA/22G-RNA duplex, and then hydrolyzes ATP as an RNA-bound dimer. While the exact role of ATP hydrolysis in secondary siRNA production is not known, one can imagine that DRH-3 dimers use ATP hydrolysis not only to regulate DRH-3 assembly (as described above), but to power translocation and ‘push’ the RdRP off the newly formed RNA. Dimers may also act to sequester 22G-RNA, preventing rebinding by the RdRP to newly synthesized 22G-RNA. This combination of DRH-3 binding and hydrolysis would result in the propagation of 22G-RNA synthesis and it would force the RdRP to turn over.
DRH-3 contains a specialized NTD that distinguishes it from related proteins, and therefore we investigated whether this adaptation plays a role in RNA length specification by the DRH-3 protein. By conducting RNA binding and ATPase kinetics experiments on a DRH-3 deletion mutant that lacks the NTD, we found that the protein was incapable of discriminating between ds12 and ds25 RNA targets (Figure 3), in striking contrast with full length DRH-3. Whether directly, or indirectly through allosteric control of the helicase domain, the NTD facilitates dimerization and functional activity of the DRH-3 protein. It is also intriguing that the NTD masks the ability of the DRH-3 CTD to recognize 5′-triphosphate groups on duplex RNA ligands. Strikingly, when the NTD is removed, DRH-3 behaves much like the RIG-I protein, having acquired the ability to bind 5′ triphosphorylated RNAs and to prefer short RNA duplexes. The known RIG-I residues important for triphosphate recognition (H847, K858, K861 and K888) (13,19) are not strictly conserved in the DRH-3 CTD, but there is a conservation of positive charge, i.e. lysine residues that have the potential to interact with the negatively charged triphosphate. Additionally, RIG-I residues K812 and K909, important for RNA duplex end-binding (12) and RNA backbone interactions (19) respectively, are conserved in DRH-3. Thus, the C-terminal domains of DRH-3 and RIG-I are similar but not identical. The binding behavior displayed by ΔN-DRH3 suggests an additional regulatory function of the NTD, and reveals that, under specific conditions, DRH-3 can recognize 5′-triphosphosphorylated RNAs. This is likely to be relevant for DRH-3 function in vivo, as 22G-RNAs contain 5′ triphosphate moieties.
In conclusion, DRH-3 uses dimerization, ATP hydrolysis and the interplay of functional domains to specifically recognize 22G-RNAs. We find that the NTD of DRH-3 plays a role in the mechanism of RNA length discrimination, and it controls the ability of DRH-3 to recognize 5′-triphosphates. We show that RNA dissociation rate constants are coupled with rate constants for ATP binding and hydrolysis, providing an important determinant in the commitment to catalysis for DRH-3. This mechanism likely plays an important role in DRH-3 assembly on 22G-RNA, and will contribute to its turnover. Perhaps most significantly, this study has provided important insights into the mechanism of RNA length discrimination by Dicer-like proteins in general.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank members of the Pyle laboratory for helpful discussions and input on the manuscript. We especially thank Dr. Srinivas Somarowthu for his help on the model of the helicase domain dimer.
REFERENCES
- 1.Zamore PD, Haley B. Ribo-gnome: the big world of small RNAs. Science. 2005;309:1519–1524. doi: 10.1126/science.1111444. [DOI] [PubMed] [Google Scholar]
- 2.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- 4.Pak J, Fire A. Distinct populations of primary and secondary effectors during RNAi in C. elegans. Science. 2007;315:241–244. doi: 10.1126/science.1132839. [DOI] [PubMed] [Google Scholar]
- 5.Sijen T, Steiner FA, Thijssen KL, Plasterk RH. Secondary siRNAs result from unprimed RNA synthesis and form a distinct class. Science. 2007;315:244–247. doi: 10.1126/science.1136699. [DOI] [PubMed] [Google Scholar]
- 6.Lee RC, Hammell CM, Ambros V. Interacting endogenous and exogenous RNAi pathways in Caenorhabditis elegans. RNA. 2006;12:589–597. doi: 10.1261/rna.2231506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smardon A, Spoerke JM, Stacey SC, Klein ME, Mackin N, Maine EM. EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr. Biol. 2000;10:169–178. doi: 10.1016/s0960-9822(00)00323-7. [DOI] [PubMed] [Google Scholar]
- 8.Aoki K, Moriguchi H, Yoshioka T, Okawa K, Tabara H. In vitro analyses of the production and activity of secondary small interfering RNAs in C. elegans. EMBO J. 2007;26:5007–5019. doi: 10.1038/sj.emboj.7601910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu W, Shirayama M, Conte D, Jr, Vasale J, Batista PJ, Claycomb JM, Moresco JJ, Youngman EM, Keys J, Stoltz MJ, et al. Distinct argonaute-mediated 22G-RNA pathways direct genome surveillance in the C. elegans germline. Mol. Cell. 2009;36:231–244. doi: 10.1016/j.molcel.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peisley A, Lin C, Wu B, Orme-Johnson M, Liu M, Walz T, Hur S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl Acad. Sci. USA. 2011;108:21010–21015. doi: 10.1073/pnas.1113651108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–422. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu C, Xu H, Ranjith-Kumar CT, Brooks MT, Hou TY, Hu F, Herr AB, Strong RK, Kao CC, Li P. The structural basis of 5′ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure. 18:1032–1043. doi: 10.1016/j.str.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, Juranek S, Sheng G, Micura R, Tuschl T, et al. Structural and functional insights into 5′-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat. Struct. Mol. Biol. 2010;17:781–787. doi: 10.1038/nsmb.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vela A, Fedorova O, Ding SC, Pyle AM. The thermodynamic basis for viral RNA detection by the RIG-I innate immune sensor. J. Biol. Chem. 2012;287:42564–42573. doi: 10.1074/jbc.M112.385146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matranga C, Pyle AM. Double-stranded RNA-dependent ATPase DRH-3: insight into its role in RNAsilencing in Caenorhabditis elegans. J. Biol. Chem. 2010;285:25363–25371. doi: 10.1074/jbc.M110.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacRae IJ, Zhou K, Doudna JA. Structural determinants of RNA recognition and cleavage by Dicer. Nat. Struct. Mol. Biol. 2007;14:934–940. doi: 10.1038/nsmb1293. [DOI] [PubMed] [Google Scholar]
- 17.Wincott F, DiRenzo A, Shaffer C, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, Usman N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res. 1995;23:2677–2684. doi: 10.1093/nar/23.14.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leatherbarrow RJ. 1998. Erithacus Software, Ltd., Staines, UK. [Google Scholar]
- 19.Luo D, Kohlway A, Vela A, Pyle AM. Visualizing the determinants of viral RNA recognition by innate immune sensor RIG-I. Structure. 2012;20:1983–1988. doi: 10.1016/j.str.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer CJ, Maluf NK, Lohman TM. Mechanism of ATP-dependent translocation of E.coli UvrD monomers along single-stranded DNA. J. Mol. Biol. 2004;344:1287–1309. doi: 10.1016/j.jmb.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 21.Fischer CJ, Lohman TM. ATP-dependent translocation of proteins along single-stranded DNA: models and methods of analysis of pre-steady state kinetics. J. Mol. Biol. 2004;344:1265–1286. doi: 10.1016/j.jmb.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 22.Peisley A, Jo MH, Lin C, Wu B, Orme-Johnson M, Walz T, Hohng S, Hur S. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc. Natl Acad. Sci. USA. 2012;109:E3340–E3349. doi: 10.1073/pnas.1208618109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henn A, Cao W, Hackney DD, De La Cruz EM. The ATPase cycle mechanism of the DEAD-box rRNA helicase, DbpA. J. Mol. Biol. 2008;377:193–205. doi: 10.1016/j.jmb.2007.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao W, Coman MM, Ding S, Henn A, Middleton ER, Bradley MJ, Rhoades E, Hackney DD, Pyle AM, De La Cruz EM. Mechanism of Mss116 ATPase reveals functional diversity of DEAD-Box proteins. J. Mol. Biol. 2011;409:399–414. doi: 10.1016/j.jmb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lau PW, Guiley KZ, De N, Potter CS, Carragher B, MacRae IJ. The molecular architecture of human Dicer. Nat. Struct. Mol. Biol. 2012;19:436–440. doi: 10.1038/nsmb.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zou J, Chang M, Nie P, Secombes CJ. Origin and evolution of the RIG-I like RNA helicase gene family. BMC Evol. Biol. 2009;9:85. doi: 10.1186/1471-2148-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JE, Heo I, Tian Y, Simanshu DK, Chang H, Jee D, Patel DJ, Kim VN. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo D, Kohlway A, Pyle AM. Duplex RNA activated ATPases (DRAs): platforms for RNA sensing, signaling and processing. RNA Biol. 2013;10:111–120. doi: 10.4161/rna.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song JJ, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat. Struct. Biol. 2003;10:1026–1032. doi: 10.1038/nsb1016. [DOI] [PubMed] [Google Scholar]
- 31.Wu B, Peisley A, Richards C, Yao H, Zeng X, Lin C, Chu F, Walz T, Hur S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell. 2013;152:276–289. doi: 10.1016/j.cell.2012.11.048. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell. 2004;118:57–68. doi: 10.1016/j.cell.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Ma E, MacRae IJ, Kirsch JF, Doudna JA. Autoinhibition of human dicer by its internal helicase domain. J. Mol. Biol. 2008;380:237–243. doi: 10.1016/j.jmb.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsutsumi A, Kawamata T, Izumi N, Seitz H, Tomari Y. Recognition of the pre-miRNA structure by Drosophila Dicer-1. Nat. Struct. Mol. Biol. 2011;18:1153–1158. doi: 10.1038/nsmb.2125. [DOI] [PubMed] [Google Scholar]
- 35.Colmenares SU, Buker SM, Buhler M, Dlakic M, Moazed D. Coupling of double-stranded RNA synthesis and siRNA generation in fission yeast RNAi. Mol. Cell. 2007;27:449–461. doi: 10.1016/j.molcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Cenik ES, Fukunaga R, Lu G, Dutcher R, Wang Y, Tanaka Hall TM, Zamore PD. Phosphate and R2D2 restrict the substrate specificity of Dicer-2, an ATP-driven ribonuclease. Mol. Cell. 2011;42:172–184. doi: 10.1016/j.molcel.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welker NC, Maity TS, Ye X, Aruscavage PJ, Krauchuk AA, Liu Q, Bass BL. Dicer's helicase domain discriminates dsRNA termini to promote an altered reaction mode. Mol. Cell. 2011;41:589–599. doi: 10.1016/j.molcel.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welker NC, Pavelec DM, Nix DA, Duchaine TF, Kennedy S, Bass BL. Dicer's helicase domain is required for accumulation of some, but not all, C. elegans endogenous siRNAs. RNA. 2010;16:893–903. doi: 10.1261/rna.2122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bochkarev A, Bochkareva E. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 2004;14:36–42. doi: 10.1016/j.sbi.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 40.Bochkarev A, Pfuetzner RA, Edwards AM, Frappier L. Structure of the single-stranded-DNA-binding domain of replication protein A bound to DNA. Nature. 1997;385:176–181. doi: 10.1038/385176a0. [DOI] [PubMed] [Google Scholar]
- 41.Lei M, Baumann P, Cech TR. Cooperative binding of single-stranded telomeric DNA by the Pot1 protein of Schizosaccharomyces pombe. Biochemistry. 2002;41:14560–14568. doi: 10.1021/bi026674z. [DOI] [PubMed] [Google Scholar]
- 42.Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP. BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science. 2002;297:1837–1848. doi: 10.1126/science.297.5588.1837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.