


Abstract
The budding yeast multi-K homology domain RNA-binding protein Scp160p binds to >1000 messenger RNAs (mRNAs) and polyribosomes, and its mammalian homolog vigilin binds transfer RNAs (tRNAs) and translation elongation factor EF1alpha. Despite its implication in translation, studies on Scp160p's molecular function are lacking to date. We applied translational profiling approaches and demonstrate that the association of a specific subset of mRNAs with ribosomes or heavy polysomes depends on Scp160p. Interaction of Scp160p with these mRNAs requires the conserved K homology domains 13 and 14. Transfer RNA pairing index analysis of Scp160p target mRNAs indicates a high degree of consecutive use of iso-decoding codons. As shown for one target mRNA encoding the glycoprotein Pry3p, Scp160p depletion results in translational downregulation but increased association with polysomes, suggesting that it is required for efficient translation elongation. Depletion of Scp160p also decreased the relative abundance of ribosome-associated tRNAs whose codons show low potential for autocorrelation on mRNAs. Conversely, tRNAs with highly autocorrelated codons in mRNAs are less impaired. Our data indicate that Scp160p might increase the efficiency of tRNA recharge, or prevent diffusion of discharged tRNAs, both of which were also proposed to be the likely basis for the translational fitness effect of tRNA pairing.
INTRODUCTION
RNA-binding proteins (RBPs) form a multilayered regulatory network that tightly controls post-transcriptional gene regulation. In the yeast Saccharomyces cerevisiae, ∼600 proteins have been predicted to possess RNA-binding activity (1). To process and regulate >6700 different transcripts within a yeast cell, combinatorial action of these proteins is required. According to the recently proposed ‘RNA-operon’ model, control occurs in a concerted manner for specific groups of messenger RNAs (mRNAs) [(2), and references therein].
Scp160p is the yeast orthologue of the evolutionary conserved vigilin protein family that is characterized by its unique repetitive array of 14 RNA-binding heterogenous ribonucleoprotein (hnRNP) K homology (KH) domains (3,4). Only seven of these represent canonical KH domains, whereas the remaining seven have insertions or deletions in a characteristic GXXG motif and are therefore termed diverged KH domains (5).
Two microarray-based studies have been carried out to identify mRNAs associated with Scp160p. A first approach discovered five mRNA targets, DHH1, BIK1, NAM8, YOR338W and YOL155C (6). A more recent study identified >1000 mRNA targets of Scp160p that cluster in groups encoding proteins of cell wall, plasma membrane, endoplasmic reticulum (ER) and nucleolus (1). However, the biological function of these Scp160p–mRNA interactions remains to be elucidated. Since its first description, Scp160p has been implicated in various processes, ranging from the control of cellular ploidy (7) to mating response (8,9), telomeric silencing (10) and control of spindle pole body biogenesis (11). In many of these processes, Scp160p seems to be involved in translational regulation of target proteins. In addition, scp160 deletion strains are sensitive against translation inhibitors such as cycloheximide and hygromycin B (12), and Scp160p is known to associate with cytosolic and membrane-bound polysomes, from which it is released by ethylenediaminetetraacetic acid treatment as component of messenger ribonucleoprotein (mRNP) complexes (13). Crosslinking experiments indicate that Scp160p binds to the ribosomal 40S subunit close to the mRNA-binding site, and that this association is partially dependent on the interaction with Asc1p, the yeast homologue of mammalian RACK1 (12). Scp160p acts in concert with the eIF4E-binding protein Eap1p in the so-called SMY2-EAP1-SCP160-ASC1 (SESA) network to inhibit translation of POM34 mRNA in response to spindle pole body duplication defects (11). In addition, there is evidence that Scp160p is involved in the elongation step of translation, as it can form chemical crosslinks with elongation factor 1 A (12). Finally, the mammalian Scp160p homolog vigilin copurifies in a complex with elongation factor 1 A and transfer RNA (tRNA) (14). The latter provides an interesting link between Scp160p and proteins that belong to the ‘hardware’ basis of translation.
When considering translation, the ambiguity of the genetic code ‘software’ enables regulation when resolving beyond the amino acid level. This means that the same amino acid sequence may be translated with varying speed, depending on the nucleic acid composition of the coding mRNA. Just as the AUG context is an important fine-tuning capacity of eukaryotic initiation (15), elongation can be boosted by choosing between different synonymous codons (16). Codon ordering also has a beneficial effect on translational fitness (17).
The two most important predictive parameters of translational fitness that work beyond the initiation stage are codon usage and tRNA autocorrelation. Highly expressed genes with large mRNA copy numbers tend to use a restricted subset of synonymous codons (18) whose decoding tRNAs are more abundant, recognized faster and incorporated during translation with higher fidelity (19,20). This can be measured with the codon adaptation index (CAI), the bias of a gene to favor ‘popular’ (or frequent) over ‘unpopular’ (infrequent) codons. On the other hand, it has been found that the re-use of the same tRNA by synonymous codons at successive occurrences of the same amino acid in an mRNA, termed ‘autocorrelation’ and measured with the tRNA pairing index (TPI), can significantly boost translational efficiency (17). Based on random walk computations compared with the distance decay of this effect, autocorrelation is thought to be caused by local molecular crowding (21) of tRNAs for recharging near the ribosome, as suggested by the substrate channelling hypothesis of translation (22). This tRNA ‘re-use’ effect is independent of the abundance of recognized codons or their number of synonyms. For example, amino acid starvation genes were found to markedly use ‘unpopular’ (infrequent) synonymous codons but autocorrelate (‘codon-cluster’) them to make use of a less depleted pool of charged aminoacyl-tRNAs (17).
To investigate the potential role of Scp160p in translational control, we followed a translational profiling approach to identify mRNAs whose translation is regulated by Scp160p. We have identified a set of mRNAs that shift their position within polyribosome gradients on Scp160p depletion, indicating changes in their translation rates. As the coding optimization of transcripts predicts their translational fitness, we hoped to elucidate possible connections between Scp160p’s action and optimized translation by bioinformatic analysis of its bound mRNAs. We found that mRNAs bound by Scp160p, and translationally affected by its depletion, show a higher frequency of ‘autocorrelated’ successive synonymous codons recognizing the same tRNA. By quantifying tRNAs associated with ribosomes in the presence and absence of Scp160p, we show that loss of Scp160p results in stronger depletion of a specific subgroup of tRNAs. Surprisingly, these tRNAs recognize codons with low autocorrelation in Scp160-bound mRNAs. This shows that Scp160p’s benefit on translation of its target mRNAs is synergistic with, but independent of, codon autocorrelation and might represent a novel mode of translation optimization. Our data thus suggest that Scp160p is required for efficient translation of a subset of mRNAs via tRNA recycling or reduction of tRNA diffusion from the ribosome.
MATERIALS AND METHODS
c-myc immunoprecipitation and ribosome affinity purification
Immunoprecipitation of She2p, Khd1p and Scp160p was carried out essentially as previously described (23) with two changes. RNasin was added to the breaking buffer at 0.8 U/µl. Instead of IgG2a beads, Protein G magnetic beads (Invitrogen) were used that had been coupled to anti-myc antibody. DNA was removed by treatment with RQ1 DNase (Promega). For reverse transcription, the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used.
Ribosome affinity purification (RAP) was performed essentially as described (24). We introduced two major changes to the protocol. Instead of 50 OD600 units, we lysed 100 OD600 units of cells and omitted tRNA as blocking agents. Quantitation of tRNAs coprecipitating with ribosomes was done by RT-qPCR. Up to five biological replicates were quantitated in duplicate technical replicates, and only observations for high (>2× Tag versus Mock) immunoprecipitation (IP) efficiency were pursued. The qPCR measurements were discarded if technical replicate crossing time (Ct) value SD exceeded 0.5 or multiple melting temperature (Tm) peaks were observed.
In both cases, mRNA was extracted using a commercially available kit (Macherey&Nagel RNA II kit, Düren, Germany). The tRNA and mRNA were separated by size using a similar system (Macherey&Nagel miRNA kit).
For tetracycline repression, appropriate strains were grown for 6 h in medium supplemented with 2 µg/ml doxycycline (25). For yef3(F605S) mutant analysis (26–28), yeast cells were grown at permissive temperature (26°C) to optical density at 600 nm of 0.7, then shifted for 1 h to restrictive temperature of 37°C before harvesting.
Quantitative RT-PCR of mRNAs following myc-IP
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) was performed using the StepOnePlus™ Real-Time PCR System and Power SYBR® Green PCR Master Mix (both from Applied Biosystems) as prescribed by the manufacturer. Primers (see Supplementary Table S3) were designed using Primer3 software version 0.4.0 (http://frodo.wi.mit.edu/primer3/input.htm). Relative quantifications were performed by the comparative CT method.
Quantitative RT-PCR of mRNAs and tRNAs following RAP-IP
The mRNA and tRNA were extracted from RAP eluates and cell lysates as described earlier in the text. Primers for tRNA were constructed based on zones of low homology using multiple ClustalW sequence alignments of tRNA genes in Molecular Genetics Evolutionary Analysis (MEGA) software. Reverse transcription of tRNA was facilitated by introducing a 95°C pre-incubation step of RNA and master mix before addition of reverse transcriptase at 4°C, to enable melting of tRNA secondary structures and annealing of primers.
Percent ribosomal occupancy (%ribocc) was calculated according to the ΔCT method as follows: the comparison between different strains (e.g. wt versus scp160Δ) was done by comparing quantitative values obtained from eluted RNA. For mRNAs, each strain’s eluate (E) was normalized to its cognate lysate (L), thus eliminating transcriptome discrepancies from consideration. In accordance with prior publications, transcriptome stability of tRNAs was assumed for further analyses. In all cases, however, each translational quantification was also normalized to its own cognate mock-IP strain to eliminate quantification resulting from unspecific adherence of ribosomes to beads.
Sucrose density gradient centrifugation
To 180 ml of logarithmically growing cells, cycloheximide was added to a final concentration of 0.1 mg/ml and incubated for 10 min at 30°C. Cells were harvested and resuspended in 400 µl polysome buffer (20 mM HEPES/KOH, pH 7.5, 75 mM KCl, 2.5 mM MgCl2, 1 mM EGTA, 0.1 mg/ml cycloheximide, 1 mM DTT) supplemented with 2.5 µl SUPERase-In RNase inhibitor (Ambion). A total of 200 µl of glass beads was added and the samples were vortexed vigorously for 5 min at 4°C. After centrifugation (5 min, 16 000g, 4°C), the supernatant was collected, snap-frozen and stored at −80°C until use.
Extract corresponding to 600 µg of RNA as determined by OD260nm was loaded onto a 12 ml 20–60% linear sucrose gradient in polysome buffer and centrifuged for 2 h at 155 000g and 4°C in an SW40 rotor. In all, 750 µl fractions were collected from top to bottom using a Biocomp Gradient Station (Fredericton, CAN). Fractions were phenol–chloroform extracted, and RNA was precipitated with isopropanol from the 1:4 diluted aqueous phase. DNase treatment and RNA cleanup were performed as described earlier in the text.
Calculations of coding determinants of translational fitness and statistics testing
The CAI (popularity of codons), TPI (codon autocorrelation across and within amino acid) and Z-scores (expressing the difference between observed counts of codon pairs and those expected as standard deviations at both codon and tRNA resolution) were computed with the Darwin package (26,28,29) as described elsewhere (17,18,30,31). The P-values were computed for each group of genes by Monte Carlo sampling in which the TPI value of the group was compared with that of 100 000 random groups of the same size chosen from the yeast genome.
ΔCt calculations, averages, standard deviations and P-values were calculated with appropriate tests, e.g. Student’s t-test, using Microsoft Excel and Prism (Graphpad Software, La Jolla, USA). Prism was also used for calculating Spearman’s non-linear correlation analyses.
Additional methods
Including plasmid and yeast strain construction as well as microarray analysis are provided in Supplementary Materials.
RESULTS
Scp160p depletion alters translation of a subset of mRNAs
Although it is known that Scp160p is an RNA- and ribosome-associated protein involved in various cellular processes, its molecular function in translation remains elusive. To assess whether a specific subset of the transcriptome is translated differentially on loss of Scp160p, we determined the changes in the translational profile for each mRNA. Because deletion of Scp160p results in alterations in ploidy (7,32), we were concerned that secondary effects caused by ploidy changes could obscure the translational profiles. Therefore, we generated a yeast strain in which Scp160p expression is under control of a repressible Tet-off operator, which allows rapid depletion of the protein on addition of doxycyclin (11,25). Repression of Scp160p resulted in a substantial reduction of the protein after three hours of depletion, and the protein was undetectable after five hours (Figure 1A). Fluorescence-assisted cell sorting (FACS) analysis revealed that the number of cells with ploidy changes after 6 h depletion is still low (Figure 1B). Therefore, we chose a 6 h depletion period of Scp160p for translational profiling experiments. Polysome profiles were recorded after separating cell lysates on a 20–60% sucrose gradient. We could not detect differences of polysome/monosome (P/M) ratios between cells expressing Scp160p (P/M = 3.7 ± 0.58) and cells depleted for Scp160p (P/M = 3.5 ± 0.37), indicating that global translation is not significantly altered on Scp160p depletion (Figure 1C).
Figure 1.

Depletion of Scp160p changes translational states of a specific set of mRNAs. (A) Depletion of Scp160p in a Tet-off SCP160 strain revealed by western blot. At times indicated, culture aliquots were removed to prepare protein extracts, and depletion of Scp160p was monitored by probing the blot with an anti-Scp160 antibody. Actin served as loading control. (B) Depletion of Scp160p finally results in an increase of ploidy. During a time course of 24 h, cells were removed at different time points, fixed and analyzed by FACS. An scp160Δ strain served as control. DNA contents corresponding to haploid (1 n), diploid (2 n) or tetraploid (4 n) state are indicated. (C) Global translation is not impaired by depletion of Scp160p. Cell extracts of non-depleted and Scp160p-depleted cells were separated by sucrose density centrifugation. From the recorded absorbance profiles (256 nm), polysome and monosome areas were defined. Polysome-to-monosome ratios (P/M) report the global translation state. Data represent averages of three independent experiments; standard deviations are indicated in brackets. (D) Calculation of translation state changes. Sucrose gradient profiles of whole cell extracts of Scp160p-depleted and control samples were monitored by ultraviolet absorbance. The positions of the small and large ribosomal subunits (asterisk: 40 S; double asterisk: 60 S), monosomes and polysomes are indicated. Light and heavy fractions of both samples were collected, and extracted mRNA was used for duplicate microarray analyses as described in the ‘Materials and Methods’ section. For each individual mRNA, translation states were calculated as the ratio of the normalized signal in heavy and light fraction. Translation state changes were then calculated as the double ratio of the translation states in Scp160p-depleted versus control sample. (E) and (F) MIPS Functional Catalogue Database classification of mRNAs with significant translation state changes (≥1.8, ≤ − 1.8) on depletion of Scp160p. Locational distribution analysis of the proteins encoded by the candidate mRNAs revealed clustering in specific classes (P-value cut-off: P < 0.01) (B). Functional distribution analysis of the proteins encoded by the candidate mRNAs showed enrichment in distinct subgroups (P-value cut-off: P < 0.01) (C).
To assess possible changes in the translational profiles of specific mRNAs, we first determined the translational state for each mRNA under control and Scp160p depletion conditions by DNA microarray analysis. Polysomes, monosomes and free ribonucleoprotein complexes (RNPs) in cell lysates were separated on sucrose density gradients. Monosomes and free RNPs were pooled as ‘light fraction’, whereas polysomes with three or more ribosomes per mRNA were pooled as ‘heavy fraction’ (Figure 1D). After extraction, RNAs were reverse transcribed and hybridized to DNA microarrays (see ‘Materials and Methods’ section). We defined the translational state of an mRNA as the ratio of its abundance in heavy versus light fractions. The translational state of each mRNA was registered in cells depleted for Scp160p and control cells, in which Scp160p expression was not shut off. From these values, ratios were calculated to obtain a translational state change (TSC) that reflects the shift in the distribution of a given mRNA within the sucrose gradient on depletion of Scp160p. Increases of the TSC value are supposed to indicate a shift from monosomes and free RNPs towards polysomes, whereas decreased TSC values should indicate an opposite shift. Using a cut-off of 1.8-fold (≥1.8 or ≤1.8), we identified 48 mRNAs with increased and 12 mRNAs with decreased polysome association (Supplementary Table S4; 2-fold cut-off: 23 and 5 mRNAs, respectively). These results indicate that depletion of Scp160p affects the translational state of a small subset of mRNAs (1.1% of the transcriptome) even before ploidy defects are detectable. The majority (80%) of the mRNAs within this set were shifted towards the heavy gradient fractions, indicating an increased association of these mRNAs with polysomes. A systematic classification of the mRNAs up- or downregulated in translation revealed that they are enriched for mRNAs encoding extracellular and cell wall proteins, as well as proteins involved in ER-to-golgi transport (Figure 1E) as classified by the MIPS Functional Catalogue Database (33,34). The targets can be functionally grouped into proteins involved in cell–cell adhesion, cell polarity establishment and filament formation, sugar binding, endocytosis and response to osmotic and salt stress (Figure 1F). A common feature of many of these proteins is their targeting to the ER, which occurs mainly cotranslationally. This is in agreement with previously published data showing that Scp160p localizes to cytoplasmic and ER-attached polyribosomes (13,30).
Scp160p binds to a subset of mRNAs
The observed changes of the translational state of some mRNAs could be a direct consequence of the loss of Scp160p, e.g. if it functions as translational regulator, or indirect by affecting the overall physiological state of the cell. A direct function on its target mRNAs is likely to be mediated by direct binding of Scp160p to these mRNAs or its binding to ribosomes that translate these mRNAs. In both cases, immunoprecipitation of myc9-tagged Scp160p in combination with qRT-PCR should allow the detection of copurifying mRNAs (Figure 2). As specificity controls, we performed the same assay with two additional RBPs that were likewise tagged with nine myc epitopes. She2p is an RBP that associates with a defined set of localized mRNAs (35). Khd1p, like Scp160p, belongs to the group of KH domain containing RBPs and binds to a large set of mRNAs with diverse functions (36). Myc-tagged versions of all three proteins were functionally expressed in yeast and western blot analysis showed that similar amounts of each protein could be immunoprecipitated (Figure 2A). RNA was extracted from the pellet, reverse transcribed and amplified with primers against 14 mRNAs showing significant TSCs (see Supplementary Table S4). We also included the nucleoporin-encoding POM34 transcript in our analysis, as it had been previously identified as an Scp160p target (11). Figure 2B summarizes the results for the four mRNAs with significant enrichment in the Scp160p immunoprecipitates. Although She2p coimmunoprecipitated with its known target mRNA ASH1, none of the other five mRNAs (Figure 2B, white bars), Scp160p immunopellets (Figure 2B, black bars) are >7-fold enriched for CCW14, AGA1 and PRY3 and >2-fold enriched for MSB2 and POM34 mRNAs. These enrichment values do not directly reflect the degree of the translation state changes of these transcripts, as the TSCs of AGA1 and PRY3 are 2.4 and 2.7, respectively, whereas both CCW14 and MSB2 have a TSC of 1.8 (Supplementary Table S4). A control mRNA, CFT1, whose translation rate did not change on Scp160p depletion, neither copurified with Scp160p nor with She2p. AGA1, PRY3 and POM34 were enriched in Khd1p immunopellets at similar levels (Figure 2, dashed bars), suggesting that these mRNAs contain binding sites for KH domain type RBPs. CCW14 and MSB2 mRNAs were also associated with Khd1p, albeit enrichment was reduced in comparison with the Scp160p immunopellets to ∼37% and 78%, respectively (P-values < 0.05). In summary, of the 14 candidate mRNAs, we found four to be enriched >2-fold in the Scp160p immunoprecipitates (Figure 2B): CCW14, encoding a cell wall glycoprotein (37), AGA1 that codes for a subunit of a-agglutinin in the cell wall (38), PRY3 that encodes a member of the PRY family of Glycosyl-phosphatidyl-inositol (GPI)-anchored cell wall proteins (1,39) and MSB2, coding for an integral plasma membrane mucin linked to osmosensing (2,40). Consistently, these four mRNAs were also identified as associated with Scp160p (log2 ≥ 1.1) in a recent large-scale survey (1,3,4). To investigate if binding of Scp160p to the mRNAs mentioned earlier is direct or mediated by an associated protein, we generated a carboxyterminally truncated version of Scp160p that lacks the last two KH domains (Scp160pΔKH13/14). Myc9-tagged Scp160pΔKH13/14 can be immunoprecipitated at similar levels as the full-length protein (Figure 2A) without copurifying any of the mRNAs (Figure 2B, grey bars).
Figure 2.
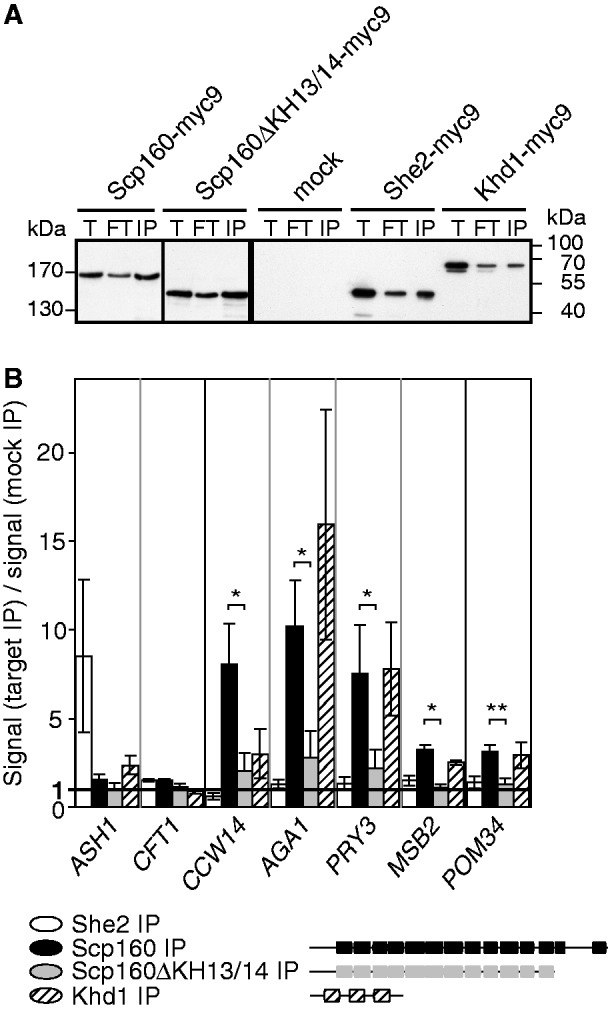
Scp160 binds to mRNAs. (A) Western blot of the immunoprecipitation of myc9-tagged Scp160p, Scp160p truncated for KH domains 13 and 14, She2p and Khd1p using anti-myc antibody. T—total extracts, FT—flowthrough, IP—immunoprecipitate. (B) Scp160p binds CCW14, AGA1, PRY3, POM34 and MSB2 mRNAs. Immunoprecipitates were subjected to qRT-PCR and analyzed for bound ASH1 RNA (positive control in She2p IP), CFT1 RNA (negative control) and candidate RNAs CCW14, AGA1, PRY3, MSB2 and POM34. Specific enrichment was calculated as ratio of the signal in the target IP to the mock IP (untagged wild-type strain). Statistical significance (Student’s t-test) compared with the KH domain truncation is indicated: *, P < 0.05; **, P < 0.01. Data are presented as mean ± standard deviation, n = 3.
Scp160p depletion results in reduced PRY3p protein levels
To test if the observed TSCs reflect altered translation, we determined protein levels for proteins before and after Scp160p depletion. According to traditional interpretations, we expected to find ‘increased’ protein production from transcripts with high TSC values due to a supposed shift into ‘fast polysomes’ on SCP160 depletion. PRY3p was chosen because it had the largest TSC ratio (2.7) of depleted/control of the known proteins as shown in Supplementary Table S4. For detection of PRY3p, a variant with six HA epitopes fused to the carboxyterminus was expressed from its genomic locus. Lysates were prepared from cells 6 h after doxycyclin addition or after 6 h incubation without doxycyclin (mock depletion). In contrast to Scp160p or phosphoglycerokinase (Pgk1p), no defined bands but a smear at >200 kDa was detected for PRY3p-6HA (Figure 3A, left panel). Interestingly, depletion of Scp160p led to a ‘reduction’ of PRY3p levels to 66% (±11%) (Figure 3A, middle panel; four independent depletion experiments). In contrast, PRY3 mRNA levels did not significantly change, whereas SCP160 mRNA dropped to 6% (±3%) of the original amount (Figure 3A, right panel). Because we suspected that the smeary western blot signal for PRY3p, which hampered quantification of the protein levels, was caused by its GPI anchor, we replaced the C-terminal 29 amino acids that include the GPI modification site as predicted by the GPI Fungal Prediction Server (5,41) by a myc9 tag (PRY3ΔGPI-9myc). This resulted in a more focused signal of PRY3p, allowing better quantification (Figure 3B and C, left panels). PRY3ΔGPI-9myc levels in scp160Δ were reduced to 53% (±7%) compared with the wild-type control (Figure 3B, middle panel). In cells depleted for Scp160p, PRY3ΔGPI-9myc levels decreased to 87% (±12%; Student's t-test: P = 0.0022) compared with non-depleted controls (Figure 3C, middle panel). In both experimental setups, mRNA levels did not drop accordingly (Figures 3B and C, right panels), suggesting a decrease in PRY3p translation or an increase in PRY3p degradation on loss of Scp160p. To rule out the latter, we performed cycloheximide chase experiments to determine the half-life of PRY3ΔGPI-9myc in Scp160p depleted, scp160Δ, and control cells. Western blotting demonstrated that the decay of PRY3ΔGPI-9myc is not accelerated after Scp160p depletion, as half-lives of PRY3ΔGPI-9myc in scp160Δ or Scp160p-depleted cells (t1/2 = 8 or 11 min, respectively) were not significantly reduced as compared with wild-type (t1/2 = 7 min) or mock-depleted (t1/2 = 14 min) cells (Supplementary Figure S1). In summary, our results demonstrate that loss of Scp160p causes translational downregulation of PRY3p. We also assayed levels of other proteins made from Scp160p target mRNAs, PTH1 and CCW14. In scp160Δ cells, myc-tagged Pth1p exhibited small but statistically significant decreases in protein level, whereas RNA levels remained constant (Supplementary Figure S2A and B). Interestingly, the smaller protein level decrease corresponds to a less pronounced polysome shift of the mRNA in sucrose gradients (TSC = 2.4). For a third target, CCW14, with yet smaller TSC (1.8) we could not detect a significant protein level decrease (not shown). In total, these three target mRNAs show that protein level ‘decrease’ shows a pronounced non-linear correlation with observed shift ‘towards’ polysomes (Spearman’s r = 1.0, P = 0.33, Figure 3D). The sum of these observations suggests that the polysome shifts found on Scp160p depletion do have implications in translational efficiency. Specifically, however, they imply that counter to traditional expectations, polysome shifts here do not seem to indicate ‘fast polysomes’.
Figure 3.

PRY3 protein levels are reduced on loss of Scp160p. (A) PRY3-6HA levels in Scp160p-depleted cells. Whole cell extracts were prepared from strain RJY3498, 6 h after addition of 2 µg/ml doxycycline or after mock depletion. Left panel: equivalent total protein amounts were used for western blotting. Probing with anti-Scp160p antibody shows the efficient depletion of Scp160p, anti-HA antibody the reduction of PRY3-6HA protein levels on depletion of Scp160p and Pgk1 serves as loading control. One representative experiment is shown. Middle panel: quantification of four experiments performed as in the left panel. Right panel: RNA was prepared from the same samples and used for qRT-PCR as described in ‘Materials and Methods’ section. Relative PRY3 and SCP160 mRNA levels were calculated as signal ratio between Scp160p-depleted and non-depleted samples. All data are presented as mean ± standard deviation, n = 4. (B) PRY3ΔGPI levels in scp160Δ cells. Whole cell extracts were prepared from yeast cells expressing PRY3ΔGPI-9myc and four independent clones of scp160Δ yeast cells expressing PRY3ΔGPI-9myc. Left panel: western blot of one representative experiment. Middle panel: quantification of three independent experiments carried out as in the left panel. Right panel: RNA was prepared from the same strains and used for qRT-PCR. Relative PRY3ΔGPI mRNA levels are represented as the signal ratio between scp160Δ and wild type (WT) background. (C) PRY3ΔGPI levels in Scp160p-depleted cells. Whole cell extracts were prepared 6 h after addition of 2 µg/ml doxycycline or after mock depletion. Left panel: representative western blot showing the efficient depletion of Scp160p (anti-Scp160p) and the reduction of PRY3ΔGPI-9myc protein levels on depletion of Scp160p (anti-myc). Pgk1 serves as loading control. Middle panel: quantification of n = 11 experiments performed as in the left panel. The decrease of the PRY3ΔGPI-9myc protein levels in the sample depleted for Scp160p is highly significant (Student’s t-test, P = 0.0022). Right panel: RNA was prepared from the same samples and used for qRT-PCR. Relative PRY3 and SCP160 mRNA levels were calculated as the signal ratio between Scp160p-depleted and non-depleted samples. All data are presented as mean ± standard deviation, n = 3. (D) Polysome shift (TSC) and protein level reduction PRY3, PTH1 and CCW14 show pronounced non-linear correlation (Spearman’s r = 1.00, P = 0.33). TSC values extracted from Supplementary Table S4 are indicated below targets for enhanced clarity.
Ribosomal occupancy of several target mRNAs of Scp160p decreases on knockout
Our observation that depletion of Scp160p results in elevated PRY3 and PTH1 mRNAs in polysomal fractions, but decreasing protein levels raises the possibility that the shift of mRNAs towards dense sucrose gradient fractions represents ‘slow polysomes’ that can be observed when elongation is decelerated on a transcript, whereas initiation is not or little affected (6,16). This is especially likely given that many Scp160p-bound mRNAs code for proteins involved in ribosome biogenesis and translation (1), a biological function that another recent study found to be significantly correlated with translational elongation instead of initiation control of transcripts (7,42).
We thus applied RAP as an alternative method to independently verify translational repression. RAP analysis results in percentages of ribocc (8,9,24), i.e. the fraction of RNA eluate that is bound to one or multiple ribosomes. For this parameter, decreased ribocc is inferred to mean less efficient translation from transcripts. We purified cycloheximide-stalled ribosomes from yeast expressing TAP-tagged Rpl6 ribosomal protein (10,24) and quantitated selected mRNAs eluted from RAP-purified ribosomes. Comparing eluted mRNAs with wild-type and Scp160-knockout cells enabled us to calculate ribocc as percent knockout versus wild-type levels for four mRNA targets of Scp160p. The ACT1, CFT1, PRY3 and CCW14 mRNAs were less abundant on ribosomes from scp160Δ compared with wild-type cells (Figure 4). It is important to note that ribocc is calculated in a way different from translation state change. Whereas TSC relates polysomal (heavy) to pooled monosomal and free RNA fractions pooled, for the calculation of %ribocc the entities of all ribosome-associated mRNAs is contrasted with the complete transcriptome. In conjunction with the observed decrease in PRY3p and Pth1p protein levels on Scp160p depletion (Figure 3D), a possible interpretation of these results is that the observed shift of mRNAs towards heavy fractions in sucrose gradients (Figure 1, Supplementary Table S4) corresponds to slow elongation-impaired polysomes for at least one mRNA target.
Figure 4.

The ribocc of several mRNAs decreases in scp160Δ cells. Normalized co-precipitation with ribosomes is shown for four mRNAs. Normalization was done as described in ‘Materials and Methods’ section and is displayed as percentage of ribocc as seen in wild-type cells. The standard deviation between up to five biological replicates with two technical replicates each is shown, while *, P <0.1 and **, P <0.01 for significance of independent average values in scp160Δ compared with wild-type cells.
Bioinformatical analysis implies a role for tRNA autocorrelation in the translational downregulation of mRNAs following Scp160 depletion
The implied translational impairment of Scp160p target mRNAs in Scp160p-deficient cells is consistent with their slow-growth phenotype, especially when grown on media supplemented with translation elongation inhibitors (11,12). This suggests that the protein or its interaction partners are at least indirectly linked to efficiency of translational elongation.
To elucidate a possible connection between the aforementioned observations, we analyzed the codon composition of mRNAs bound by Scp160p [Figure 2 and (1,12)] or translationally altered on its knockdown (Supplementary Table S4).
First, we asked if codon popularity or tRNA re-use would be a characteristic of mRNAs interacting with Scp160p. Therefore, we computed TPI to measure the autocorrelation or ‘clustering’ of codons across all amino acids, in mRNAs found to be reproducibly bound by Scp160p in a recent large-scale survey [Supplementary Table S5 and (1,13)]. We found a marked increase in the average TPI (12,17) of the entire group, with high significance (0.527 compared with 0.124 genomic average, P = 0.005 for 100 000 random pickings of equal-sized groups), whereas the CAI (11,18) was not significantly changed (results not shown). This result suggests that Scp160p interacting mRNAs might be biased towards optimized translation not by containing ‘popular codons’ fitting to globally high tRNA abundance, but because ‘codon clustering’ in them could cause locally high tRNA abundance.
We next asked if the observed deviation from genomic averages for ‘codon clustering’ would be directional above the average non-randomness proven earlier in the text. We thus created subgroups of these mRNAs ordered by rising binding reproducibility, subjected them to within-group average calculations and asked by t-tests if the subgroups are significantly distinct. We found that candidate subgroups ordered for binding reproducibility have a pronounced non-linear correlation of high significance with codon autocorrelation (Spearman’s r = 0.833, P = 0.015; Supplementary Figure S3), and the subgroups’ averages are significantly distinct (Supplementary Table S6). This means that the more reproducibly mRNAs associate with Scp160p, the more they are optimized for efficient translation.
Next, we asked whether the bias of Scp160p-interacting mRNAs to be skewed for ‘codon clustering’ could be a key to explaining their shift to presumably ‘slow polysomes’ in sucrose gradients. To this end, we calculated the TPI of mRNAs (Supplementary Table S7) shifted to different degrees towards heavy polysome fractions after loss of Scp160p. The data were grouped into subgroups constructed by taking all members within distinct thresholds of TSC. This way, a rising ‘translational depletion’ threshold would select subgroups of decreasing size, raising the statistical threshold they would have to overcome to prove the significance of their elevated ‘codon clustering’: The smaller a set of genes, the less likely they are skewed by chance. For each subgroup, we plotted average TSC (Supplementary Table S4) against average TPI as a measure of across-amino acid autocorrelation and found a pronounced non-linear correlation of high significance (Spearman’s r = 0.893, P = 0.012, Figure 5A) where the subgroups’ averages are significantly distinct (Figure 5B). The same tendency was observed for select amino acids when autocorrelation was resolved at within-amino acid level for the subgroups of mRNAs differently affected by Scp160p depletion (Supplementary Table S8), albeit with the TPI only being significantly distinct from random genomic pickings of equal-sized groups in some cases. This means that the mRNA targets bound by Scp160p, and translationally affected more drastically by its knockdown, rely more heavily on ‘codon clustering’ (autocorrelation), which has been proposed to work by promoting tRNA recycling and/or preventing tRNA diffusion (12,17).
Figure 5.

Codon autocorrelation is a characteristic feature of mRNAs affected by Scp160p depletion. (A) Average use of isoaccepting codons within subgroups of Scp160p-bound mRNAs shows pronounced non-linear correlation with average translational repression threshold in polysome assay. (B) Average TPI (equates to codon autocorrelation) and CAI (equates to codon frequency above genomic average) at mRNA level across all amino acids, for rising TSC (equivalent to degree of mRNA distribution shift in polysome gradients) threshold. P-values indicate significance by comparison with 100 000 random genomic pickings of equally sized groups. TPI and CAI were calculated for the whole set, respectively, up to the indicated TSC threshold.
RAP followed by RT-qPCR enables quantitative detection of different tRNAs at the ribosome
The results described earlier in the text indicate that Scp160p might recruit specific mRNAs with unique coding optimization to ribosomes. Alternatively, it would be conceivable that Scp160p is implicated in the correct execution of autocorrelation itself, i.e. at its mechanistic basis. Because both scenarios are in line with our bioinformatical metastudies (see earlier in the text), we designed an experimental setup to distinguish between both models. We hypothesized that if the protein functioned by recruiting mRNAs of biased codon composition to ribosomes, or ensuring correct tRNA ‘re-use’ for these, loss of Scp160p should decrease the ribosome occupancy of such tRNAs that have a potential for pronounced recycling on target mRNAs (14,17,43). Successful measurement of such changes would speak in favour of the substrate channelling hypothesis, which holds that re-use of discharged tRNAs and their circulation near the ribosome boosts translational efficiency (15,28).
Therefore, we established a protocol for detection of tRNAs co-precipitating with ribosomes to verify in vivo that the translational decrease observed in mRNAs on Scp160p depletion is interconnected with tRNA autocorrelation, as predicted by bioinformatics. Translating ribosomes were arrested by cycloheximide before applying RAP. The tRNAs co-precipitating with ribosomes were reverse transcribed and quantitated by qPCR. To cover a broad spectrum of different tRNA classes, we included one ‘synonym-poor’ tRNA (that decodes only one codon) and one ‘synonym-rich’ tRNA (that decodes more than one codon), for four different amino acids (Ser, Leu, Thr, Val), respectively. For each amino acid, certain combinations of tRNAs using ‘rare’ [low relative synonymous codon usage (RSCU), i.e. low genomic frequency] versus ‘popular’ (high RSCU, i.e. high genomic frequency) codons with or without capacity to recognize different synonymous iso-decoding codons were chosen. Rare or popular is defined using the RSCU (16,18). The RSCU was computed and the highest and the lowest taken as popular or non-popular, respectively. Figure 6A sums up the array of tRNA gene classes investigated, together with the number and popularity of synonymous codons. For each homologous tRNA class, one representative member was used as the eponym for the whole block used in multiple sequence alignments to construct qPCR primers (see ‘Materials and Methods’ section). tS_E and tL_L represent high-copy gene families for tRNAs (Ser2, Leu1) recognizing a popular codon, whereas tS_F and tL_G denote low-copy gene families encoding tRNAs (Ser4, Leu3) each recognizing several unpopular codons. tT_J and tV_M represent high-copy gene families for tRNAs (Thr1, Val1) recognizing several popular codons, whereas tT_K and tV_D denote low-copy gene families encoding tRNAs (Thr3, Val2) recognizing an unpopular codon. Thus, all possible combinations were assessed to distinguish between effects due to codon popularity and/or synonym-richness.
Figure 6.
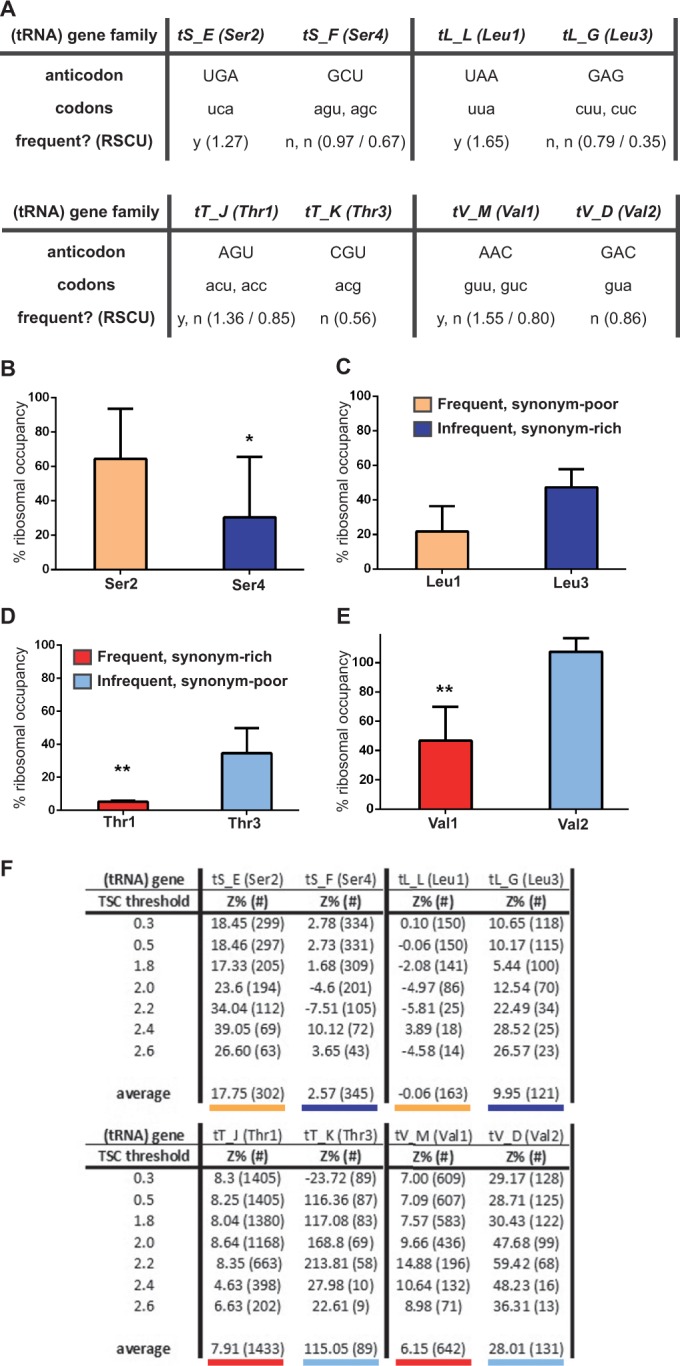
The tRNAs recognizing codons less autocorrelated in Scp160p target mRNAs are more depleted from ribosomes following Scp160 depletion. This effect in independent both of the number of synonymous codons, or their frequency in the genome. (A) The tRNA gene family names, common tRNA names, anticodon used, codons recognized and RSCU for the candidate pairs contrasting within the four amino acids experimentally assessed. (B–E) Residual ribocc of tRNA pairs for amino acids on Scp160p depletion, displayed as percentage of wild-type levels. The standard deviation between at least two biological replicates with two technical replicates each is indicated, while *, P <0.1 and **, P <0.01 for significance of independent average values between knockdown and wild-type (above columns). (B) Serine, comparing Ser2 and Ser4: tRNAs recognizing only one frequent (orange) versus several infrequent (dark blue) codons. (C) Leucine, comparing Leu1 and Leu3: tRNAs recognizing only one frequent versus several infrequent codons. (D) Threonine, comparing Thr1 and Thr3: tRNAs recognizing several frequent (red) versus only one infrequent (light blue) codon. (E) Valine, comparing Val1 and Val2: tRNAs recognizing several frequent versus only one infrequent codon. (F) Z-score percentage (equals to codon autocorrelation at the tRNA level) in mRNA target subgroups ordered by TSC (equivalent to degree of mRNA distribution shift in polysome gradients) threshold. Z-scores were calculated individually for each indicated set up to the indicated TSC threshold. All tRNAs analyzed are shown. For enhanced clarity, underlinings of average Z-score values correspond to color-coding of the respective tRNA species analyzed experimentally in the above figures, with red versus blue shades for popular or unpopular codons, and bold versus faint shades for synonym-rich or synonym-poor tRNAs.
As will be elaborated later in the text, in contrast to synonym-richness, just like codon popularity, codon clustering cannot be inferred from the tRNA, but has to be computed from the usage in mRNA targets of interest. As codon clustering, in contrast to codon popularity, is a ‘local’ effect, the respective values stem from a subset of mRNAs affected by SCP160 depletion (Supplementary Table S4) and were amended at a later point in the study (Figure 6F). This setup made the investigation blind towards bias by expectations.
As a preliminary experiment, yeast cells expressing a temperature-sensitive mutant of the gamma subunit of translational elongation factor eEF1B [yef3-F650S; (17,27)] was grown at restrictive temperature and the ribocc of tRNAs from this mutant was compared with that of wild-type cells to assess the impact of impaired translation elongation on ribocc. As a general pattern, we found a more drastic decrease in ribocc for popular codon-reading tRNAs than for unpopular ones within a given amino acid. At the same time, ribocc across all investigated amino acids dropped uniformly compared with wild-type as shown for serine and leucine (Supplementary Figure S4A and B).
To ascertain that this is a general trend, we also quantitated ribosome-associated tRNAs coding for two amino acids from our set, applying an alternative translational stress condition, amino acid depletion in the growth medium. Again, this led to a uniformly higher drop in ribosome association of tRNAs recognizing popular codons as opposed to unpopular codons for threonine and valine (Supplementary Figure S4C and D). As the most highly expressed genes are also uniformly biased towards popular codons (18), this experiment confirms that global translational deficits are mirrored in our tRNA-RAP measurements.
Ribosomal occupancy of most synonym-rich tRNAs decreases on Scp160p depletion, regardless of popularity of cognate codons
Next, we applied the method to monitor tRNA ribocc to Scp160p-depleted cells. Again, in comparison with wild-type the level of both assayed tRNA species of a given amino acid dropped on depletion of Scp160p (Figure 6). Yet, the pattern was different than the one observed for yef3-F650S cells. For three of the four amino acids investigated (serine, threonine, valine), loss of Scp160p more drastically decreased ribocc of ‘synonym-rich’ tRNAs recognizing several synonymous codons. Conversely, ‘synonym-poor’ tRNAs read by only one synonymous codon were still co-precipitating with ribosomes at higher levels with no significant deviation from wild-type.
This effect was independent of the popularity of the codons recognized by said tRNAs, in contrast to the pattern observed in the yef3-F650S mutant. For instance, for serine, the unpopular synonym-rich Ser4 species was significantly (P < 0.1) more depleted (30.4 ± 3.3% residual occupancy) than the popular synonym-poor Ser2 species (64.3 ± 18.7% residual occupancy, Figure 6B). In contrast, for valine, the popular synonym-rich Val1 species showed a highly significant (P < 0.01) greater depletion (46.8 ± 1.2% residual occupancy) than the unpopular synonym-poor Val2 species (107.2 ± 7.9% residual occupancy, Figure 6E).
In summary, following Scp160p depletion, the decrease in ribocc of synonym-rich tRNAs was much more pronounced than the respective decrease for synonym-poor tRNAs. This unique pattern of tRNA depletion from the ribosome cannot be explained by a general translational depletion and speaks in favour of a specialized role for Scp160p in translational efficiency.
Scp160p can boost tRNA ‘re-use’ independent of mRNA optimization at the codon level
Intuitively, one might speculate that synonym-rich tRNAs might have a higher chance to ‘recycle’ and thus would be more prone to be used in ‘codon clusters’. This might lead to the premature assumption that the ribocc drop of synonym-rich tRNAs on knockdown means that Scp160p has a function in autocorrelation itself. However, no such clear-cut prediction of codon clustering from synonym-richness is possible, as autocorrelation is an evolutionarily entrenched effect (17,19,20).
To elucidate the basis of how Scp160p is beneficial to optimal translation that is not caused by use of ‘popular’ codons, we thus had to improve the analysis at the amino acid average level (Supplementary Table S8) and performed a similar analysis at the resolution of individual tRNAs (Figure 6F) to find out which tRNAs are the most and least dependent on coding-optimization by ‘codon clustering’.
When calculating the autocorrelation (‘codon clustering’) of mRNA subgroups that are to varying degrees translationally affected by Scp160p depletion at a tRNA resolution, it turned out that the tRNAs ‘least impaired’ in ribocc are ‘most codon-clustering’ within the mRNA groups specifically affected by Scp160p depletion, i.e. those with a high TSC. Conversely, the tRNAs ‘most affected’ by Scp160p depletion are in contrast the ones that are the ‘least codon-clustering’ in the mRNA groups most affected—despite a general tendency of these targets to be ‘re-use’—optimized at within-amino acid resolution, presumably by virtue of the other tRNA (Supplementary Table S8). At the same time, the setup of tRNA classes assessed rules out contributions imparted by other effects, i.e. codon popularity or synonym-richness of tRNAs.
Thus, the observed ribocc changes of tRNAs cannot be explained by impaired recruitment of clustering-biased mRNAs to the ribosome. This finding also immediately rejects another hypothesis, namely, that Scp160p would be involved in the correct execution of robust autocorrelation. In contrast, it indicates that Scp160p could provide the same benefits in translational fitness as autocorrelation optimization does, i.e. by preventing used tRNAs’ diffusion or aiding used tRNAs’ recycling. Furthermore, autocorrelation seems to be a robust independent mechanism, which, however, relies on an intact molecular setup of the ribosome with accessory beneficial factors, of which Scp160p might be a part.
DISCUSSION
We have shown that Scp160p is a yeast protein determining the translational state of several target mRNAs, and that at least for one such mRNA, a shift towards polysome fractions on knockdown corresponds to less protein being produced. Ribosomal affinity purification and detection of other target mRNAs indicate that this may be a general pattern.
These results shed new light on prior observations, such as the indirect implication of Scp160p in the control of ploidy, chromatin remodelling or cell wall integrity. The most parsimonious interpretation is that decreased translation of Scp160p’s mRNA targets with ontologies fitting these processes is the basis of its indirect influence in these fields, without direct physical interactions.
We found a correlation between the translational shift of target mRNAs and their coding potential for improved translation by tRNA autocorrelation, which led to a further investigation of the link between Scp160p function, translational fitness and tRNA recycling.
Based on the hypothesis that ordering of synonymous codons in a transcript prevents tRNA species change, presumably increasing ribosomal economy and thus the mRNA’s translational fitness (17), and on our findings that Scp160p mRNA targets are enriched for mRNAs with coding optimization, we initially hypothesized that the preferential recruitment of ontologically important codon-optimized mRNA targets to the ribosome depends on the presence of Scp160p. This might especially be the case in times of ample resources and exponential growth, as Scp160p is recruited to the ribosome by the adapter protein Asc1p/RACK1 (12,21), which itself is found to associate with the ribosome only in exponentially growing cells. An alternative model could be a direct involvement of Scp160p in tRNA recycling (26,28).
To address the question which of these models is valid, we developed a protocol to detect tRNAs that co-precipitate with translating ribosomes and successfully measured global depletion of tRNAs from ribosomes in elongation factor mutants and amino acid starvation. To our knowledge, this is the first PCR-based quantitative measurement of tRNAs on the translating ribosome using non-spectrometric methods. When we monitored tRNA ribocc in Scp160p-depleted cells, it turned out that the tRNA species the most depleted from the ribosome are the ones that are less optimized for tRNA recycling by codon order autocorrelation within Scp160p target mRNAs. This pattern was independent of the number of synonymous codons, or their frequency of use. A likely interpretation of these findings is that Scp160p has a beneficial effect on translational fitness that is similar to, but independent of, tRNA autocorrelation at the mRNA coding level.
From the observation that tRNAs that are more optimized for re-use were less depleted from ribosomes, it can be interpreted that autocorrelation is robust and deterministic. However, autocorrelation relies on synergism with independent accessory factors that maximize its effect. Our results indicate that Scp160p might constitute one of these factors. Scp160p-associated mRNAs are already partially optimized for autocorrelation of codons (Supplementary Table S7), which boosts efficiency synergistic with but largely independent of Scp160p. Loss of Scp160p might therefore decrease translation of target mRNAs due to impaired autocorrelation-independent tRNA recycling. Scp160p’s mode of action, just as codon autocorrelation itself, may make use of molecular crowding phenomena. Together with the proposed substrate channelling hypothesis for the ribosome (17,26,28), a role for a tight association between ribosomes, tRNAs and their aminoacyl synthetases as tRNA recycling factors has been proposed by physical modelling to be the basis of autocorrelation [Cannarrozzi, 2010 (42)], and supported by experimental evidence (23,30). We hypothesize that Scp160p may exert its benefits on translation by either consolidating these associations or preventing the dissociation of tRNAs from the ribosome (Figure 7). Our quantitation of tRNAs at the ribosome in the presence and absence of Scp160p in combination with the bioinformatics analysis of the codon composition of mRNAs rules out a third hypothesis that Scp160p recruits only those mRNAs to ribosomes, which are optimized for translational efficiency by tRNA re-use due to codon autocorrelation. This is because, due to the general optimization of Scp160p target mRNAs for tRNA re-use, abrogation of hypothetical ribosomal recruitment caused by Scp160p depletion would be expected to more drastically affect tRNA species thus optimized.
Figure 7.
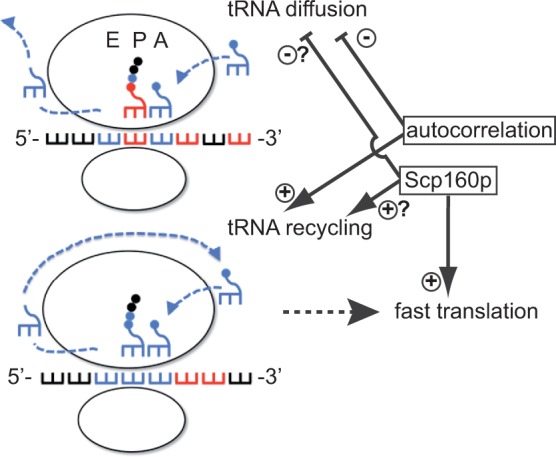
Proposed model for Scp160p function in translation. See ‘Discussion’ section for further explanation.
Our data provide corroborating evidence for two hypothetical models of cooperation between RBPs and the ribosome. The RNA operon model (24,32) holds that shifting coalitions of RBPs’ interactions might be the key to understanding the complex networks of post-transcriptional regulation phenomena. Scp160p has already been implicated in one such network, the SESA (SMY2/EAP1/SCP160/ASC1) network, in the function of a translational activator (11,25). It binds ribosomes via its interaction partner Asc1p, whose mammalian homolog RACK1 binds activated kinases and might integrate growth signalling cues (27,33). In an unrelated second model, efficiency of translation in vivo is explained by the observation of active aminoacyl tRNA synthetase recruitment to translating ribosomes by interaction with the ribosome itself or ribosome-associated proteins (29,30). Along these lines, Scp160p with its multitude of RNA-binding domains could serve as a facilitator of tRNA recycling, possibly by preventing diffusion from an associated ribosome, or ‘catching’ tRNAs given off by a ribosome that has advanced further on the same transcript. Its adapter Asc1p on the other hand has been shown to interact with aminoacyl tRNA synthetases, although these recharge tRNAs different from those assayed by us (17,18,30,31). Both proteins could function hand-in-hand to facilitate fast tRNA recycling, Asc1 by recruiting aminoacyl-tRNA synthetases and Scp160p by ensuring that empty tRNAs are not lost from one translating ribosome to the next. In this scenario, Scp160p might also aid mRNAs not-yet perfectly composed to make the most use of autocorrelation because evolutionary entrenchment has not yet succeeded in consolidating their optimal coding.
Conclusively, our observations add weight to an emerging view of the ribosome governed by the laws of supply and demand (7,44), possibly due to kinetic effects imposed by the substrate channelling (25,28) and molecular crowding phenomena (21,34). Our work also describes the first in vivo method to prove the predictions of the bioinformatical parameter of ‘tRNA Pairing Index’ (13,17) both on the mRNA and tRNA level, reinforcing it as a novel predictor of translational fitness.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Central funds of the research cluster Munich Center for Integrated Protein Science (CIPSM); Deutsche Forschungsgemeinschaft [SFB646 to R.-P.J. and P.C.]. Funding for open access charge: University of Tübingen, Interfaculty Institute of Biochemistry.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Matthias Seedorf for providing the anti-Scp160 antibody, to Terry Goss Kinzy for providing plasmid pYEF3(F650S), to Enrique Herrero for providing plasmid pCM182 and to Ingrid Fetka for generating CCW14-myc9 strains. They thank Eli van der Sluis for introducing them to the Biocomp gradient fractionation systems. Special thanks go to T. Strittmatter, J. Ristau, J. Overbeck and B. Singer-Krüger for helpful discussion and suggestions during this project.
REFERENCES
- 1.Hogan DJ, Riordan DP, Gerber AP, Herschlag D, Brown PO. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mansfield KD, Keene JD. The ribonome: a dominant force in co-ordinating gene expression. Biol. Cell. 2009;101:169–181. doi: 10.1042/BC20080055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dodson RE, Shapiro DJ. Vigilin, a ubiquitous protein with 14 K homology domains, is the estrogen-inducible vitellogenin mRNA 3′-untranslated region-binding protein. J. Biol. Chem. 1997;272:12249–12252. doi: 10.1074/jbc.272.19.12249. [DOI] [PubMed] [Google Scholar]
- 4.Weber V, Wernitznig A, Hager G, Harata M, Frank P, Wintersberger U. Purification and nucleic-acid-binding properties of a Saccharomyces cerevisiae protein involved in the control of ploidy. Eur. J. Biochem. 1997;249:309–317. doi: 10.1111/j.1432-1033.1997.00309.x. [DOI] [PubMed] [Google Scholar]
- 5.Brykailo MA, Corbett AH, Fridovich-Keil JL. Functional overlap between conserved and diverged KH domains in Saccharomyces cerevisiae SCP160. Nucleic Acids Res. 2007;35:1108–1118. doi: 10.1093/nar/gkl1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li A-M, Watson A, Fridovich-Keil JL. Scp160p associates with specific mRNAs in yeast. Nucleic Acids Res. 2003;31:1830–1837. doi: 10.1093/nar/gkg284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wintersberger U, Kühne C, Karwan A. Scp160p, a new yeast protein associated with the nuclear membrane and the endoplasmic reticulum, is necessary for maintenance of exact ploidy. Yeast. 1995;11:929–944. doi: 10.1002/yea.320111004. [DOI] [PubMed] [Google Scholar]
- 8.Guo M, Aston C, Burchett SA, Dyke C, Fields S, Rajarao SJR, Uetz P, Wang Y, Young K, Dohlman HG. The yeast G protein alpha subunit Gpa1 transmits a signal through an RNA binding effector protein Scp160. Mol. Cell. 2003;12:517–524. doi: 10.1016/s1097-2765(03)00307-1. [DOI] [PubMed] [Google Scholar]
- 9.Gelin-Licht R, Paliwal S, Conlon P, Levchenko A, Gerst JE. Scp160-dependent mRNA trafficking mediates pheromone gradient sensing and chemotropism in yeast. Cell Rep. 2012;1:483–494. doi: 10.1016/j.celrep.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marsellach F-X, Huertas D, Azorín F. The multi-KH domain protein of Saccharomyces cerevisiae Scp160p contributes to the regulation of telomeric silencing. J. Biol. Chem. 2006;281:18227–18235. doi: 10.1074/jbc.M601671200. [DOI] [PubMed] [Google Scholar]
- 11.Sezen B, Seedorf M, Schiebel E. The SESA network links duplication of the yeast centrosome with the protein translation machinery. Genes Dev. 2009;23:1559–1570. doi: 10.1101/gad.524209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baum S, Bittins M, Frey S, Seedorf M. Asc1p, a WD40-domain containing adaptor protein, is required for the interaction of the RNA-binding protein Scp160p with polysomes. Biochem. J. 2004;380:823–830. doi: 10.1042/BJ20031962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frey S, Pool M, Seedorf M. Scp160p, an RNA-binding, polysome-associated protein, localizes to the endoplasmic reticulum of Saccharomyces cerevisiae in a microtubule-dependent manner. J. Biol. Chem. 2001;276:15905–15912. doi: 10.1074/jbc.M009430200. [DOI] [PubMed] [Google Scholar]
- 14.Kruse C, Grünweller A, Willkomm DK, Pfeiffer T, Hartmann RK, Müller PK. tRNA is entrapped in similar, but distinct, nuclear and cytoplasmic ribonucleoprotein complexes, both of which contain vigilin and elongation factor 1 alpha. Biochem J. 1998;329:615–621. doi: 10.1042/bj3290615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozak M. Regulation of translation in eukaryotic systems. Annu. Rev. Cell Biol. 1992;8:197–225. doi: 10.1146/annurev.cb.08.110192.001213. [DOI] [PubMed] [Google Scholar]
- 16.Plotkin JB, Kudla G. Synonymous but not the same: the causes and consequences of codon bias. Nat. Rev. Genet. 2011;12:32–42. doi: 10.1038/nrg2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cannarrozzi G, Schraudolph NN, Faty M, Rohr, Von P, Friberg MT, Roth AC, Gonnet P, Gonnet G, Barral Y. A role for codon order in translation dynamics. Cell. 2010;141:355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 18.Sharp PM, Li W-H. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Percudani RR, Pavesi AA, Ottonello SS. Transfer RNA gene redundancy and translational selection in Saccharomyces cerevisiae. J. Mol. Biol. 1997;268:322–330. doi: 10.1006/jmbi.1997.0942. [DOI] [PubMed] [Google Scholar]
- 20.Charneski CA, Hurst LD. Positively charged residues are the major determinants of ribosomal velocity. PLoS Biol. 2013;11:e1001508. doi: 10.1371/journal.pbio.1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput. Biol. 2010;6:e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negrutskii BS, Stapulionis R, Deutscher MP. Supramolecular organization of the mammalian translation system. Proc. Natl Acad. Sci. USA. 1994;91:964–968. doi: 10.1073/pnas.91.3.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Böhl F, Kruse C, Frank A, Ferring D, Jansen RP. She2p, a novel RNA-binding protein tethers ASH1 mRNA to the Myo4p myosin motor via She3p. EMBO J. 2000;19:5514–5524. doi: 10.1093/emboj/19.20.5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halbeisen RE, Scherrer T, Gerber AP. Affinity purification of ribosomes to access the translatome. Methods. 2009;48:306–310. doi: 10.1016/j.ymeth.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Bellí G, Garí E, Piedrafita L, Aldea M, Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic Acids Res. 1998;26:942–947. doi: 10.1093/nar/26.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stapulionis R, Deutscher MP. A channeled tRNA cycle during mammalian protein synthesis. Proc. Natl Acad. Sci. USA. 1995;92:7158–7161. doi: 10.1073/pnas.92.16.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anand M, Chakraburtty K, Marton MJ, Hinnebusch AG, Kinzy TG. Functional interactions between yeast translation eukaryotic elongation factor (eEF) 1A and eEF3. J. Biol. Chem. 2003;278:6985–6991. doi: 10.1074/jbc.M209224200. [DOI] [PubMed] [Google Scholar]
- 28.Negrutskii BS, Deutscher MP. Channeling of aminoacyl-tRNA for protein synthesis in vivo. Proc. Natl Acad. Sci. USA. 1991;88:4991–4995. doi: 10.1073/pnas.88.11.4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gonnet GH, Hallett MT, Korostensky C, Bernardin L. Darwin v. 2.0: an interpreted computer language for the biosciences. Bioinformatics. 2000;16:101–103. doi: 10.1093/bioinformatics/16.2.101. [DOI] [PubMed] [Google Scholar]
- 30.David A, Netzer N, Strader MB, Das SR, Chen CY, Gibbs J, Pierre P, Bennink JR, Yewdell JW. RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 2011;286:20688–20700. doi: 10.1074/jbc.M110.209452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Friberg MT, Gonnet P, Barral Y, Schraudolph NN, Gonnet GH. Algorithms in Bioinformatics, Lecture Notes in Computer Science. Vol. 4175. Berlin, Heidelberg: Springer; 2006. Measures of Codon Bias in Yeast, the tRNA Pairing Index and Possible DNA Repair Mechanisms; pp. 1–11. [Google Scholar]
- 32.Keene JD. Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome. Proc. Natl Acad. Sci. USA. 2001;98:7018–7024. doi: 10.1073/pnas.111145598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun. Signal. 2011;9:22. doi: 10.1186/1478-811X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, et al. The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res. 2004;32:5539–5545. doi: 10.1093/nar/gkh894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shepard KA, Gerber AP, Jambhekar A, Takizawa PA, Brown PO, Herschlag D, DeRisi JL, Vale RD. Widespread cytoplasmic mRNA transport in yeast: identification of 22 bud-localized transcripts using DNA microarray analysis. Proc. Natl Acad. Sci. USA. 2003;100:11429–11434. doi: 10.1073/pnas.2033246100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasegawa Y, Irie K, Gerber AP. Distinct roles for Khd1p in the localization and expression of bud-localized mRNAs in yeast. RNA. 2008;14:2333–2347. doi: 10.1261/rna.1016508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moukadiri I, Armero J, Abad A, Sentandreu R, Zueco J. Identification of a mannoprotein present in the inner layer of the cell wall of Saccharomyces cerevisiae. J. Bacteriol. 1997;179:2154–2162. doi: 10.1128/jb.179.7.2154-2162.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roy A, Lu CF, Marykwas DL, Lipke PN, Kurjan J. The AGA1 product is involved in cell surface attachment of the Saccharomyces cerevisiae cell adhesion glycoprotein a-agglutinin. Mol. Cell. Biol. 1991;11:4196–4206. doi: 10.1128/mcb.11.8.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin QY, de Groot PWJ, Dekker HL, de Jong L, Klis FM, de Koster CG. Comprehensive proteomic analysis of Saccharomyces cerevisiae cell walls: identification of proteins covalently attached via glycosylphosphatidylinositol remnants or mild alkali-sensitive linkages. J. Biol. Chem. 2005;280:20894–20901. doi: 10.1074/jbc.M500334200. [DOI] [PubMed] [Google Scholar]
- 40.O'Rourke SM, Herskowitz I. A third osmosensing branch in Saccharomyces cerevisiae requires the Msb2 protein and functions in parallel with the Sho1 branch. Mol. Cell. Biol. 2002;22:4739–4749. doi: 10.1128/MCB.22.13.4739-4749.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eisenhaber B, Schneider G, Wildpaner M, Eisenhaber F. A sensitive predictor for potential GPI lipid modification sites in fungal protein sequences and its application to genome-wide studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae and Schizosaccharomyces pombe. J. Mol. Biol. 2004;337:243–253. doi: 10.1016/j.jmb.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 42.Ciandrini L, Stansfield I, Romano MC. Ribosome traffic on mRNAs maps to gene ontology: genome-wide quantification of translation initiation rates and polysome size regulation. PLoS Comput. Biol. 2013;9:e1002866. doi: 10.1371/journal.pcbi.1002866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruse C, Grünweller A, Notbohm H, Kügler S, Purschke WG, Müller PK. Evidence for a novel cytoplasmic tRNA-protein complex containing the KH-multidomain protein vigilin. Biochem. J. 1996;320:247–252. doi: 10.1042/bj3200247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pechmann S, Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2012;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.