

Abstract
Although XRN2 proteins are highly conserved eukaryotic 5′→3′ exonucleases, little is known about their function in animals. Here, we characterize Caenorhabditis elegans XRN2, which we find to be a broadly and constitutively expressed nuclear protein. An xrn-2 null mutation or loss of XRN2 catalytic activity causes a molting defect and early larval arrest. However, by generating a conditionally mutant xrn-2ts strain de novo through an approach that may be also applicable to other genes of interest, we reveal further functions in fertility, during embryogenesis and during additional larval stages. Consistent with the known role of XRN2 in controlling microRNA (miRNA) levels, we can demonstrate that loss of XRN2 activity stabilizes some rapidly decaying miRNAs. Surprisingly, however, other miRNAs continue to decay rapidly in xrn-2ts animals. Thus, XRN2 has unanticipated miRNA specificity in vivo, and its diverse developmental functions may relate to distinct substrates. Finally, our global analysis of miRNA stability during larval stage 1 reveals that miRNA passenger strands (miR*s) are substantially less stable than guide strands (miRs), supporting the notion that the former are mostly byproducts of biogenesis rather than a less abundant functional species.
INTRODUCTION
XRN2 proteins constitute a family of eukaryotic 5′→3′ exoribonucleases that have various RNA substrates (1). For instance, in yeast, where XRN2 has been particularly well studied and is commonly known as Rat1p, it is involved in processing of ribosomal RNAs and small nucleolar RNAs (2–6), transcriptional termination (7) and degradation of aberrant transfer RNAs (8), among other functions. The diversity of substrates in vivo is reflected by relaxed substrate specificity in vitro where Rat1p processively degrades 5′ monophosphorylated RNAs that lack strong secondary structures to mononucleotides (9,10). The catalytic site of XRN2/Rat1p contains seven acidic amino acids, which form a pocket for a divalent cation (Mg2+ or Mn2+) required for the exoribonuclease activity (11).
A paralogous enzyme, Xrn1p, exists in the yeast cytoplasm (12), where it is involved in degradation of decapped mRNAs (13). Single orthologues of XRN1 and XRN2, respectively, are also found in animals, and it is assumed that distinct localization and the resulting division of labor that characterize yeast Xrn1p and Rat1p (14) also apply to their orthologues in other organisms, although this has not yet been investigated systematically. A nuclear localization signal present in Rat1p is not conserved in XRN2 orthologues of other species (14), but nuclear RNAs such as pre-mRNAs and 5.8S and 18S ribosomal RNAs have been reported as common substrates of XRN2 in yeast and other species [reviewed in (15)]
In Caenorhabditis elegans, the single XRN2-type protein was found to function in degradation of mature microRNAs (miRNAs) (16). These short (∼22 nt) non-coding RNAs are derived from longer precursor transcripts, from which two successive processing steps release a ∼22 nt duplex RNA consisting of an miRNA guide (miR) bound to an miRNA passenger (miR*) strand (17). This duplex is loaded onto an Argonaute protein and the guide strand retained, whereas the passenger strand is released and presumably discarded. The designation of miR and miR* was initially based on their relative abundance, with the more abundant strand assumed functional and thus designated miR. However, individual miR*s have also been shown to be functional [reviewed in (18)], so that in recent times the use of two suffixes indicating the ‘arm’ of the precursor transcript from which an miRNA is derived, i.e. −3p or −5p, has become more common. At any rate, miRNA-Argonaute complexes can bind to partially complementary sequences in 3′-untranslated regions (3′-UTRs) of mRNAs to repress their translation and induce their degradation (19). They thus regulate a large number of genes, affording them, as a class, important roles in animal development and pathology (20).
Two lines of evidence support a function of XRN2 in the degradation of mature miRNAs (16). First, C. elegans lysates containing wild-type levels of XRN2 were more active in decay of naked synthetic and Argonaute-associated miRNAs than XRN2-depleted lysates. Second, depletion of XRN2 by RNA interference (RNAi) yielded increased steady-state levels of a number of endogenous miRNAs. In these latter experiments, however, the levels of some miRNAs were unchanged. Because RNAi may be inefficient in certain tissues or at certain times, it remained unknown whether this reflected true substrate specificity or a technical limitation of the experiment.
Despite prominent molecular functions, the roles of XRN2 in animal development largely remain to be explored (15). In mice and humans, over-expression of XRN2 has been implicated as a risk factor for a specific type of lung cancer (21), but a molecular basis remains to be established. In C. elegans, XRN2, encoded by the xrn-2 gene, was found in a genome-wide RNAi screen for factors involved in molting (22), the process in which worms synthesize a new and shed their old cuticle. Molting occurs once at the end of each of the four larval stages, L1 through L4, (23) and Frand et al. (22) found that xrn-2 depleted animals were unable to shed the cuticle from the pharynx at the final (L4) molt. Consistent with this phenotype, a putative xrn-2 promoter, with only limited spatial activity as assayed by a Green Fluorescent Protein (GFP) reporter, was active in myoepithelial cells that secrete the pharyngeal cuticle (22). Promoter activity also occurred in other cells implicated in molting, including a particular pharyngeal neuron and intestinal cells. How XRN2 affects molting is unknown, although this function may involve regulation of expression of MoLTing Defective 10 (MLT-10), another molting factor, in a direct or indirect manner, through an unknown mechanism. RNAi against xrn-2 also causes slow growth and sterility (16), but again the basis of these phenotypes remains unknown.
To obtain a better understanding of the developmental functions of XRN2 and its role in miRNA turnover, we have characterized xrn-2 null mutant C. elegans. We find that these animals arrest at the L2 stage, following a failed molt from the L1 to the L2 stage. The unanticipated ability to complete embryogenesis was not due to the absence of an essential embryonic function of XRN2, but reflected masking of the null phenotype due to maternal contribution. We demonstrate this through an xrn-2ts allele, which we generated by transplanting conditional mutations from yeast to C. elegans. We can thus show that XRN2 is essential during several stages of C. elegans development, including embryogenesis. These broader functions are consistent with a revised picture of xrn-2 expression that we obtained using a rescuing transgene and detection of the endogenous protein by western blotting. Using small RNA deep sequencing to determine miRNA decay rates, we find that miR*s are generally less stable than miRs. Strikingly, among the small group of unstable miRs, only some become stabilized by inactivation of XRN2. We conclude that XRN2 has unanticipated miRNA substrate specificity in vivo and diverse developmental functions.
MATERIALS AND METHODS
Strains
Caenorhabditis elegans strains were cultured by standard methods described previously (24). The Bristol N2 strain was used as wild-type. Animals heterozygous for xrn-2(tm3473) were obtained from Dr Shohei Mitani, backcrossed three times and balanced. Strains used are shown in Supplementary Table S1.
Cloning and site-directed mutagenesis
Cloning and site-directed mutagenesis were performed by PfuUltra II Fusion HS DNA Polymerase (Agilent Technologies, Santa Clara, CA, USA) according to the supplier’s protocol using specific primers (Supplementary Table S2). The codon-optimized xrn-2 with three artificial introns (Supplementary Table S3) was designed according to a previous report (25) and synthesized using a commercial service (GenScript, Piscataway, NJ, USA).
Single-copy transgene insertion
DNA fragments were inserted into pCFJ210 (for chromosome I) or pCFJ201 (for chromosome IV) vectors by Multisite Gateway Technology (Life Technologies, Carlsbad, CA, USA) according to the supplier’s protocol. Mos1-mediated single-copy transgene insertion was performed according to previous reports (26,27). Following confirmation of correct insertion by polymerase chain reaction (PCR), transgenic strains were backcrossed at least three times to the N2 strain.
Multicopy transgene arrays
The multisite gateway cloning system (Invitrogen) was used to insert transgenes into the pCG150 destination vector (containing unc-119 rescuing fragment), which was transformed into young adult unc-119(ed3) worms by microparticle bombardment using the Biolistic PDS-1000/He particle delivery system (BioRad) (28). For each bombardment, 16 µl of 0.5 µg/µl pCG150 and 4 µl of 0.8 µg/µl pCFJ90 (co-injection marker containing Pmyo-2::mCherry) were coupled to 1-µm microcarrier gold beads (BioRad, Cat#165-2263). Worms were allowed to recover for 1 h at 15°C after bombardment and were then grown at 25°C on NG 2% plates seeded with OP50 bacteria for ca. 2 weeks before screening for wild-type moving worms and mCherry-fluorescence from the co-injection marker. Transgenes containing wild-type or D234A-D236A double mutant xrn-2 sequences were stably transmitted and expressed in the germline, suggesting integration into the genome.
Antibodies and western blotting
Recombinant full-length C. elegans XRN2 was prepared as described (16) and used to immunize rats (Charles River Laboratories, Kisslegg, Germany), to obtain an anti-XRN2 antibody. A mouse monoclonal anti-actin antibody (clone C4) was purchased from Millipore (Billerica, MA, USA). The anti-XRN2 antibody and anti-actin antibody were used with 1000- and 3000-fold dilutions, respectively, followed by horseradish peroxidase-conjugated secondary antibody (GE Healthcare, Little Chalfont, UK) reaction. The membranes were treated with ECL Western Blotting Detection Reagents, and protein bands were detected using Amersham Hyperfilm ECL (Figure 3C) or by an ImageQuant LAS 4000 hemiluminescence imager (all GE Healthcare) (Figure 4C). Band intensities were quantified using the ImageJ software (NIH, Bethesda, MD, USA).
Figure 3.

XRN2 is ubiquitously and constitutively expressed. (A) Schematic depiction of the xrn-2 genomic locus and promoters used. The arrows indicate the direction of transcription. (B) Micrographs showing GFP signal of single-copy-integrated, codon-optimized and gfp-tagged xrn-2 expressed under the control of the 1413-bp long promoter region. The GFP signal is ubiquitously detected. Examples of hypodermal and intestinal cells are marked with arrowheads. Insets: DIC images of the same worms. (C) Western blot showing a time-course for endogenous XRN2. ‘e’, ‘m’ and ‘l’ stands for early, mid and late, respectively; ‘YA’ and ‘GA’ for young and gravid adult, respectively. An asterisk indicates an apparent proteolytic fragment of XRN2, which did not occur consistently in other western blots. (D) Single-copy-integrated, codon-optimized and gfp-tagged xrn-2 expressed under the control of the 1413-bp long xrn-2 promoter region rescues the phenotypes of xrn-2(tm3473), but the 132-bp long xrn-2 promoter region does not. Scale bar, 20 µm (B) and 50 µm (D).
Figure 4.

Characterization of an improved xrn-2ts strain reveals reduced XRN2 levels at restrictive temperature. (A) Schematic representation of xrn-2ts phenotypes at different temperature. xrn-2ts embryos or worms were cultured under the indicated conditions. Phenotypes observed are described on the right. For less-penetrant phenotypes, a number indicating worms affected/worms scored is shown in brackets. (B) The wild-type (‘wt’; N2) and xrn-2ts worms were cultured from L1-stage at 20 or 26°C as indicated for 72 h (wt) and 93 h (xrn-2ts), respectively. The worms were observed by stereo microscopy at the same magnification. (C, D) The wt, xrn-2(+) and xrn-2ts worms were cultured from mid L3- to late L4-stage at 15°C or 26°C and harvested. (C) XRN2, XRN2/GFP and actin protein levels were examined by western blotting. XRN2 and XRN2/GFP levels were normalized to actin levels and shown with values of wt at 15°C defined as 100. (D) The mRNA levels of the xrn-2::gfp transgenes in xrn-2(+) and xrn-2ts worms were quantified by RT-qPCR, normalized to actin mRNA levels and shown with values of xrn-2(+) at 15°C as 1 (n = 2, means + SEM). ‘xrn-2(+)’ denotes xrn-2(tm3473) homozygous animals expressing a wild-type xrn-2 transgene.
Microscopy
Differential Interference Contrast (DIC) and fluorescent images were obtained using an Axio Observer Z1 microscope and AxioVision SE64 (release 4.8) software (Carl Zeiss, Oberkochen, Germany). Stereoscopic images were obtained by M205 A stereo microscope (Leica, Solms, Germany).
RNA preparation, sequencing and RT-qPCR
Gravid N2 or xrn-2ts worms were treated with bleaching solution [30% sodium hypochlorite (5% chlorine) reagent (Thermo Fisher Scientific, Waltham, MA, USA), 750 mM potassium hydroxide] to extract eggs, which were then incubated in M9 medium overnight to hatch. The resulting synchronized L1 larvae were cultured with Escherichia coli OP50 in S-medium supplemented with trace metal solution (29) at a concentration of 1 × 104 worms/ml with shaking (180 rpm) at 25°C for 2 h. Subsequently, α-amanitin (Sigma-Aldrich, St. Louis, MO, USA) was added to a final concentration of 50 µg/ml, which blocks transcription and stalls larval development (Supplementary Figure S2). A total of 1.5 × 104 worms were harvested at each sampling time point during the next 8 h, washed three times with M9 medium, resuspended in 700 µl of TRIzol reagent (Life Technologies) and frozen in liquid nitrogen. Worms were broken open by five repeats of freeze and thaw using liquid nitrogen and a 42°C heating block, before RNA was extracted and purified according to the supplier’s protocol with the modification that RNA was incubated with 50% 2-propanol at −80°C overnight for efficient precipitation of small RNA.
Small RNA (15–30 nt) libraries were prepared from extracted total RNA using TruSeq Small RNA Sample Prep Kit (Illumina, San Diego, CA, USA) according to the supplier’s protocol. All samples were multiplexed and 13 pM of the multiplexed libraries sequenced on two lanes of an Illumina HiSeq 2000 instrument using RTA 1.13.48. Individual reads were assigned to their sample based on the TruSeq barcode using the Illumina software Casava v1.8.0.
Quantification of individual miRNAs by reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was done using TaqMan MicroRNA Assays (Life Technologies) and StepOnePlus Real-time PCR Systems (Applied Biosystems, Foster City, CA, USA) according to the suppliers’ protocols. Forty nanogram of total RNA was used as a template for reverse transcription reaction (15 µl), and 1.3 µl of the reaction was used for qPCR reaction (25 µl). The miRNA levels were normalized to the small nucleolar RNA sn2841 levels.
For mRNA quantification, complementary DNA (cDNA) was generated from total RNA by ImProm-II Reverse Transcription System (Promega, Fitchburg, WI, USA) using oligo(dT)15 primers (for Figure 4D) or random primers (for Supplementary Figure S2B, C) according to the supplier’s protocol. RT-qPCR was performed with specific primers (Supplementary Table S2), a SYBR Green PCR Master Mix (Applied Biosystems) and a StepOnePlus Real-time PCR System. Primer sequences for pre-eft-3 mRNA and 18S ribosomal RNA were taken from (30) and (31), respectively.
Analysis of the miRNA sequencing data
For each read, the 3′ adaptor TGGAATTCTCGGGTGCCAAGG was removed by aligning it to the read allowing one or two mismatches in prefix alignments of at least 7 or 10 bases, respectively. Reads with low complexity were filtered out based on their dinucleotide entropy (removing <1% of the reads). Only reads with a minimum length of 14 nt were retained. Alignments to the miRNA database miRBase release 18 (http://www.mirbase.org/) were performed by the software bowtie (version 0.9.9.1) (32) with parameters -v 2 -a -m 100, tracking up to 100 best alignment positions per query and allowing at most two mismatches. Reads that mapped to a miRNA but at the same time also mapped with fewer mismatches to the genome (ce6) were filtered out. The expression of each miRNA was determined by counting the number of associated reads. To compensate for differences in the read depths of the individual libraries, each sample was divided by its total number of counts and multiplied by the average sample size. The resulting values were log2 transformed using a pseudo-count of 1 (y = log2(x + 1)). To obtain relative decay rates for the time window t = 1 h to t = 8 h, the change in expression of each miRNA over time was determined by the slope of a linear fit performed in R (www.r-project.org). Slopes for the two replicates were calculated separately and then averaged for further use.
Release 18 of miRBase does no longer provide identifiers that label a miRNA as a mature or a star form. We thus identified the star forms by firstly pairing the 5p and 3p forms using the miRNA name (without the −5p and −3p extensions) and then assigning the star label to the form with the lower expression level in the untreated sample.
Determination of miRNA half-life
We assumed miRNAs to decay exponentially according to the following equation:
where t is the time, N(t) is the concentration of the miRNA at time point t, N0 is the starting concentration and τ is the half-life of the miRNA.
From this follows a linear relationship between the logarithmic concentration (measured as delta-Ct values) and the half-time τ:
τ can be obtained from the slope of a linear regression by the following equation:
The intercept term captures differences in the starting concentration; for visualization, the term was subtracted from delta-Ct values.
The miRNA half-lives were calculated for individual replicate experiments. The half-life of stable miRNAs that decreased <20% (the detection limit) over the course of the 8-h experiment was set to 30 h, which is the τ resulting from a 20% decrease in 8 h and corresponds to a lower limit estimate for the half-life of such miRNAs.
The significance of differences in half-lives between worm strains was calculated using a two sample t-test assuming equal variances.
RESULTS
tm3473 is a bona fide null allele of xrn-2
Previous studies on xrn-2 mutant phenotypes relied on its depletion by RNAi (16,22). However, knock-down of genes by RNAi is usually incomplete and may vary across tissues. Therefore, we set out to characterize the xrn-2 mutant xrn-2(tm3473), provided by Dr Shohei Mitani. The tm3473-allele is a deletion of 278 bases in exon 3 leading to a frame shift at amino acid position 278 and a premature stop codon at position 308 (Figure 1A). Western blotting using an antibody against XRN2 confirmed absence of full-length XRN2 protein in the xrn-2(tm3473) background (Figure 1B). This strain, and a wild-type strain included for comparison, contains a transgene to express full-length GFP-tagged XRN2 to achieve wild-type development (see later in the text). We also failed to detect a band corresponding to the predicted size of a potential truncated translation product (data not shown). Although we cannot formally exclude that the polyclonal antiserum that we used would fail to cross-react with such a truncated product despite the fact that it was raised against recombinant full-length protein, the data suggest that the mutant mRNA may be degraded through nonsense-mediated decay. We conclude that xrn-2(tm3473) is a bona fide null allele.
Figure 1.

xrn-2(tm3473) is a bona fide null allele that causes molting defects and developmental arrest. (A) Schematic representation of wild-type and mutant XRN2. Conserved regions are shown in light grey. Dark grey indicates sequence unique to the xrn-2(tm3473) mutant due to a frame shift. Point mutations investigated in this study are indicated. (B) Western blotting confirms absence of endogenous XRN2 in the xrn-2(tm3473) background (lane 3). xrn-2(+) denotes the N2 wild-type strain. Note the presence of an XRN2/GFP-encoding transgene in the strains shown in lane 2 and 3, used to restore development of the xrn-2(tm3473) mutant strain. (C) DIC micrographs of worms grown at 25°C; gonads are outlined to facilitate staging. (i, ii) After 18 h, both xrn-2/+ (tm3473 heterozygous) and xrn-2/xrn-2 (tm3473 homozygous) worms are at the L2 stage. (iii, iv) After 29 h, xrn-2/xrn-2 worms remain arrested at the L2 stage (iv), whereas the heterozygous siblings have reached the L4 stage (iii). (v, vi) Larval arrest is accompanied by molting defects. xrn-2/xrn-2 worms are unable to shed the pharyngeal cuticle (v, arrow head), which leads to superposition of the old and newly synthesized cuticle (vi, arrow heads). Scale bar, 20 µm.
xrn-2(0) mutant animals fail to molt and arrest during L2
Worms exposed to xrn-2(RNAi) from L1 stage arrest as L4 larvae that are unable to ecdyse, i.e. shed the cuticle (22). By contrast, xrn-2(tm3473) animals already displayed penetrant defects in the L1-to-L2 molt (Figure 1C), the first molt during development. Ecdysis starts with loosening of the cuticle at the pharynx followed by rotations around the longitudinal axis that loosen the body cuticle (33). XRN2 appears to be involved in the early shedding of the cuticle taking place at the pharynx as the mouth of worms homozygous for tm3473 remained attached to the old cuticle through a string-like structure [Figure 1C(v)]. The rest of the cuticle around the head and the body was at least partially detached [Figure 1C(v and vi)], and a new cuticle was already visible beneath the old one, indicating that XRN2 is predominantly involved in ecdysis rather than cuticle synthesis. Finally, following failure to shed the L1 cuticle, and possibly as a direct consequence (33), the mutant worms arrested during the L2 stage [Figure 1C(iv and iii)].
XRN-2 catalytic activity is required for molting
Although XRN2 is an RNase, it was not evident that the RNase activity was actually required for the developmental functions of this protein. XRN1 and XRN2 proteins share a conserved three amino acid motif, DXD, that is essential for exonuclease activity in vivo (11,34). The aspartic acids (D) in this motif are important for coordination of Mg2+ ions that are required for RNA hydrolysis. We thus constructed cDNA-based transgenes that encoded either the wild-type XRN2 or the catalytic dead D234A-D236A double mutant protein, where A stands for alanine. Both transgenes were driven from a promoter region covering 1.4 kb of upstream sequence and carried the xrn-2 3′-UTR as well as a C-terminal triple GFP/His6/Flag-tag (Figure 2). As expected, the wild-type transgene efficiently rescued both the molting defect and larval arrest when introduced as a stable multicopy array (Figure 2C). By contrast, the mutant transgene was incapable of rescuing molting defect and larval arrest (Figure 2D), although mutant and wild-type protein accumulated at equivalent levels in vivo (Figure 2E). We conclude that the RNase activity of XRN2 is essential for its function in early larval development.
Figure 2.

XRN2 catalytic activity is required for molting and growth beyond the L2 stage. (A) Wild-type worms develop into gravid adults, whereas (B) xrn-2(tm3473) homozygous worms arrest development. (C) Transgenic extrachromosomal xrn-2 expressed under the control of the xrn-2 1413-bp promoter and xrn-2 3′-UTR rescues xrn-2(tm3473) mutant animals, whereas (D) a catalytically inactive version of xrn-2 with two point mutations (D234A and D236A) does not. Both transgenes contain a C-terminal GFP tag, permitting their detection with an anti-GFP antibody. (E) Western blotting reveals equivalent accumulation of wild-type (lane 1) and mutant (lane 2) protein in vivo. Scale bar, 50 µm. xrn-2(+) denotes the N2 wild-type strain.
xrn-2 is expressed broadly and constitutively
Frand et al. (22) previously analysed the ability of a 132 bp sequence upstream from the xrn-2 start codon to drive expression of gfp when present in a multicopy extrachromosomal array, and concluded that xrn-2 expression was limited, occurring mostly in the pharyngeal myoepithelium, the intestine and certain neurons. This seemed surprising given that, based on our understanding of yeast and human Rat1p/XRN2 proteins, C. elegans XRN2 would be expected to be broadly involved in RNA processing and decay processes. Moreover, the Wormbase database annotates xrn-2 as the second gene in a two-gene operon where rpl-43 is the upstream gene, 132 bp away (Figure 3A). In generating the rescuing transgene described earlier in the text, we had therefore used an extended sequence of 1413 bp upstream of the xrn-2 start codon, reaching the 5’-end of the Y48B6A.1 ORF (Figure 3A). This construct revealed widespread, possibly ubiquitous expression, with XRN2/GFP signal being detectable from early embryo through adulthood (Figure 3B). This expression was further validated through a time course that followed endogenous XRN2 protein by western blotting, and equally revealed continuous xrn-2 expression throughout the C. elegans life cycle (Figure 3C).
Complementation of mutant phenotypes can provide a functional test for the authenticity of a putative promoter, and we found that xrn-2 transgenes driven by the xrn-2 ‘long’ promoter could rescue the xrn-2(tm3473) strain. This was true both when xrn-2 cDNA was used (Figure 2C), which resulted in protein levels that were reduced relative to the endogenous protein (Figure 1B), and when a codon-optimized variant with synthetic introns was used (Figure 3D), which generated protein levels more similar to endogenous levels (see later in the text). By contrast, the xrn-2 ‘short’ promoter failed to rescue the xrn-2(tm3473) mutation, although the optimized transgene was used (Figure 3D). Taken together, our results demonstrate that xrn-2 is expressed broadly, perhaps ubiquitously, across tissues and developmental stages, and that expression beyond previously reported tissues is important for its role in molting.
A xrn-2 temperature-sensitive allele generated de novo reveals additional XRN2 functions
Our finding of a molting defect as the predominant phenotype of xrn-2 null mutant animals was consistent with a previously reported molting defect in xrn-2(RNAi) animals (22). However, given the broad expression of XRN2, which extends to the embryo, we wondered whether earlier phenotypes were obscured due to maternal contribution of mRNA or protein from xrn-2/+ heterozygous mothers to their xrn-2/xrn-2 homozygous daughters. Rapidly inactivatable, conditional alleles would permit addressing this issue, but such alleles can currently not be generated in a targeted manner, for a specific gene of interest, in C. elegans. However, temperature-sensitive (ts) alleles have been described in Saccharomyces cerevisiae for Rat1p (35) and the Rat1p/XRN2 paralogue Xrn1p (36). Individual mutation of either aspartate of the DXD motif mentioned earlier in the text to alanine (A) may further impair but not abrogate Mg2+ binding and render the protein function ts (34). We thus went to test whether the corresponding mutations in C. elegans xrn-2-elicited temperature sensitivity within the worm’s physiological temperature window, ∼10°C below that of yeast. We introduced single-copy integrated xrn-2 transgenes with appropriate mutations into strains that were homozygous for xrn-2(tm3473), i.e. lacked endogenous XRN2. Among three distinct mutations that we tested (Figure 1A), P107L, corresponding to S. cerevisiae xrn1-10(P90L) (36), conferred temperature sensitivity, supporting viability at 15°C but not at 25°C. By contrast, a Y594C-mutant transgene supported viability at either temperature, whereas the D234A mutant transgene rescued at neither temperature. In the following, we will refer to the mutant strain that expresses xrn-2P107L as xrn-2tscDNA to distinguish it from an optimized version described later in the text. An analysis of different temperature regimens revealed numerous phenotypes of xrn-2tscDNA animals beyond the molting defect observed with the xrn-2 null strain, including arrest in embryonic development and sterility (Supplementary Figure S1). These mutant strains thus revealed multiple functions of XRN2 beyond molting, which had been obscured in the null mutant animals.
Although the xrn-2tscDNA transgene permitted rapid and tight inactivation of xrn-2 (Supplementary Figure S1), it failed to provide full XRN2 activity at the permissive temperature as illustrated by slow growth and small brood sizes small (∼25 relative to ∼250 for wild-type animals) relative to wild-type animals. This reduced the strain's utility for molecular or biochemical studies or genetic screens. Because low-protein levels relative to the endogenous protein (Figure 1B) might account for the reduced functionality, we introduced artificial introns into the xrn-2 cDNA and optimized its codon composition (25). For the wild-type protein, these nucleotide changes increased XRN2/GFP levels as determined by epifluorescence microscopy (data not shown). Moreover, xrn-2(tm3473) animals expressing the sequence-optimized xrn-2P107L::gfp single-copy transgene, which we will henceforth call xrn-2ts, grew better (although still more slowly than wild-type animals) and had an increased brood size. At the same time, we could still rapidly and efficiently inactivate the optimized transgene by raising the temperature (Figure 4A, B), although a fully penetrant embryonic or L1 arrest now necessitated incubation at 26°C, rather than 25°C. Viability and development of N2 wild-type animals remained unimpaired at this temperature (Figure 4B) (37).
The P107L mutation induces temperature sensitivity by reducing XRN2 stability
To test whether destabilization of the protein by elevated temperature contributed to the ts behavior of xrn-2P107L, we examined steady-state levels of XRN2 at 15 and 26°C. We observed that XRN2 levels were substantially lower in the xrn-2ts mutant strain than either N2 or a strain carrying the wild-type transgene (Figure 4C). We note that wild-type XRN2/GFP levels were also reduced relative to endogenous XRN2 concentration in N2, particularly at 26°C, but the decrease was less than that seen with XRN2P107L/GFP. Hence, it seems likely that the P107L mutation renders XRN2 ts by destabilizing it, consistent also with its location directly adjacent to an unusually long α-helix, previously termed ‘tower domain’ (11). To test this possibility further, we quantified xrn-2 mRNA levels in the two xrn-2 transgenic strains. Unlike XRN2 protein levels, the xrn-2 mRNA levels were not reduced in the mutant strain. In fact, xrn-2ts mRNA accumulated at increased concentrations relative to the wild-type mRNA, particularly at 26°C (Figure 4D). Hence, these results not only confirm that the P107L mutation causes temperature sensitivity by reducing XRN2 protein stability but also indicate the existence of an auto-regulatory mechanism that promotes transcription or stabilization of xrn-2 mRNA when XRN2 activity is low.
The miR* strands decay more rapidly than guide strands
Our previous studies (16,38) had implicated C. elegans XRN2 in miRNA turnover by revealing increased steady-state levels of certain endogenous miRNAs in xrn-2(RNAi) worms and XRN2-dependent degradation of naked or Argonaute-loaded miRNAs in worm lysates. However, a formal demonstration that XRN2 depletion slowed miRNA degradation in vivo was missing. Moreover, certain endogenous miRNAs appeared unchanged on XRN2 depletion, but whether due to substrate specificity, or technical limitations, e.g. in the kinetics or tissue distribution of RNAi-mediated XRN2 depletion, remained unknown. To address these two issues, we examined miRNA decay globally in vivo in wild-type N2 animals. We performed a time-course experiment in which we inhibited transcription in L1 stage larvae by addition of α-amanitin (Supplementary Figure S2) and surveyed miRNAs at several subsequent time points over the next 8 h by deep sequencing (Figure 5A). We chose the L1 stage because these larvae had previously been reported to be sensitive to treatment with α-amanitin (39), and we confirmed that this treatment efficiently blocked transcription by assaying eft-3 pre-mRNA levels (Supplementary Figure S2B, C). For each time point, we calculated the levels of each miRNA as reads normalized to average library size (‘Materials and Methods’ section), which means that these numbers can go up or down or stay unchanged for a given miRNA depending on whether it decays less rapidly, more rapidly or just as rapidly as the average miRNA in this pool. Accordingly, the fold changes per hour in log2 can be positive, negative or 0, with negative values indicating less stable miRNAs. However, these values cannot be translated into absolute decay rates.
Figure 5.
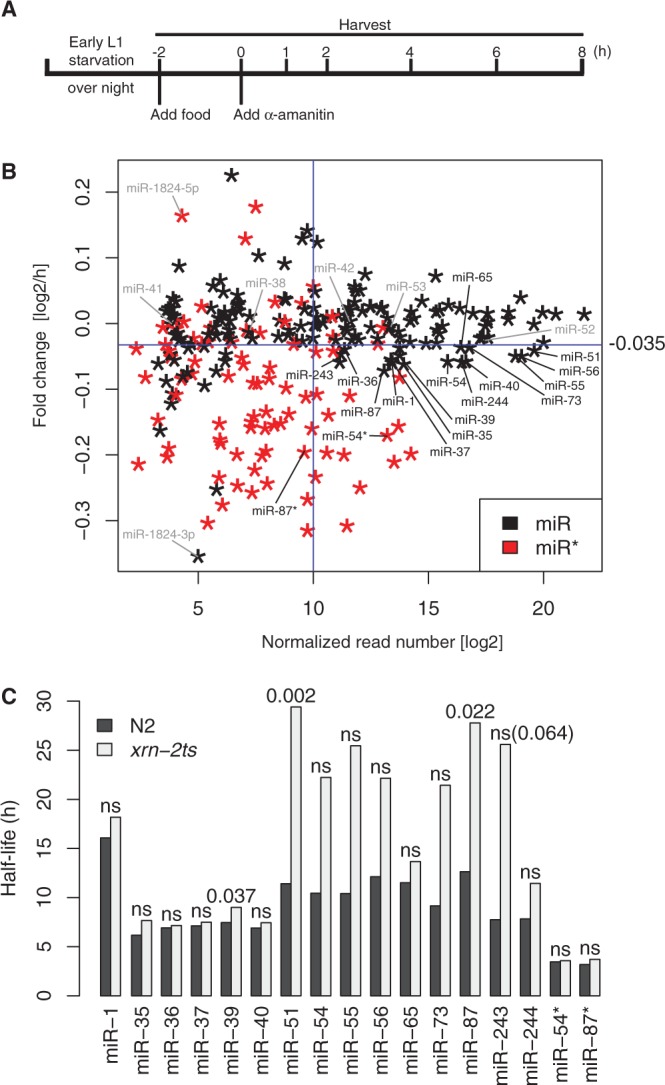
Specific miRNAs are stabilized on XRN2 depletion. (A) Experimental design for miRNA decay analysis. RNA was extracted for (B) deep sequencing and (C) RT-qPCR analyses. (B) Relative decay rates of miRNAs are plotted against normalized reads for miRNAs with sufficient expression (‘Materials and Methods’ section). Black stars, miRs; red stars, miR*s. Fifteen miRs that showed high-read numbers and fast relative decay as indicated by the blue cut-off lines. (Because the plot shows fold changes per hour, not decay constants, unstable miRNAs are those below the cut-off line.) These and two miR*s, indicated in black, were further examined by RT-qPCR. Other miRs discussed in the main text are shown in grey. (C) The miRNA levels at each time point in wt and xrn-2ts worms were quantified by RT-qPCR, and their half-lives were calculated as described in ‘Materials and Methods’ section. Relevant P-values are shown. ns, not significant.
The fold changes per hour thus calculated for two independent biological replicates correlated well (Supplementary Figure S3) and their averages were used for subsequent analysis. A scatter plot displaying fold changes versus read numbers revealed that decay rates were broadly distributed with a subset of miRNAs displaying a strikingly faster decay than average (Figure 5B). The effect was particularly pronounced for miRNAs of lower abundance. Strikingly, when we coloured miR*s, operationally defined as the one of two miRNA strands derived from a pre-miRNA that is less abundant, in red, and miRs in black, a clear separation of colours became apparent (Figure 5B). Hence, highly unstable RNAs were almost exclusively miR*s (Figure 5B). This result makes immediate and intuitive sense when considering miR*s as biogenesis byproducts. It is also consistent with generally long miRNA half-lives observed in a microarray-based study that was confined to a survey of annotated miRs (40).
Specific miRNAs are stabilized on XRN2 inactivation
Although the fast decay rates were preferentially seen for miR*s, some miRs exhibited unusually low stability, most notably several members, though not all, of each of the miR-35 (miR-35 through miR-42) and the miR-51 (miR-51 through miR-56) families. To test whether this was due to decay by XRN2, we repeated the α-amanitin time-course experiment for wild-type and xrn-2ts worms. Under the conditions that we use, α-amanitin completely blocks development at the early L1 stage (Supplementary Figure S2; Supplementary Materials and Methods), so that wild-type and xrn-2ts animals are equally arrested in development.
For this analysis, we focused on miRNAs with low stability (apparent log2 fold change of less than −0.035/h and thus below the blue cut-off line in Figure 5B) and moderately high, to high, expression levels (>210 normalized reads, to the right of the cut-off line). We determined the levels of individual miRNAs by RT-qPCR and normalized them to sn2841, a small nucleolar RNA whose level is stable during the time course (data not shown). When testing the five rapidly decaying members of the miR-35 family, all of them displayed comparable half-lives in wild-type and xrn-2ts animals (Figure 5C and Supplementary Figure S4). Similarly, xrn-2 inactivation had little effect on the decay of miR-1, miR-65 and miR-244. By contrast, the decay of miR-51 and miR-87 was substantially and significantly delayed in xrn-2ts animals. The miR-54, miR-55, miR-56, miR-73 and miR-243 showed a similar trend, although differences failed to reach statistical significance (Supplementary Figure S4). We also examined decay of the highly expressed and unstable miR-54* and found it to be unaffected by XRN2 inactivation. Similarly, miR-87*, unlike miR-87, continued to decay rapidly when XRN2 was inactive. As the passenger and guide strand derive from the same precursor, this directly confirms that the decreased apparent half-lives of the guide strands truly reflects stabilization of this guide strand and not a secondary effect of altered processing of residual pre-miRNAs. Taken together, our data reveal that XRN2 is essential for rapid decay of a subset of miRNAs during the first larval stage.
DISCUSSION
xrn-2 is broadly expressed and functions in processes beyond molting
Although molecular functions of XRN2 proteins have been studied extensively, particularly in yeast and cultured human cells, their developmental functions have remained virtually unexplored (15). An RNAi-based screen had implicated XRN2 in molting in C. elegans, consistent also with its expression in tissues important for cuticle generation or shedding (22), and in agreement with this idea, we find that an xrn-2 null mutation causes a penetrant L1 molting defect and subsequent L2 stage arrest. However, by generating a conditional allele, we could demonstrate that this only represents the tip of the iceberg; XRN2 in C. elegans is required for numerous events during embryonic and post-embryonic development as demonstrated for instance by embryonic lethality and sterility under appropriate regimens.
In yeast, where Rat1p/XRN2 is essential for viability, mutations cause a diverse array of defects in various RNA metabolic processes, such as transcriptional termination, ribosomal RNA processing, intron degradation and aberrant transfer RNA degradation (15). However, it remains to be determined which of these processes constitutes the essential function of Rat1p or whether it is any one process. Similarly, it remains to be established for C. elegans whether the requirements for functional XRN2 in different tissues and developmental stages reflect a core underlying theme, or whether the respective targets and processes that become dysregulated on XRN2 depletion vary. We also note that although we have focused here on miRNAs as the only currently known substrate of C. elegans XRN2, it is highly likely that numerous additional substrates exist, and any of these, individually or in combination, may be relevant for the xrn-2 mutant phenotypes. Nonetheless, our demonstration that mutations inactivating the XRN2 catalytic site also abrogate its ability to complement an xrn-2 null mutation argue that it is processing or degradation of one or several RNA substrates that are important for the function of XRN2 in molting. Modulator, i.e. enhancer and suppressor, screens may offer a way forward to identify specific targets and pathways affected by xrn-2 deficiency and have been initiated in our laboratory.
XRN2 substrate preferences
In vitro, XRN2 proteins can degrade various RNA sequences, provided they are 5′-monophosphorylated and devoid of stable structures (9,10). However, we find here that in the L1 stage, only a subset of miRNAs is stabilized on XRN2 inactivation. We cannot formally rule out that XRN2 activity at the restrictive temperature is not fully eliminated in the xrn-2ts strain and that complete loss of activity would stabilize all miRNAs. Nonetheless, the available data demonstrate that, minimally, some miRNAs are more dependent on XRN2 for degradation than others.
The mechanisms that provide specificity remain to be elucidated. On the XRN2 side, the enzyme may either contain previously unrecognized intrinsic specificity, or its substrate range may be restricted specifically in vivo through the action of protein binding partners, such as the newly identified PAXT-1 (41). Similarly, features of the miRNA that render them sensitive or insensitive to XRN2 remain to be identified. Although we lack enough examples of miRNAs that are stabilized by mutation of xrn-2 to confidently comment on the involvement of sequence features, we note that there is almost no overlap in sequence between miR-51 and miR-87, and they even differ in their 5' ends, with miR-51 sporting the miRNA-characteristic U and miR-87 and miR-243 a more unusual G and C, respectively. Hence, it seems possible that instead of, or in addition to, sequence, the site of expression of an miRNA might affect its sensitivity to degradation by XRN2. Because our expression analysis of XRN2 indicates widespread, possibly ubiquitous expression of xrn-2, such a model would imply the existence of additional factors that either promote degradation of specific miRNAs by XRN2 in some tissues or prevent it in others. Targets of miRNAs might be one such factor. We previously reported that target RNAs protected their cognate miRNAs from degradation (16,38). At this point, it is not known whether any target can do this, for any miRNA, or if specific miRNA-target duplex architectures are required. Nonetheless, differences in the levels of either the entire group of target RNAs, or only individual targets, might thus alter XRN2 activity towards miRNAs in a tissue-specific manner.
Finally, intracellular localization of miRNAs may affect their susceptibility to degradation by XRN2. This notion is based on our finding that XRN2 accumulates preferentially, perhaps exclusively in the nucleus [this study and (42)]. By contrast, miRNAs are thought to function in the cytoplasm, where they would thus be shielded from XRN2 activity. At the same time, a number of mature C. elegans miRNAs have recently been detected in both nucleus and cytoplasm, with individual miRNAs apparently differing in their nucleocytoplasmic distribution (43). However, because we have so far been unable to achieve sufficiently clean fractionation of nuclei versus cytoplasm, it remains to be determined whether XRN2-sensitive miRNAs partition more extensively to the nucleus than those that are XRN2-insensitive.
miRs and miR*s differ in their stabilities
Initially, it was assumed that miRNA precursors give rise to only one functional molecule, the mature miRNA or guide strand/miR. A second partially complementary molecule derived from the opposite strand of the pre-miRNA, the passenger strand/miR*, might be visible at much lower levels and constitute merely a biogenesis intermediate. More recently, however, several examples of functional miR*s have been described, and it has emerged that in some cases the ratio of miR to miR* may be variable and change with site of expression or development (18). Accordingly, a different nomenclature that identifies miRNA molecules based on their provenance from either the 5′ or the 3′ arm of the pre-miRNA has been adopted. Although there can be little doubt on the functionality of certain miR*s, our decay data strongly suggest that at least in our system most of them accumulate only transiently, supporting their designation as processing intermediates. Although ours is the first demonstration of this phenomenon on a global scale, Winter and Diederichs previously examined the half-lives of a small number of miRs and miR*s in human cells and equally observed reduced half-lives of the latter (44). Moreover, they noted that over-expression of Argonaute proteins could stabilize two miR*s that were investigated, suggesting that it is lack of Argonaute loading that renders miR*s unstable, which would also deprive them of a functional miRNA status.
We note that the least stable of all miRNAs that we observe is annotated as miR, miR-1824-3p, rather than miR*. However, deep sequencing is subject to sequence-dependent biases that prevent exact quantification of distinct small RNAs [(45) and our unpublished data]. The miR-1824-3p displays only marginally (∼1.6-fold) more reads than its presumed miR*, miR-1824-5p, which is much more stable (log2 fold change of 0.16/h versus −0.35/h for 5p versus 3p). Hence, we predict that absolute quantification would reveal that miR-1824-5p is more abundant than miR-1824-3p and thus the true miR by our criterion.
De novo generation of a conditional xrn-2 allele
Genetic mutations are invaluable tools in assigning function to genes. However, if a gene has multiple consecutive functions in development, it can be difficult or impossible to study all of them with ‘constitutive’ mutations especially when an early function is essential during development. At the same time, for essential genes, homozygously mutant animals by necessity need to be derived from heterozygous parents, which may contribute mRNA or protein to their offspring so that early phenotypes can be masked (46). RNAi may be used to deplete such maternal mRNAs, but usually results in only partial depletion of transcripts and protein products. Similarly, although RNAi may be applied such that an early terminal phenotype in development is bypassed (47,48), it can usually not be timed precisely. Although xrn-2(RNAi) phenocopies the sterile phenotype of xrn-2ts animals, none of the conditions we tried so far were able to elicit embryonic lethality.
Conditional alleles, encoding rapidly inactivatable gene products, would permit addressing both of the aforementioned issues. The ts alleles are widely used for instance in yeast, and screens have been conducted in C. elegans to identify ts alleles for specific processes. However, because it has not been possible to predict a priori which mutations will generate a ts allele, targeted approaches for generation of conditional alleles of specific genes have been lacking.
We provide here proof of principle that a C. elegans ts mutation can be generated de novo by exploiting information from a different organism, yeast, despite major differences in their physiological temperature ranges. We note that our approach is not easily scalable and its generality remains to be established. However, many yeast ts alleles exist, and new ones can easily be generated, e.g. by complementing yeast deletion mutant cells with randomly mutagenized transgenes expressing the genes of interests. Hence, ours may be a fertile approach for other researchers interested in generating conditionally mutant C. elegans strains, complementing transcriptional (49,50), co-transcriptional (51) or post-transcriptional (52) approaches that modulate mRNA levels and thus, indirectly, protein activity.
ACCESSION NUMBERS
The small RNA sequencing data discussed in this study have been deposited at GEO and can be accessed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=xdwvjqcwsguaozo&acc=GSE46753.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement number [241985] (European Research Council ‘miRTurn’). Novartis Research Foundation through the FMI and the Swiss National Science Foundation [SNF 31003A_127052 and SNF 31003A_143313]. Boehringer Ingelheim Fonds PhD fellowship (to S.R.). Funding for open access charge: European Union Seventh Framework Programme.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Dr Iskra Katic, Dr Rafal Ciosk and Matyas Ecsedi for helpful comments on the manuscript. The authors thank Kirsten Jacobeit and Sophie Dessus-Babus of the FMI Functional Genomics Facility for library preparation and sequencing, which was performed at the Basel Deep Sequencing Facility, Dr Iskra Katic for help with C. elegans transgenesis and Dr David T. Harris and Dr Robert H. Horvitz for strain MT16418. The authors are particularly grateful to Dr Shohei Mitani and the National Bioresource Project for C. elegans (Japan) for the tm3473 allele, and to Dr Saibal Chatterjee for generating XRN2 protein used to raise the anti-XRN2 antibody.
REFERENCES
- 1.Miki TS, Großhans H. The multifunctional RNase XRN2. Biochem. Soc. Trans. 2013;41:825–830. doi: 10.1042/BST20130001. [DOI] [PubMed] [Google Scholar]
- 2.Henry Y, Wood H, Morrissey JP, Petfalski E, Kearsey S, Tollervey D. The 5′ end of yeast 5.8S rRNA is generated by exonucleases from an upstream cleavage site. EMBO J. 1994;13:2452–2463. doi: 10.1002/j.1460-2075.1994.tb06530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petfalski E, Dandekar T, Henry Y, Tollervey D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol. Cell. Biol. 1998;18:1181–1189. doi: 10.1128/mcb.18.3.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villa T, Ceradini F, Presutti C, Bozzoni I. Processing of the intron-encoded U18 small nucleolar RNA in the yeast Saccharomyces cerevisiae relies on both exo- and endonucleolytic activities. Mol. Cell. Biol. 1998;18:3376–3383. doi: 10.1128/mcb.18.6.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qu LH, Henras A, Lu YJ, Zhou H, Zhou WX, Zhu YQ, Zhao J, Henry Y, Caizergues-Ferrer M, Bachellerie JP. Seven novel methylation guide small nucleolar RNAs are processed from a common polycistronic transcript by Rat1p and RNase III in yeast. Mol. Cell. Biol. 1999;19:1144–1158. doi: 10.1128/mcb.19.2.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geerlings TH, Vos JC, Raue HA. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′–>3′ exonucleases. RNA. 2000;6:1698–1703. doi: 10.1017/s1355838200001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004;432:517–522. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- 8.Chernyakov I, Whipple JM, Kotelawala L, Grayhack EJ, Phizicky EM. Degradation of several hypomodified mature tRNA species in Saccharomyces cerevisiae is mediated by Met22 and the 5′-3′ exonucleases Rat1 and Xrn1. Genes Dev. 2008;22:1369–1380. doi: 10.1101/gad.1654308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kenna M, Stevens A, McCammon M, Douglas MG. An essential yeast gene with homology to the exonuclease-encoding XRN1/KEM1 gene also encodes a protein with exoribonuclease activity. Mol. Cell. Biol. 1993;13:341–350. doi: 10.1128/mcb.13.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevens A, Poole TL. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J. Biol. Chem. 1995;270:16063–16069. doi: 10.1074/jbc.270.27.16063. [DOI] [PubMed] [Google Scholar]
- 11.Xiang S, Cooper-Morgan A, Jiao X, Kiledjian M, Manley JL, Tong L. Structure and function of the 5′–>3′ exoribonuclease Rat1 and its activating partner Rai1. Nature. 2009;458:784–788. doi: 10.1038/nature07731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyer WD, Johnson AW, Reinhart U, Kolodner RD. Regulation and intracellular localization of Saccharomyces cerevisiae strand exchange protein 1 (Sep1/Xrn1/Kem1), a multifunctional exonuclease. Mol. Cell. Biol. 1995;15:2728–2736. doi: 10.1128/mcb.15.5.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu CL, Stevens A. Yeast cells lacking 5′–>3′ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5' cap structure. Mol. Cell. Biol. 1993;13:4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson AW. Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol. Cell. Biol. 1997;17:6122–6130. doi: 10.1128/mcb.17.10.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nagarajan VK, Jones CI, Newbury SF, Green PJ. XRN 5'–>3' exoribonucleases: Structure, mechanisms and functions. Biochim. Biophys. Acta. 2013;1829:590–603. doi: 10.1016/j.bbagrm.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chatterjee S, Großhans H. Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature. 2009;461:546–549. doi: 10.1038/nature08349. [DOI] [PubMed] [Google Scholar]
- 17.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010;11:597–610. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 18.Mah SM, Buske C, Humphries RK, Kuchenbauer F. miRNA*: a passenger stranded in RNA-induced silencing complex? Crit. Rev. Eukaryot. Gene Expr. 2010;20:141–148. doi: 10.1615/critreveukargeneexpr.v20.i2.40. [DOI] [PubMed] [Google Scholar]
- 19.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat. Struct. Mol. Biol. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 20.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. doi: 10.1016/j.cell.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Liu P, James M, Vikis HG, Liu H, Wen W, Franklin A, You M. Genetic variants cis-regulating Xrn2 expression contribute to the risk of spontaneous lung tumor. Oncogene. 2010;29:1041–1049. doi: 10.1038/onc.2009.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frand AR, Russel S, Ruvkun G. Functional genomic analysis of C. elegans molting. PLoS Biol. 2005;3:e312. doi: 10.1371/journal.pbio.0030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnstone IL. Cuticle collagen genes. Expression in Caenorhabditis elegans. Trends Genet. 2000;16:21–27. doi: 10.1016/s0168-9525(99)01857-0. [DOI] [PubMed] [Google Scholar]
- 24.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Redemann S, Schloissnig S, Ernst S, Pozniakowsky A, Ayloo S, Hyman AA, Bringmann H. Codon adaptation-based control of protein expression in C. elegans. Nat. Methods. 2011;8:250–252. doi: 10.1038/nmeth.1565. [DOI] [PubMed] [Google Scholar]
- 26.Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, Jorgensen EM. Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet. 2008;40:1375–1383. doi: 10.1038/ng.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frokjaer-Jensen C, Davis MW, Hollopeter G, Taylor J, Harris TW, Nix P, Lofgren R, Prestgard-Duke M, Bastiani M, Moerman DG, et al. Targeted gene deletions in C. elegans using transposon excision. Nat. Methods. 2010;7:451–453. doi: 10.1038/nmeth.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Praitis V, Casey E, Collar D, Austin J. Creation of low-copy integrated transgenic lines in Caenorhabditis elegans. Genetics. 2001;157:1217–1226. doi: 10.1093/genetics/157.3.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis JA, Fleming JT. Basic culture methods. Methods Cell Biol. 1995;48:3–29. [PubMed] [Google Scholar]
- 30.Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S. Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature. 2010;465:1097–1101. doi: 10.1038/nature09095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sumitani M, Kasashima K, Matsugi J, Endo H. Biochemical properties of Caenorhabditis elegans HMG-5, a regulator of mitochondrial DNA. J. Biochem. 2011;149:581–589. doi: 10.1093/jb/mvr008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh RN, Sulston JE. Some observations on molting in Caenorhabditis elegans. Nematologica. 1978;24:63–71. [Google Scholar]
- 34.Solinger JA, Pascolini D, Heyer WD. Active-site mutations in the Xrn1p exoribonuclease of Saccharomyces cerevisiae reveal a specific role in meiosis. Mol. Cell. Biol. 1999;19:5930–5942. doi: 10.1128/mcb.19.9.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amberg DC, Goldstein AL, Cole CN. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992;6:1173–1189. doi: 10.1101/gad.6.7.1173. [DOI] [PubMed] [Google Scholar]
- 36.Page AM, Davis K, Molineux C, Kolodner RD, Johnson AW. Mutational analysis of exoribonuclease I from Saccharomyces cerevisiae. Nucleic Acids Res. 1998;26:3707–3716. doi: 10.1093/nar/26.16.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alvarez-Saavedra E, Horvitz HR. Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol. 2010;20:367–373. doi: 10.1016/j.cub.2009.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee S, Fasler M, Bussing I, Großhans H. Target-mediated protection of endogenous microRNAs in C. elegans. Dev. Cell. 2011;20:388–396. doi: 10.1016/j.devcel.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 39.Sanford T, Golomb M, Riddle DL. RNA polymerase II from wild type and alpha-amanitin-resistant strains of Caenorhabditis elegans. J. Biol. Chem. 1983;258:12804–12809. [PubMed] [Google Scholar]
- 40.Lehrbach NJ, Castro C, Murfitt KJ, Abreu-Goodger C, Griffin JL, Miska EA. Post-developmental microRNA expression is required for normal physiology, and regulates aging in parallel to insulin/IGF-1 signaling in C. elegans. RNA. 2012;18:2220–2235. doi: 10.1261/rna.035402.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miki TS, Richter H, Rüegger S, Großhans H. PAXT-1 promotes XRN2 activity by stabilizing it through a conserved domain. Mol. Cell. 2014;53:351–360. doi: 10.1016/j.molcel.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 42.Bosse GD, Ruegger S, Ow MC, Vasquez-Rifo A, Rondeau EL, Ambros VR, Großhans H, Simard MJ. The decapping scavenger enzyme DCS-1 controls microRNA levels in Caenorhabditis elegans. Mol. Cell. 2013;50:281–287. doi: 10.1016/j.molcel.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zisoulis DG, Kai ZS, Chang RK, Pasquinelli AE. Autoregulation of microRNA biogenesis by let-7 and Argonaute. Nature. 2012;486:541–544. doi: 10.1038/nature11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winter J, Diederichs S. Argonaute proteins regulate microRNA stability: Increased microRNA abundance by Argonaute proteins is due to microRNA stabilization. RNA Biol. 2011;8:1149–1157. doi: 10.4161/rna.8.6.17665. [DOI] [PubMed] [Google Scholar]
- 45.Hafner M, Renwick N, Brown M, Mihailovic A, Holoch D, Lin C, Pena JT, Nusbaum JD, Morozov P, Ludwig J, et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17:1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jorgensen EM, Mango SE. The art and design of genetic screens: Caenorhabditis elegans. Nat. Rev. Genet. 2002;3:356–369. doi: 10.1038/nrg794. [DOI] [PubMed] [Google Scholar]
- 47.Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding XC, Slack FJ, Großhans H. The let-7 microRNA interfaces extensively with the translation machinery to regulate cell differentiation. Cell Cycle. 2008;7:3083–3090. doi: 10.4161/cc.7.19.6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei X, Potter CJ, Luo L, Shen K. Controlling gene expression with the Q repressible binary expression system in Caenorhabditis elegans. Nat. Methods. 2012;9:391–395. doi: 10.1038/nmeth.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bacaj T, Shaham S. Temporal control of cell-specific transgene expression in Caenorhabditis elegans. Genetics. 2007;176:2651–2655. doi: 10.1534/genetics.107.074369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calixto A, Ma C, Chalfie M. Conditional gene expression and RNAi using MEC-8-dependent splicing in C. elegans. Nat Methods. 2010;7:407–411. doi: 10.1038/nmeth.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaudet J, Mango SE. Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science. 2002;295:821–825. doi: 10.1126/science.1065175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.