

Abstract
Immunoglobulin (Ig) diversification by somatic hypermutation in germinal center B cells is instrumental for maturation of the humoral immune response, but also bears the risk of excessive or aberrant genetic changes. Thus, introduction of DNA damage by activation-induced cytidine deaminase as well as DNA repair by multiple pathways need to be tightly regulated during the germinal center response to prevent lymphomagenesis. In the present study, we show that DNA damage checkpoint signaling via checkpoint kinase 1 (Chk1) negatively regulates somatic hypermutation. Chk1 inhibition in human B cell lymphoma lines as well as inactivation of Chk1 alleles by gene targeting in DT40 B cells leads to increased somatic hypermutation. This is apparently due to changes in DNA repair pathways regulated by Chk1, such as a decreased homologous recombination efficiency that also leads to decreased Ig gene conversion in DT40. Our data show that Chk1 signaling plays a crucial role in regulation of Ig diversification and sheds unexpected light on potential origins of aberrant somatic hypermutation in B cell lymphomagenesis.
INTRODUCTION
Maintenance of genome integrity is based on multiple DNA repair pathways that may act in a complementary or redundant manner, depending on the type and timing of the DNA lesion (1). In eukaryotes, these repair pathways are subject to control by DNA damage checkpoints, which halt the cell cycle and activate DNA repair to ensure adequate restoration of the original DNA sequence (2). Genome maintenance and checkpoint control play a crucial role in disease prevention in the adaptive immune system, in which antigen receptor variability is based on targeted genetic changes introduced into receptor genes of lymphocytes (3).
While DNA double-strand breaks lead to the activation of the ATM/checkpoint kinase 2 (Chk2) pathway, replication protein A (RPA)-coated single-stranded DNA arising, e.g. due to collapsed replication forks, induces the ATR/checkpoint kinase 1 (Chk1) pathway. Despite being structurally distinct, the effector kinases Chk1 and Chk2 have, in part, overlapping functions, converging both in cell cycle arrest by inducing the degradation or inhibition of CDC25 phosphatases. However, Chk1 and Chk2 also independently phosphorylate and activate many additional target proteins important for DNA repair, cell cycle control, cell proliferation, survival or apoptosis (4).
During secondary immunoglobulin (Ig) diversification in germinal centers via somatic hypermutation and class switch recombination, DNA lesions are inserted by activation-induced cytidine deaminase (AID) and processed by various repair pathways (5). AID is recruited to certain loci by binding to stalled RNA polymerase II (6) and converts cytidine to uracil in single-stranded DNA (7). Replication over these uracils leads to transition mutations. Alternatively, excision by uracil glycosylase (UNG) leads to either translesion synthesis over the resultant abasic site and hence transversion mutations, or its cleavage by apurinic/apyrimidinic endonuclease (APE1) and processing of the strand breaks for class switch recombination or Ig gene conversion (5). An alternative uracil-processing mechanism apparently relies on non-canonical mismatch repair, which activates PCNA ubiquitination and recruitment of polymerase η, leading to mutations at A and T residues (8,9). Overall, multiple repair pathways act in an error-prone manner in gene loci undergoing somatic hypermutation, while functioning in an error-free mode in other genes of the cell (10). It is presently unknown whether the employment of repair pathways with a divergent error rate is regulated by several distinct mechanisms, or whether upstream regulatory processes affect multiple repair pathways involved in somatic hypermutation.
Checkpoint signaling affects multiple repair pathways. Intriguingly, in germinal center B cells, the entire ATR/Chk1/p53/p21 checkpoint axis is subject to negative regulation by the key germinal center transcription factor Bcl-6 (11–14), suggesting that modulation of checkpoint responses is an important part of a successful germinal center response. It has been speculated that this dampening of checkpoint responses is instrumental to allow survival of the DNA damaging challenges imposed by Ig diversification (13). However, potential effects of decreased checkpoint signaling on AID-induced mutagenesis during germinal center-derived lymphomagenesis have not been investigated before.
In the present study, we have investigated the effect of Chk1 on Ig diversification. We show that chemical inhibition of Chk1 leads to enhanced somatic hypermutation in two Burkitt lymphoma cell lines. Also, partial inactivation of Chk1 by gene targeting in DT40 B cells leads to increased somatic hypermutation, which was not due to changes in AID levels but rather to interference of Chk1 with DNA repair pathways involved in the processing of AID-induced lesions. In particular, decreased Ig gene conversion in Chk1-depleted DT40 B cells indicates a defect in homologous recombination. Our data imply that dampening of checkpoint responses may be required for efficient somatic hypermutation in germinal center B cells.
MATERIALS AND METHODS
Antibodies and inhibitors
The following antibodies and inhibitors were used: anti-AID (EK2 5G9), anti-CDC25A (F-6, Santa Cruz), anti-Chk1 (G-4, Santa Cruz), anti-Actin (A2066, Sigma-Aldrich), anti-human IgM (P9295, Sigma-Aldrich), UCN-01 (U6508, Sigma-Aldrich) and TCS2312 (TOC-3038, Tocris).
Cell culture
Raji and RAMOS cells were cultured at 37°C and 5% CO2 in RPMI 1640 medium (GIBCO) supplemented with 10% fetal calf serum (Biochrom AG), 100 µg/µl penicillin/streptomycin, 2 mM glutamine and 1 mM sodium pyruvate (all GIBCO). The DT40Cre1 and DT40ΨV− cell lines were cultured at 41°C, and the medium was supplemented additionally with 1% chicken serum and 0.1 µM 2-mercaptoethanol (both Sigma-Aldrich). Transfections were performed with a BioRad gene pulser set at 50 µF and 800 V for DT40 cells and at 850 µF and 250 V for Raji cells. Deletion of loxP-flanked AID in DT40ΨV− cell lines was achieved by overnight culture in 1 µM 4-OHT (Sigma-Aldrich) followed by limiting dilution subcloning.
Inactivation and reconstitution of Chk1 in DT40 cells
The targeting vectors for Chk1 were provided by D. Gillespie (15). The original resistance cassettes were exchanged with loxP-flanked resistance cassettes [from ploxpuro, ploxbsr, ploxgpt and ploxbleo (16)] using BamHI restriction sites. In DT40ΨV−, the first Chk1 allele was disrupted with a blasticidin-, the second allele with a gpt- and the third allele with a bleomycin-based targeting vector. In DT40Cre1, the first Chk1 allele was disrupted with a blasticidin- and the second allele with a puromycin-based targeting vector. Drug-resistant single cell clones were selected with 0.8 µg/ml puromycin (Sigma-Aldrich), 5 µg/ml blasticidin S HCl (Mobitec GmbH), 30 µg/ml mycophenolic acid (gpt; VWR) or 200–300 µg/ml zeocin (bleomycin; Invitrogen). Subclones were screened for targeted integration by polymerase chain reaction (PCR) using a primer binding outside of the homologous arms (5′-CCA CGC TTT ATT GAA CCC ATA-3′) in combination with primers binding in the respective resistance cassette (Supplementary Table S4). For Southern blot analysis, 20 µg of DNA was cleaved with BamHI, and the membrane was incubated with a probe binding in the 3′ homologous arm amplified from DT40 with the primers 5′-CAA GAT GCT GAA CCA CGA GA-3′ and 5′-CGA AAT CGG AGA TCT TCA GG-3′. For reconstitution, the Chk1 coding region was amplified using Phusion polymerase (Finnzymes) and tagged with HA on the C-terminus with the primers 5′-GGG AAG CTT GCC GCC ACC ATG GCG GTG CCC TTC GTG GAG-3′ and 5′-GGG CCA TGG TCA TGC ATA GTC CGG GAC GTC ATA GGG ATA GGG TGG GGG CAG CCA CAC CT-3′ and cut with HindIII and NcoI for cloning into pExpress (16). Subsequently, the cDNA expression cassette was excised with SpeI and cloned into the NheI site of ploxgpt.
Analysis of somatic hypermutation in human B cell lymphoma lines
Somatic hypermutation using a GFP reporter in Raji cells was measured as described before in (17). For each condition, 2 × 107 cells were transfected with 20 µg of the hypermutation reporter. Three days after electroporation, the cells were selected with 200 µg/ml hygromycin (Invitrogen). After completion of selection (typically between day 13 and 16), the culture was split, and aliquots were treated continuously until day 30 with dimethyl sulfoxide, UCN-01 (75 nM) or TCS2312 (250 nM). Specificity of these Chk1 inhibitors has been assessed before; UCN-01 shows some side effects, while TCS2312 is a highly specific Chk1 inhibitor (18–20). Every 2–4 days, ∼106 living cells were analyzed for GFP+ cells on a FACSCalibur (Becton Dickinson) and analyzed by Cellquest and/or FlowJo software. Viability of the cells was determined by FACS forward scatter analysis and propidium iodide exclusion.
Hypermutation analyses of endogenous Ig genes in RAMOS cells were essentially performed as described in (21), with minor alterations. Cells were seeded as single cells in a medium containing the different inhibitors (TCS2312 125 nM, UCN-01 35 nM and 75 nM) or dimethyl sulfoxide. After 30 days, IgM-loss of the single cell clones was measured by FACS analysis. At day 40, DNA of representative single cell clones was isolated, and a fragment of the VH rearrangement between framework region 1 and the JH6 gene was amplified with the primers 5′-GAC CCT GTC CCT CAC CTG CRC TGT C-3′ and 5′-ACC TGA GGA GAC GGT GAC CGT GGT-3′, cloned into a pGEM®-T vector and sequenced.
Analyses in mice
C57BL/6 host mice were obtained from Jackson laboratories and bred and maintained in an SPF facility. Immunizations of mice were performed intraperitoneally at 8–12 weeks of age using 100 µg of the T-cell-dependent antigen nitrophenylacetyl chicken gamma globulin (NP36-CGG, Biosearch Technologies). Fourteen days post-immunization, B220+CD95+PNAhigh and B220+CD95-PNAlow cells from the spleen were sorted on a FACSAria. RNA was isolated (peqGOLD TriFast™, PEQLAB Biotechnology GmbH), reversely transcribed using the First Strand cDNA Synthesis Kit (Roche) and analyzed by quantitative real-time PCR using the Light Cycler and the SybrGreen technique (Roche). The following primers were used: GAPDH (5′-TCG TCC CGT AGA CAA AAT G-3′ and 5′-GAG ATG ATG ACC CTT TTG G-3′), Bcl-2 (5′-CCC TGT GGA TGA CTG AGT ACC-3′ and 5′-CAG CCA GGA GAA ATC AAA CAG-3′), Chk1 (5′-CCA ACT CAT GGC AGG GGT GGT TT-3′ and 5′-GGG CTG GTC CCA CGG CAA TT-3′) and AID (5′-CAA CAG CAC TGA AGC AGC CTT GC-3′ and 5′-TCC ACG TGG CAG CCA GAC TTG TTG-3′). The experiments were performed in compliance with the German animal welfare law and have been approved by the institutional committee on animal experimentation and the government of Upper Bavaria.
Analysis of proliferation, drug survival and immunoblots
Cell proliferation was measured by carboxyfluorescein succinimidyl ester staining according to the instructions of the manufacturer (VybrantTM CFDA SE Cell Tracer Kit, V-12883, Invitrogen). Survival in the presence of methyl methanesulfonate (MMS) was measured with a colony-forming assay. Methylcellulose medium (1.5%, Sigma-Aldrich; supplemented with 11.9 g Dulbecco’s modified Eagle medium, 2.44 g NaHCO3, 1 mM sodiumpyruvate, 2 mM l-glutamine, 100 µg/µl penicillin/streptomycin, 1% chicken serum, 10% fetal calf serum and 0.1 µM 2-mercaptoethanol) was stirred overnight with different concentrations of MMS (Sigma-Aldrich). In all, 2 × 104, 2 × 103 and 2 × 102 cells were added to 5 ml of methylcellulose medium in 6-well plates, followed by 10–14 days of culture. For the analysis of CDC25A degradation, 1.5 × 107 cells were washed, resuspended in phosphate-buffered saline and ultraviolet (UV)-irradiated with 15 J/m2 (UV Stratalinker, Lab Vista) in cell culture plates without lid. Subsequently, the cells were washed, resuspended in medium and incubated for 1 h at 41°C, 5% CO2. The cells were then washed once with phosphate-buffered saline, resuspended in lysis buffer (20 mM Hepes, 350 mM NaCl, 20% glycerin, 1 mM MgCl2, 0.5 mM ethylenediaminetetraacetic acid, 0.1 mM EGTA, 1% NP-40) supplemented with Complete Protease Inhibitor (Roche), incubated for 15 min on ice, centrifuged at 14 000 rpm and used for immunoblotting.
Analysis of Ig diversification in DT40 cells
For analysis of somatic hypermutation, DT40ΨV− cells were subcloned by limiting dilution and cultured for ∼2 weeks. In all, 30–50 subclones per cell line were stained with anti-chicken-IgM-PE (8310-09, Southern Biotech) before FACS analysis. Representative subclones were cultured for 6 weeks before sequence analysis. For analysis of Ig gene conversion, DT40Cre1 cells were subcloned by limiting dilution, cultured for 2–4 weeks and stained with anti-chicken-IgM-FITC (A30-102F, Bethyl) before FACS analysis. Representative subclones were cultured for 4 weeks and analyzed by sequencing. Genomic DNA was isolated to amplify the rearranged light chain λ locus with the primers 5′-TGG GAA ATA CTG GTG ATA GGT GGA T-3′ and 5′-CCT CCAT TTT TTG ACA GCA CTT ACC TGG ACA GCT G-3′ using Phusion polymerase (Finnzymes). Products were cloned into the pGEM®-T vector (Promega), and sequencing was performed with the primer 5′-GAG CGC AGG GAG TTA TTT GCA TAG-3′. Sequence alignments were performed using Geneious software, and hypermutation frequencies and pattern were acquired manually from non-unique mutations and confirmed with the SHMTool (22) (http://scb.aecom.yu.edu/cgi-bin/p1). The 5′ region of the bcl6 gene was amplified with the primers 5′-CGC GGC CGC TCC AGG AAT AG-3′ and 5′-CGG GGC CTT TTC CGC ATG GT-3′, cloned into the pGEM®-T vector and sequenced. Sequence analysis of Ig gene conversion was performed as previously described (23). In brief, the database of pseudogene donor sequences was taken from (24), and sequence changes were categorized as Ig gene conversion events (when two or more mutations could be assigned to a donor sequence of ≥9 bp), untemplated point mutations (when the change could not be found in a donor sequence) and ambiguous mutations (when only one isolated mutation could be assigned to a donor sequence).
RESULTS
Role of Chk1 in hypermutating human B cell lymphoma lines
To assess the effect of Chk1 on somatic hypermutation, we used a functional assay based on reversion of a premature stop codon in an episomally replicating GFP reporter system (25). In the hypermutating human B cell lymphoma line Raji, an increase in the relative frequency of GFP-positive cells in the culture after completion of selection for vector-bearing cells is indicative of ongoing AID-dependent somatic hypermutation. On addition of two different chemical inhibitors of Chk1 kinase activity to aliquots of the assay, a clear increase in GFP-positive cells was detectable (Figure 1A), which was not linked to changes in cell proliferation or cell death (Figure 1B and C). Functionality of the inhibitors was confirmed by CDC25A accumulation (Supplementary Figure S1). Thus, we conclude that Chk1 activity negatively regulates somatic hypermutation activity in this human model cell system.
Figure 1.
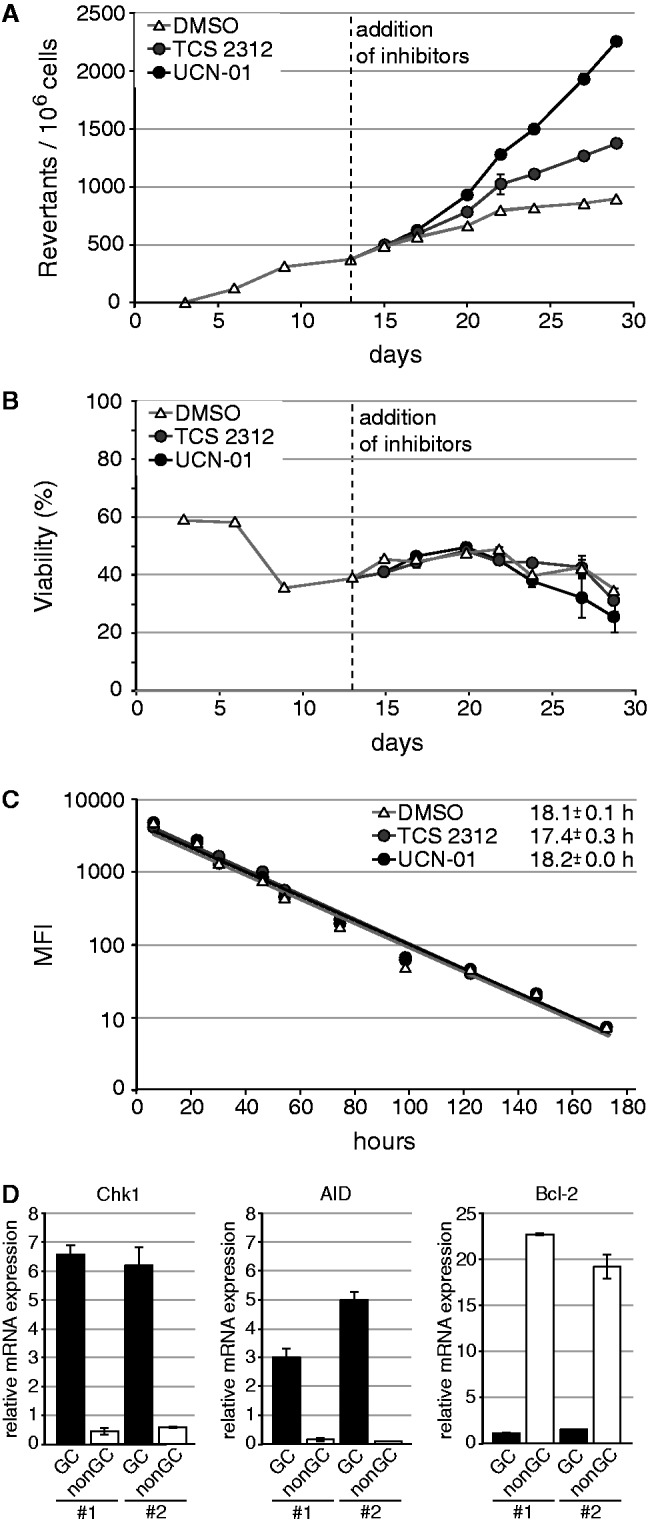
Inhibition of Chk1 leads to increased somatic hypermutation in Raji. (A) Somatic hypermutation was monitored with a reporter measuring reversion of a premature stop codon in a GFP gene. The dashed line indicates the time point of addition of dimethyl sulfoxide or the Chk1 inhibitors UCN-01 and TCS2312 to aliquots of the culture. (B) The influence of the applied UCN-01 and TCS2312 concentrations on cellular viability was assessed by propidium iodide staining. (C) The influence of the applied UCN-01 and TCS2312 concentrations on proliferation was measured by carboxyfluorescein succinimidyl ester staining. The decrease in mean fluorescence intensity of the cells and the average time per cell division are indicated. (D) In mice, Chk1 expression was analyzed in sorted germinal center (GC; B220+CD95+PNAhigh) and non-GC B cells (B220+CD95-PNAlow) by quantitative real-time RT-PCR analysis and normalized to GAPDH. AID and Bcl-2 expression confirm purity of the cell populations. Data from two representative mice (designated with #1 and #2) out of six are shown.
Chk1 transcription was reported to be negatively transcriptionally regulated by Bcl-6 in germinal center B cells, as is also true for other members of this signaling pathway: ATR, p53 and p21 (11–14). To assess whether an analysis of Chk1 function in somatic hypermutation is at all plausible for the in vivo situation, we sorted naïve and germinal center B cells from C57BL/6 mice 14 days after immunization and analyzed expression of key genes by quantitative real-time PCR. As expected, AID mRNA levels were much higher in germinal center B cells than in naïve B cells, while bcl-2 gene expression behaved vice versa. The expression levels of Chk1 mRNA paralleled those of AID (Figure 1D), indicating that Chk1 is highly expressed in normal germinal center B cells despite high Bcl-6 expression (14). This is consistent with previous findings concerning the influence of B cell activation on DNA repair factor expression (26). We were thus encouraged to analyze the effect of Chk1 on somatic hypermutation in a clear-cut genetic model, but refrained from using mice, as Chk1 deletion has previously been shown to be embryonic lethal (27).
Role of Chk1 in somatic hypermutation in DT40 cells
In the DT40 B cell lymphoma line, Chk1 inactivation by gene targeting has previously been achieved, but its effect on Ig diversification has not been tested (15). Therefore, we used the same targeting vectors used before to inactivate Chk1 by deletion of exons coding for an essential part of its kinase domain (Figure 2A). Instead of the DT40 clone used in previous studies (15), we used the DT40ΨV− cell line characterized by high hypermutation activity (28), which was generated from an AID-overexpressing variant of the DT40Cre1 cell line by deletion of the pseudogenes required for Ig gene conversion. Much to our surprise, Southern blot analyses after removal of two alleles revealed that DT40ΨV− cells apparently contain four Chk1 alleles (Figure 2B and Supplementary Figure S2), only two of which could be inactivated with reasonable efficiencies in DT40ΨV− cells (Supplementary Table S1). However, even partial deletion of Chk1 causes substantially decreased Chk1 protein expression (Figure 2C) as well as functional defects in methylcellulose survival assays on treatment with MMS (Figure 2D). Also, CDC25A degradation on UV irradiation was impaired in these cells (Figure 2E). Because partial deletion of Chk1 in DT40ΨV− showed an impairment of Chk1 function as observed in Chk1+/−mice (27,29), and complete Chk1 inactivation appeared impossible, we decided to use cells lacking two alleles for the analysis of Ig diversification.
Figure 2.

Generation of DT40ΨV− cells with reduced Chk1 activity. (A) Schematic illustration of the Chk1 gene targeting strategy of Zachos et al. (15). Black boxes represent exons, and white boxes highlight the deleted region. (B) Southern blot of DT40ΨV− Chk1 wild-type cells and two independent clones with one and two targeted Chk1 alleles, respectively, using BamHI and the 3' probe shown in (A). The probe hybridizes to a 6.0- and 4.6-kb genomic fragment derived from the wild-type and Chk1-targeted alleles. (C) Immunoblot analysis of Chk1 and AID expression in the clones analyzed in (B). Two different exposures are shown for Chk1. (D) Clonogenic survival of Chk1-targeted cells following exposure to MMS. (E) Confirmation of Chk1 depletion by assessment of CDC25A degradation on UV irradiation. Two different exposures are shown for CDC25A.
To investigate the effect of reduced Chk1 levels on somatic hypermutation, we measured the loss of surface Ig (sIg) expression due to hypermutation-derived deleterious mutations in the Ig genes (Supplementary Figure S3A). In cells lacking two alleles of Chk1, a statistically significant increase in sIg loss was detectable (Figure 3A) that was also reproducible in multiple experiments (Figure 3B). To confirm this functional effect of reduced Chk1 expression, we reconstituted Chk1 function with an overexpression vector and could now detect a decreased hypermutation activity (Figure 3A and B).
Figure 3.

Chk1 depletion in DT40ΨV− cells leads to increased somatic hypermutation. (A) Percentage of sIgM− cells in individual subclones of the indicated genotype. P-values are indicated for cell clones deriving from the same original population. rec. = reconstituted. (B) Summary of the relative mean value of sIgM− cells determined in 4–8 experiments. In each experiment, 30–50 subclones per cell line were analyzed. (C) Percentage of sIgM− cells and (D) GFP+ cells in individual subclones of the indicated cell lines. AID negative cells of the indicated genotype were transfected with an AID WT-GFP fusion construct or equivalent fusion constructs containing the H56R/E58Q (HE) or R19E/R24E (RR) mutations. Statistically significant differences are marked in (A–C) with brackets, and P-values are given (Student’s t-test).
To investigate at which stage of the hypermutation process Chk1 is interfering, we analyzed its effect on AID protein levels. These were comparable between DT40 clones with different Chk1 allele status (Figure 2C and Supplementary Figure S4B), but we also directly analyzed the link between Chk1, AID and hypermutation by transfection of AID–GFP fusion constructs into AID-negative variants of the DT40ΨV− cell set generated above. The expression levels of the transfected fusion proteins were similar in all genotypes (Figure 3D), but they caused increased somatic hypermutation in cells lacking two Chk1 alleles, which was dependent on AID function as indicated by the use of two AID mutants with inactivating mutations of the catalytic center (Figure 3C). This indicates that the effect of Chk1 on hypermutation occurs at a step beyond regulation of AID expression, e.g. its localization or function, or alternatively at the level of DNA repair.
Sequencing of subclones of a representative hypermutation assay confirmed that more mutations are introduced in clones with two deleted Chk1 alleles, but not in the respective reconstituted derivatives (Figure 4A). We could not observe an increase of the low level of mutagenesis of the bcl-6 gene in Chk1-depleted cells (Supplementary Table S2), implying that Chk1 inactivation does not simply impair overall genetic stability of the cells. In the Ig locus, we observed changes in the hypermutation pattern, suggesting that the different repair pathways involved in somatic hypermutation may be differentially affected by decreased Chk1 function. Specifically, transition mutations at C and G residues were increased to an even stronger extent than transversion mutations, leading to a shift in the relative ratio of transitions to transversions in the hypermutation pattern (Figure 4B). This phenotype cannot be easily explained by perturbed base excision repair or translesion synthesis, but rather suggests that a major alternative error-free repair pathway, such as homologous recombination, is affected by Chk1 deficiency.
Figure 4.

Chk1 depletion in DT40 cells affects DNA repair pathways. (A) Mutation frequencies in the VJ-region of the Igλ locus (VJλ) in DT40ΨV− wild-type, Chk1 +/+/−/−, Chk1 rec. and AID −/− cell lines. The total number of sequences analyzed is indicated in the center of the chart, and the number of point mutations per DNA sequence is given in the periphery of the pie chart segments. (B) Pattern of nucleotide substitutions detected in the VJλ-region. Tables in the upper part show numbers of independent mutational events and the lower part gives percentages of total mutations. Below each table, the ratio of transition:transversion substitutions at dC/dG is indicated. Significance analysis: X2 test. ***P < 0.001, **P < 0.01, *P < 0.05. (C) Percentage of sIgM+ cells determined by FACS in individual subclones of the indicated genotypes in the DT40Cre1 cell line. A logarithmic scale is used for better visualization of differences. P-values are given for comparison of cell clones deriving from the same original population. rec.: reconstituted. sIgM: surface IgM. Significance analysis: Student’s t-test. (D) Sequence analysis of the VJλ-region in representative clones analyzed in (C). GCM: Ig gene conversion events; AM: ambiguous mutations; PM: point mutations; TV: transversion mutations; TS: transition mutations; AT: transversion and transition mutations at dAdT. Significance analysis: X2 test. Mutational analysis in (A–D) was performed with two representative subclones of each genotype.
To confirm this notion, we set out to assess Ig gene conversion, where defects in lesion processing by base excision repair or homologous recombination would also be detectable (23,28,30). This form of Ig diversification is measured in DT40Cre1 cells containing the pseudogenes required for Ig gene conversion, so these can be used to repair a frameshift mutation in the λ gene locus, leading to sIg re-expression (31) (Supplementary Figure S3B).
We observed a decrease in Ig gene conversion when we deleted two Chk1 alleles in DT40Cre1 cells (Figure 4C and Supplementary Figure S4), strengthening our conclusion that increased hypermutation is not simply caused by increased AID function. Sequencing of representative subclones confirmed a decrease in Ig gene conversion events on Chk1 depletion, and a concomitant increase in untemplated nucleotide substitutions, in particular, transversion mutations (Figure 4D and Supplementary Figure S5). This indicates that a defect in homologous recombination is the basis for the changes in Ig diversification on Chk1 depletion. Direct assessment of the capacity of homologous recombination via analysis of gene targeting efficiency in cells lacking Chk1 alleles indicated a negative impact on this pathway on inactivation of two of the four Chk1 alleles (Supplementary Table S3).
Role of Chk1 in somatic hypermutation of the endogenous Ig locus in human B cells
Our study implies that Chk1 inhibition or inactivation leads to decreased homologous recombination activity and hence increased somatic hypermutation. To show directly that this also holds true for somatic hypermutation of the endogenous Ig locus gene in human B cells, we used RAMOS cells. Inhibition of Chk1 activity in RAMOS leads to a clear increase in surface Ig loss, indicative of an increased introduction of deleterious mutations during somatic hypermutation (Figure 5A). Direct sequencing of a region of the rearranged Ig-gene also revealed an increase in mutation frequency on Chk1 inhibition (Figure 5B). Thus, we conclude that mutagenesis of endogenous Ig genes in human cells is affected on decrease of Chk1 function in a similar way as shown for our GFP reporter and DT40 cells.
Figure 5.
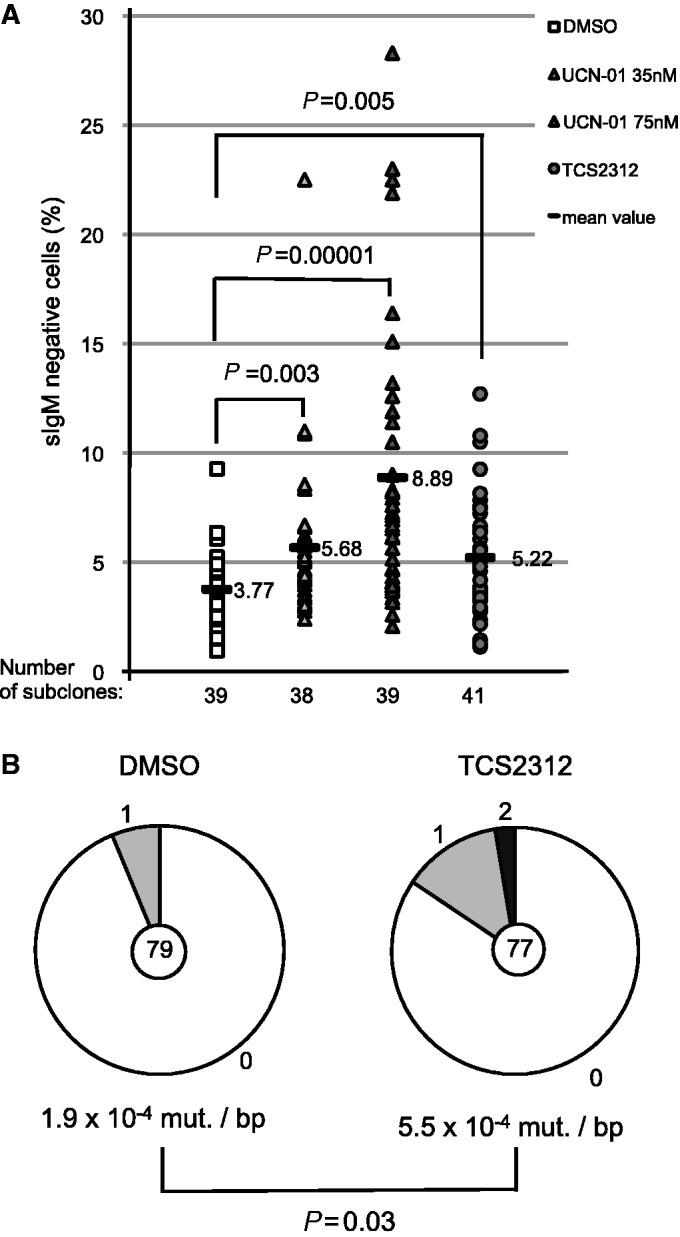
Chk1 inhibition leads to increased hypermutation of the endogenous Ig locus in RAMOS cells. (A) IgM loss on culture of RAMOS cells in the absence or presence of the Chk1 inhibitors (TCS2312 125 nM, UCN-01 35 nM and 75 nM). Statistically significant differences are marked with brackets and P-values are given (Student’s t-test). (B) Sequencing analyses of a segment of the Ig locus, spanning from framework region 1 to the JH gene, of representative subclones of the analysis shown in (A). The total number of sequences analyzed is indicated in the center of the chart and the number of point mutations per DNA sequence is given in the periphery of the pie chart segments. Significance analysis: X2 test.
DISCUSSION
In the present study, we have shown that defects in Chk1 function lead to increased somatic hypermutation. Mechanistic analyses revealed that a limited promotion of homologous recombination by Chk1 is responsible for these effects. As proficient homologous recombination is required for genome maintenance in AID-expressing cells (32), our findings have potential implications for causes of genetic instability in the germinal center B cell precursors of human B cell lymphoma entities.
Previous studies have shown that Chk1 phosphorylation can be induced by AID activity (33), and that Chk1 directly phosphorylates Rad51 and BRCA2, thus affecting the efficiency of strand exchange during homologous recombination (34,35). Inhibition of homologous recombination has previously been shown to trigger or increase somatic hypermutation, in particular in in vitro models (23). In primary B cells, a defect in homologous recombination increases aberrant processing of double-strand breaks induced by AID (30), which may explain the deletions we observe in B cells deficient in Chk1 (Figure 4). Although we cannot directly assess the efficiency of Rad51 phosphorylation in the DT40 mutant cells due to lack of suitable reagents, both our sequencing and targeting data clearly indicate a defect in the process of homologous recombination. It is notable that gene-targeting efficiencies are affected for some loci but not for others, a phenomenon observed before in certain DT40 mutants (36–38).
The observed homologous recombination phenotype may in fact also be the basis for our inability to completely inactivate Chk1 in the DT40 model cell lines used, as homologous recombination has previously been shown to protect AID-expressing cells from genomic instability and cell death (32). Along these lines, one may speculate that Chk1 locus amplification has occurred during generation of the DT40Cre1 cell line, to compensate for its increased AID-dependent Ig diversification via improved activation of homologous recombination repair by Chk1. The Chk1 locus is on microchromosome 24, for which trisomy has been reported in DT40 (39), but our Southern blot analyses indicate that more than three copies, apparently four, of the Chk1 gene are present in the DT40 model cell lines we used (Figure 2 and Supplementary Figure S2).
Within the context of a germinal center B cell, an effect of Chk1 on somatic hypermutation by interference with homologous recombination is highly interesting, as the activity of the entire ATR/Chk1 axis appears to be dampened by Bcl-6 (13,14). We have shown that Chk1 mRNA itself is highly expressed in total germinal center B cells, and previous studies have also found upregulation of Chk1 expression on B cell activation (26) implying that several regulatory pathways differentially affect the components of this axis during the germinal center reaction. Detailed analyses of the expression and activity of Chk1 and its upstream inducers in germinal center B cells are certainly required, but the available data would be consistent with low Chk1 expression/activity in centroblasts [due to Bcl-6 (13,14)] and high Chk1 expression in centrocytes [due to B cell activation stimuli (26)]. One may surmise that limitation of Chk1 activity may be required for efficient somatic hypermutation. However, if excessive suppression of this signaling pathway occurs, homologous recombination may be inhibited to a much more substantial extent, resulting in genome-wide genetic instability (32). Such a scenario resembles the aberrant somatic hypermutation observed before in diffuse large B cell lymphoma characterized by Bcl-6 deregulation, in which multiple gene loci may be subject to hypermutation-like mutagenesis as well as chromosomal translocations (40). It will thus be highly interesting to investigate in vivo whether inhibition of Chk1 activity or of homologous recombination leads to increased hypermutation of Ig genes, or to aberrant hypermutation of other loci, and hence genome instability leading to lymphomagenesis.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The Deutsche Forschungsgemeinschaft [TRR54-TPA4, JU2690/1-2 and JU-2690/4-1 to B.J.]. Funding for open access charge: In house funding.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank D. Gillespie for providing the Chk1 targeting vectors, L. Strobl for insights in gene expression data, D. Eick for persistent support and R. Küppers for stimulating discussion and for the gift of primers for human V gene analysis.
REFERENCES
- 1.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 2.Warmerdam DO, Kanaar R. Dealing with DNA damage: relationships between checkpoint and repair pathways. Mutat. Res. 2010;704:2–11. doi: 10.1016/j.mrrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Callen E, Nussenzweig MC, Nussenzweig A. Breaking down cell cycle checkpoints and DNA repair during antigen receptor gene assembly. Oncogene. 2007;26:7759–7764. doi: 10.1038/sj.onc.1210873. [DOI] [PubMed] [Google Scholar]
- 4.Stracker TH, Usui T, Petrini JH. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst.) 2009;8:1047–1054. doi: 10.1016/j.dnarep.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 6.Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143:122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl Acad. Sci. USA. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Langerak P, Nygren AO, Krijger PH, van den Berk PC, Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 10.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, Schatz DG. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451:841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 11.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 12.Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat. Immunol. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- 13.Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J, Green R, Carroll M, Melnick A. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat. Immunol. 2007;8:705–714. doi: 10.1038/ni1478. [DOI] [PubMed] [Google Scholar]
- 14.Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol. Dis. 2008;41:95–99. doi: 10.1016/j.bcmd.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zachos G, Rainey MD, Gillespie DA. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003;22:713–723. doi: 10.1093/emboj/cdg060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scheller H, Tobollik S, Kutzera A, Eder M, Unterlehberg J, Pfeil I, Jungnickel B. c-Myc overexpression promotes a germinal center-like program in Burkitt’s lymphoma. Oncogene. 2010;29:888–897. doi: 10.1038/onc.2009.377. [DOI] [PubMed] [Google Scholar]
- 18.Zhao B, Bower MJ, McDevitt PJ, Zhao H, Davis ST, Johanson KO, Green SM, Concha NO, Zhou BB. Structural basis for Chk1 inhibition by UCN-01. J. Biol. Chem. 2002;277:46609–46615. doi: 10.1074/jbc.M201233200. [DOI] [PubMed] [Google Scholar]
- 19.Teng M, Zhu J, Johnson MD, Chen P, Kornmann J, Chen E, Blasina A, Register J, Anderes K, Rogers C, et al. Structure-based design and synthesis of (5-arylamino-2H-pyrazol-3-yl)-biphenyl-2’,4’-diols as novel and potent human CHK1 inhibitors. J. Med. Chem. 2007;50:5253–5256. doi: 10.1021/jm0704604. [DOI] [PubMed] [Google Scholar]
- 20.Sato S, Fujita N, Tsuruo T. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine) Oncogene. 2002;21:1727–1738. doi: 10.1038/sj.onc.1205225. [DOI] [PubMed] [Google Scholar]
- 21.Sale JE, Neuberger MS. TdT-accessible breaks are scattered over the immunoglobulin V domain in a constitutively hypermutating B cell line. Immunity. 1998;9:859–869. doi: 10.1016/s1074-7613(00)80651-2. [DOI] [PubMed] [Google Scholar]
- 22.Maccarthy T, Roa S, Scharff MD, Bergman A. SHMTool: a webserver for comparative analysis of somatic hypermutation datasets. DNA Repair (Amst.) 2009;8:137–141. doi: 10.1016/j.dnarep.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sale JE, Calandrini DM, Takata M, Takeda S, Neuberger MS. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature. 2001;412:921–926. doi: 10.1038/35091100. [DOI] [PubMed] [Google Scholar]
- 24.Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- 25.Ruckerl F, Busse B, Bachl J. Episomal vectors to monitor and induce somatic hypermutation in human Burkitt-Lymphoma cell lines. Mol. Immunol. 2006;43:1645–1652. doi: 10.1016/j.molimm.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 26.Wu X, Tschumper RC, Gutierrez AJ, Mihalcik SA, Nowakowski GS, Jelinek DF. Selective induction of DNA repair pathways in human B cells activated by CD4+ T cells. PLoS One. 2010;5:e15549. doi: 10.1371/journal.pone.0015549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, Ikeda K, Nakayama K, Nakanishi M, Nakayama K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes Dev. 2000;14:1439–1447. [PMC free article] [PubMed] [Google Scholar]
- 28.Arakawa H, Saribasak H, Buerstedde JM. Activation-induced cytidine deaminase initiates immunoglobulin gene conversion and hypermutation by a common intermediate. PLoS Biol. 2004;2:E179. doi: 10.1371/journal.pbio.0020179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 30.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 31.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 32.Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, Wilpan RY, He Y, King BL, Mills KD. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat. Immunol. 2010;11:820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gourzi P, Leonova T, Papavasiliou FN. A role for activation-induced cytidine deaminase in the host response against a transforming retrovirus. Immunity. 2006;24:779–786. doi: 10.1016/j.immuni.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 34.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, Helleday T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell. Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 35.Bahassi EM, Ovesen JL, Riesenberg AL, Bernstein WZ, Hasty PE, Stambrook PJ. The checkpoint kinases Chk1 and Chk2 regulate the functional associations between hBRCA2 and Rad51 in response to DNA damage. Oncogene. 2008;27:3977–3985. doi: 10.1038/onc.2008.17. [DOI] [PubMed] [Google Scholar]
- 36.Takao N, Kato H, Mori R, Morrison C, Sonada E, Sun X, Shimizu H, Yoshioka K, Takeda S, Yamamoto K. Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene. 1999;18:7002–7009. doi: 10.1038/sj.onc.1203172. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Seki M, Otsuki M, Tada S, Takao N, Yamamoto K, Hayashi M, Honma M, Enomoto T. The absence of a functional relationship between ATM and BLM, the components of BASC, in DT40 cells. Biochim. Biophys. Acta. 2004;1688:137–144. doi: 10.1016/j.bbadis.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M. Fanconi anemia FANCG protein in mitigating radiation-and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol. Cell. Biol. 2003;23:5421–5430. doi: 10.1128/MCB.23.15.5421-5430.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neiman PE, Burnside J, Elsaesser K, Hwang H, Clurman BE, Kimmel R, Delrow J. Analysis of gene expression, copy number and palindrome formation with a Dt40 enriched cDNA microarray. Subcell. Biochem. 2006;40:245–256. doi: 10.1007/978-1-4020-4896-8_14. [DOI] [PubMed] [Google Scholar]
- 40.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.