

Abstract
Melanocortin 4 receptors (MC4R) are hypothesized to mediate the central nervous system actions of leptin to enhance the satiety effects of cholecystokinin (CCK). To further elucidate this mechanism, we confirmed that peripheral administration of CCK-8 is less effective in producing this effect in MC4R-deficient mice (MC4R−/−). Whereas intraperitoneal (ip) CCK-8 at 0.75 nmol/kg lean body mass (lbm) suppressed food intake in wild-type mice, CCK-8 doses of 7.5 nmol/kg lbm were required to attenuate food intake in MC4R−/− mice. To determine whether melanocortin signaling in the hypothalamic paraventricular nucleus (PVN) participates in regulating this CCK satiety response, we administered the MC3/MC4R antagonist, SHU9119, into the PVN of rats before ip CCK-8 administration. PVN administration of SHU9119 attenuated the ability of CCK-8 to reduce 30-min food intake by 20%. To determine whether MC4R are expressed by PVN neurons that project directly to hindbrain nuclei involved in the satiety response to ip CCK-8, the retrograde tracer fluorescent cholera toxin subunit B was injected into the nucleus tractus solitarius (NTS) of the hindbrain. After 4 days, labeled PVN neurons were collected by laser capture microdissection and found to express MC4R mRNA by quantitative RT-PCR analysis. These data provide evidence for a neuroanatomical link between hypothalamic melanocortin signaling in the PVN and NTS neurons that regulate food intake. These findings highlight the contribution of melanocortin signaling in the PVN toward regulating the satiety effects of CCK-8 while acknowledging that melanocortin-dependent pathways in other brain regions and/or melanocortin-independent mechanisms are also important in this mechanism.
Keywords: paraventricular nucleus, retrograde tracer, cholecystokinin
the central nervous system (CNS) melanocortin system plays a critical role in the regulation of energy homeostasis. Indeed, effects of leptin, glucose, and many other humoral inputs on food intake and autonomic function are dependent on activation of melanocortin neurons situated in the arcuate nucleus (ARC). Neuronal signaling mediated by melanocortins and their receptors have effects similar to those produced by leptin on food intake and body weight, and intact melanocortin signaling is required for leptin-mediated food intake suppression (12, 43). Activation of central melanocortin receptors (MC3R and MC4R) by melanotan II (MTII), a synthetic surrogate for the endogenous ligand α-melanocotye-stimulating hormone (α-MSH), reduces meal size in rodents (4, 49, 53) and evidence suggests that these effects are largely due to activation of the MC4R (3). Conversely, genetic ablation of melanocortin signaling in MC4R-deficient mice (MC4R−/−) results in hyperphagia and increased meal size (3), obesity (23), and inability to respond to the melanocortin MC3/MC4R agonist MTII (28). Blockade of central melanocortin signaling by intracerebroventricular administration of either the endogenous melanocortin receptor antagonist agouti-related peptide (AGRP) or the synthetic MC3/MC4 antagonist SHU9119 produces hyperphagia and obesity (19, 35, 38) and attenuates the anorexic response to leptin (14, 43). Taken together, the evidence supports the hypothesis that the CNS melanocortin system is a major contributor to the regulation of food intake.
One mechanism whereby melanocortins may attenuate meal size is by increasing the CNS sensitivity to gastrointestinal satiety signals such as cholecystokinin (CCK) that participate in meal termination. CCK plays a physiological role to limit the size of individual meals by activating vagal pathways from the GI tract to the hindbrain (20, 46, 53). MC4R−/− mice do not reduce chow intake following intraperitoneal (ip) injections of CCK-8 (20) and, furthermore, intracerebroventricular administration of the MC3R/MC4R antagonist SHU9119 into either the third or fourth cerebral ventricles of normal rats attenuates the ability of peripheral CCK-8 to reduce food intake (20). These findings suggest that central melanocortin signaling is required for an intact satiety response to CCK, but do not identify the nuclei containing the melanocortin-sensitive neurons that mediate this effect.
Among the many brain nuclei that contain MC4 receptors, the hypothalamic paraventricular nucleus (PVN) is especially well-positioned to mediate melanocortin regulation of CCK responsiveness. The PVN is richly supplied by axons that contain the endogenous melanocortin ligand, α-MSH (32), and PVN neuronal cell bodies express MC4 receptors (31). Moreover, both the endogenous MC3/MC4R agonist α-MSH and the synthetic MC3/MC4R agonist MTII inhibit food intake when injected into this nucleus (51).
Delineating the specific neuronal subsets that transduce input from melanocortins into heightened sensitivity to satiety signals will allow an improved understanding of energy homeostasis circuitry and may shed light on the pathogenesis of common forms of obesity. Toward this end, we sought to extend previous evidence that CNS melanocortin signaling is required for an intact satiety response induced by peripheral CCK in mice and rats. First, we compared the dose-response effects of peripheral CCK-8 on food intake in wild-type and MC4R−/− mice, which lack endogenous MC4R signaling. To test the hypothesis that endogenous melanocortin signaling in the PVN contributes to the intact satiety response to CCK, we measured the effects of PVN administration of the MC3/MC4R antagonist SHU9119 in combination with peripheral administration of CCK-8 on food intake in rats. To address the question of whether MC4R-expressing neurons in the PVN project directly to hindbrain nuclei sensitive to peripheral CCK, we injected a retrograde neuronal tracer, fluorescent cholera toxin subunit B (CTB), into the nucleus tractus solitarius (NTS) of rats to label parvocellular PVN (pPVN) neurons that project directly to the NTS. We then used laser capture microdissection (LCM) in combination with quantitative RT-PCR to determine whether MC4R mRNA is expressed by pPVN neurons that project to the NTS.
MATERIALS AND METHODS
Experimental animals.
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the VA Puget Sound Medical Center and the University of Washington. Adult male Wistar rats (272–417 g) from Charles River Laboratories (Wilmington, MA) were used in the rat studies. For mouse studies, adult male and female MC4R null mice (MC4R−/−) and wild-type (MC4R+/+) mice on a mixed B6/129 background from the original colony described by Huszar and colleagues (23) (Millenium Pharmaceuticals, Cambridge, MA) were bred as heterozygotes and genotyped from tailsnips at 5–6 wk of age (20.7–50.5 g). All animals were housed individually in Plexiglas cages in a temperature-controlled room (22 ± 2°C) under a 12:12-h light-dark cycle. Rats and mice were adapted to lights-off at 1500 and 1300, respectively, and were fed ad libitum a standard rat chow and mouse chow diet unless otherwise noted. Water was freely available.
MC4R null mice.
Age- and sex-matched mice deprived of food for 18 h received an injection of CCK-8 [0, 0.75, 2.5, and 7.5 nmol/kg of lean body mass (lbm) in a final volume of 0.2 ml] or vehicle (0.1% BSA) in randomized order immediately before the start of the dark cycle. Animals were given at least 72 h between injections. LBM was determined before the experiment in all mice using the Echo MRI whole body composition analyzer and Magnetic Resonance Spectroscopy (MRS) by the Body Composition and Energy Expenditure Core at the University of Washington Clinical Nutrition Research Unit. The average percentage fat mass for the MC4R+/+ and MC4R−/− mice was 14.8 ± 0.8 and 31.8 ± 1.6 g, respectively. The average percentage lean mass in the MC4R+/+ and MC4R−/− mice was 78.1 ± 0.9 and 64.2 ± 1.5 g, respectively. As in previous studies, here we opted not to adjust dose based on body weight when comparing obese and lean animals (30), judging that lean mass is strongly correlated with body water content and thus adjusting the CCK-8 dose to lean mass is more apt to achieve comparable blood levels.
Surgery and stereotaxic procedures.
The detailed procedure for implantation of guide cannulas into specific rat brain sites has been described previously (45). Briefly, animals were anesthetized with a cocktail of ketamine (71.4 mg/kg), xylazine (3.57 mg/kg), and acepromazine (1.1 mg/kg) (8). A 26-gauge bilateral guide cannula (Plastics One, Roanoke, VA) was stereotaxically implanted 1 mm dorsal to the PVN (6.9 mm anterior to the interaural line, 0.4 mm lateral to the midline, and 7.3 mm ventral to the skull surface); for 4V cannulation, the coordinates were 3.5 mm posterior to the interaural line, 1.4 mm lateral to the midline, and 6.2 mm ventral to the skull surface.
Retrograde labeling.
Animals received unilateral injections of a fluorescent retrograde tracer, CTB, conjugated to Alexa 488 fluorochrome (Invitrogen, Carlsbad, CA), through a cannula directed toward the medial NTS (cmNTS). The stereotaxic coordinates for the cmNTS injections were −5.5 mm posterior to interaural line, +0.7 mm to cerebral vein, and −7.1 mm ventral to surface of skull (33). A 0.5 μl volume of 0.2% CTB in saline was injected via a 33-gauge injector connected by polyethylene 20 tubing to a 10-μl Hamilton syringe. Injections were completed over 30 s; the injector was held stationary for another 30 s and slowly removed followed by removal of the guide cannula. Four days later, animals received a lethal dose of ketamine (214.3 mg/kg), xylazine (10.7 mg/kg), and acepromazine (3.3 mg/kg) cocktail before start of the dark cycle (8). Brains were collected, snap-frozen for 10–15 s in isopentane, and stored at −80°C.
PVN injections.
After being food deprived for 6 h before the start of the dark cycle, rats were injected through indwelling PVN cannulae as described above. The 33-gauge injector was held stationary at the end of the injection for ∼30 s and the injection volume was 0.5 μl/side. Animals were immediately returned to their cages at the conclusion of each injection protocol.
4V injections.
After being food deprived for 6 h before the start of the dark cycle, rats were injected through an indwelling unilateral 4V cannula as described above. The 33-gauge injector was held stationary at the end of the injection for ∼30 s and the injection volume was 2.0 μl. Animals were immediately returned to their cages at the conclusion of each injection protocol.
Experimental treatments.
SHU9119 (Bachem/Peninsula, Belmont, CA; 0.08 nmol/0.5 μl) was solubilized in saline for infusion into the PVN 1 h before the start of the dark cycle. CCK-8 (Bachem/Peninsula) was dissolved in saline with 0.1% BSA and was given as an ip injection (1 ml/kg injection volume) immediately before the start of the dark cycle, when the animals normally begin eating and when CCK-8 has its most potent effect to reduce food intake. The four treatments groups for studies in rats were as follows: 1) PVN SHU9119 vehicle (saline) administered 1 h before a dose of CCK-8 vehicle (0.1% BSA, saline), 2) PVN SHU9119 vehicle 1 h before CCK-8, 3) PVN SHU9119 1 h before CCK-8 vehicle, and 4) PVN SHU9119 1 h before CCK-8. Doses of CCK-8 and SHU9119 were selected on the basis of preliminary studies from our laboratory and published reports (10, 50, 51). We intentionally selected a dose of SHU9119 that was subthreshold for feeding effects to avoid the confounding effect of an independent feeding response from the SHU9119. For 4V administration of SHU9119, SHU9119 was administered in saline 75 min before the start of the dark cycle.
Food intake studies.
Food intake was measured 30 min after animals received injections of CCK-8 or vehicle and given access to food at the onset of the dark cycle as previously described (9). Examination of cage bottoms revealed negligible spillage, and there were no visually apparent differences in spillage within or between treatment groups. The dose of CCK-8 used in determining the effect of PVN administration of SHU9119 on CCK-8-elicited inhibition of food intake in rats was 1.30 nmol/kg.
Behavioral assessment of 4V cannula placement.
Assessment of 4V cannula placement was done with injections of 15 pmol/2.0 μl bombesin (Bachem/Peninsula) immediately before the start of the dark cycle in rats that were fasted for 6 h. All animals used in the subsequent analysis of the data reduced their food intake by at least 20% within 1 h, an established criterion for correct placement of these cannulas (2).
Histological verification of injection sites.
Brains were frozen by submerging for 10–15 s in isopentane and then covered with crushed dry ice. Coronal cryostat sections (60 μm) at the level of the ventral extent of the cannula track were mounted on microscope slides and dried 24 h before staining with Cresyl violet. A Nikon SMZ-U ×10 stereo microscope was used to locate the most ventral portion of the scar tract made by the injector needle within the PVN, defined as the injection site. Data from an animal were excluded when the injection site extended 0.3 mm beyond the PVN boundary.
A subset of animals (n = 5) that received unilateral CTB injections into the NTS was also analyzed to validate injection site location. Coronal cryostat sections (14 μm) at the level of the ventral extent of the cannula tract were mounted on microscope slides. Slides were analyzed with a Zeiss Axioplan fluorescence microscope and all injection sites were analyzed using a ×20 objective lens. Identification of anatomic landmarks was assisted by staining cell nuclei with Hoechst 33258 (Sigma, St. Louis, MO), which was added to the mounting medium and observed with a conventional DAPI filter set. CTB was clearly visible in the dorsal vagal complex of all five brains that were examined. As expected, CTB diffused throughout the dorsal vagal complex in all five of these brains and was not restricted solely to the NTS.
Laser capture microdissection and RT-PCR of cells in containing retrograde tracer.
Slide-mounted cryostat sections (from unperfused rat brains) were thawed briefly, followed by graded dehydration in consecutive 1-min immersions of 75, 95, and 100% (2×) ethanol and xylene (1×) for 5 min, then air-dried for 10 min. The sections were then visualized on the Arcturus AutoPix Fluorescent laser capture microdissection (LCM) System (Molecular Devices, Union City, CA). To obtain PVN cells for PCR measurement of MC4R mRNA, slide-mounted cryostat sections prepared from nonperfused brains that had previously been injected 4 days earlier with CTB retrograde tracer into the cmNTS were dehydrated and air-dried as described above. In the pPVN, ∼250 neurons that were labeled with Alexa-488-labeled CTB (cells that projected to the NTS) were collected from three to four anatomically matched coronal sections of the PVN from each rat. As a negative control, 250 cells were picked by LCM from the suprachiasmatic nucleus (SCN), a region that expresses low levels of oxytocin (24) and corticotropin-releasing hormone (CRH) as well as MC4 receptors (31). In addition, ∼250 neurons that did not contain fluorescent CTB were also collected from the pPVN. RNA was extracted from the captured cells using an Arcturus Picopure RNA Isolation Kit (Molecular Devices) followed by reverse transcription into cDNA using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). Analysis for MC4R, oxytocin, and CRH mRNA in the RNA extracts was performed by quantitative RT-PCR on an Applied Biosystems Prism 7000 Sequence Detection System. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA in each sample was measured as an internal standard. Each sample was measured in triplicate. Primers and probes were designed using Primer Express (version 2.0.0) from TaqMan (Applied Biosystems). The primer sequences used for RT-PCR were rat GAPDH forward primer: 5′-GCCAGCCTCGTCTCATAGACA-3′; rat GAPDH reverse pri mer: 5′-GTCCGATACGGCCAAATCC-3′; rat GAPDH probe primer: VIC-5′-ATGGTGAAGGTCGGTGTG-3′; rat oxytocin, forward: 5′-TGACCTCCGCCTGCTACATC-3′; rat oxytocin reverse: 5′-AGGGAAGACACTTGCGCATATC-3′; rat oxytocin probe primer: 6-carboxyfluorescein (FAM)-5′-CTGGGCGGCAAGAG-3′, rat melanocortin-4 receptor forward primer: 5′-CAACATGAAGGGCGCAATT-3′; rat melanocortin-4 receptor reverse primer: 5′-GCCCAGCAGACAACAAACACT-3′; rat melanocortin-4 receptor probe primer: FAM-5′-CCTTGACCATTCTGATTG-3′.
The probe and primer for rat CRH were acquired from Applied Biosystems (cat. no. Rn01462137_m1). PVN and SCN expression levels of MC4R, oxytocin, and CRH were normalized to GAPDH mRNA content.
The mRNA measurements in the SCN were a control to determine whether PCR data produced by LCM picking of the subpopulation of pPVN cells that contained the CTB tracer from the NTS could detect meaningful levels of MC4R mRNA rather than just background levels. MC4R are known to be expressed at low levels in the PVN relative to the SCN. Thus, oxytocin and CRH mRNA, which like MC4R are highly expressed in the PVN but not in the SCN, were also probed as they would also be expected to show lower mRNA levels in the SCN, if the MC4R differences produced by the LCM/PCR procedure are valid.
Statistics.
The data for each group are expressed as means ± SE. Comparisons between multiple groups in a within-subjects design were made using a two-way repeated-measures ANOVA followed by Fisher's least-significant-difference test as a post hoc test. Comparisons between multiple groups as a between-subjects design also were made using a two-way ANOVA followed by Fisher's least-significant-difference test as a post hoc test. Comparisons in a within-subjects design were made using a one-way repeated-measures ANOVA followed by Fisher's least-significant-difference test as a post hoc test. Analyses were performed using the statistical program SYSTAT (Richmond, CA) and Statistica (StatSoft, Tulsa, OK). Differences were considered significant if P < 0.05. Animals that did not meet the acceptance criterion for proper cannula placement were eliminated from the analysis.
RESULTS
Effect of CCK-8 on food intake in MC4R null mice.
To extend previous findings suggesting that mice lacking MC4R have reduced sensitivity to the satiety effect to a single dose of CCK on standard mouse chow (20), we determined the effects of multiple doses of CCK-8 in chow-fed wild-type and MC4R−/− mice. Our aim was to evaluate the dose-response function within each of the two groups relative to the baseline of the respective genotypes, rather than comparing the baseline differences between groups. MC4R−/− mice consumed nearly 26% less food over a 30-min period compared with wild-type mice after a 24-h fast (P < 0.05). CCK-8 produced a 22, 29, and 43% suppression of 30-min food intake in wild-type mice at doses of 0.75 (P < 0.05), 2.5 (P < 0.01), and 7.5 (P < 0.01) nmol/kg lbm compared with the same animals that received vehicle alone (Fig. 1A). There was a significant main effect of CCK-8 to reduce 30-min food intake in wild-type mice [F(3,15) = 6.92, P < 0.01]. When administered to MC4R−/− mice, CCK-8 did not suppress 30-min food intake at 0.75 nmol/kg (P = NS), 2.5 nmol/kg (P = NS), but CCK-8 produced a 36% suppression of 30-min food intake at 7.5 nmol/kg lbm (P < 0.01) compared with the same animals that received vehicle alone (Fig. 1B). There was a significant main effect of CCK-8 to reduce 30-min food intake in the MC4R−/− mice [F(3,33) = 4.83, P < 0.01]. Two-way repeated-measures ANOVA revealed a significant main effect on 30-min food intake for strain [F(1,16) = 7.94, P < 0.05] and CCK-8 [F(3,48) = 10.52, P < 0.01], but there was no significant interaction between strain*CCK (P = NS). Overall, the data showed that there is a decreased sensitivity to CCK-8 in MC4R−/− mice at doses that were effective in wild-type mice, but the response to a large dose of CCK-8 in MC4R−/− mice is not attenuated.
Fig. 1.
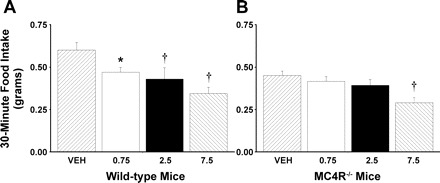
Effect of intraperitoneal (ip) administration of CCK-8 (0, 0.75, 2.5, 7.5 nmol/kg lean body mass) on 30-min food intake in age- and sex-matched MC4R−/− and wild-type mice. Data represent means ± SE. Age- and sex-matched mice deprived of food for 18 h received an injection of CCK-8 or vehicle (0.1% BSA) in randomized order immediately before the start of the dark cycle. Food intake was measured 30 min after wild-type (n = 6–8) and MC4R−/− mice (n = 12–13) received injections of CCK-8 or vehicle and access to food at the onset of the dark cycle. A: response to CCK-8 in wild-type mice. B: response to CCK-8 in MC4R−/− mice. *P < 0.05 vs. vehicle (VEH). †P < 0.01 vs. VEH.
Effect of PVN SHU9119 and CCK-8 on food intake.
To determine whether endogenous melanocortin signaling through MC3/MC4Rs in the PVN contributes to the normal feeding response to CCK, we measured the feeding response to PVN injections of SHU9119 in combination with ip injections of CCK-8. Due to technical challenges inherent in targeting the PVN in mice, the effects of SHU9119 were examined in the PVN in rats, where the feasibility of placing bilateral cannulas in the PVN is well-established. In the presence of PVN pretreatment with SHU9119, the ability of CCK-8 to inhibit 30-min food intake was attenuated by 20% compared with controls that received PVN pretreatment with vehicle (P < 0.05; Fig. 2). CCK-8 inhibited 30-min food intake by 61% compared with the same animals that received PVN injections of vehicle alone (P < 0.05). SHU9119 alone did not increase 30-min food intake relative to animals that received PVN injections of vehicle alone (P = NS). Two-way repeated-measures ANOVA revealed a significant main effect of CCK-8 to inhibit 30-min food intake [F(1,48) = 56.397, P < 0.01], without a main effect of SHU9119 on 30-min food intake [F(1,48) = 0.624, P = NS]. There was a significant interactive effect of SHU9119 and CCK-8 on 30-min food intake [F(1,48) = 4.516, P < 0.05]. A representative photomicrograph of the injection site in the PVN and the distribution of injection sites within the PVN are shown in Figs. 3 and 4, respectively. One rat whose injection site was beyond the boundary of the PVN did not show a decreased response to CCK-8 following PVN injections of SHU9119 compared with CCK-8 alone. Two additional animals who showed an enhanced response to CCK-8 following injections of SHU9119 were deleted from the data analysis because the histological verification was compromised from bad morphology.
Fig. 2.
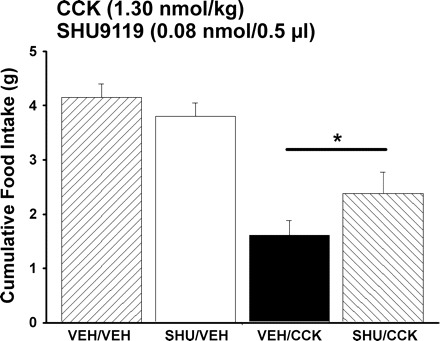
Effect of paraventricular nucleus (PVN) administration of SHU9119 on ip CCK-induced inhibition of food intake. Data represent means ± SE. SHU9119 or SHU9119 vehicle (saline) was administered 1 h before a dose of CCK-8 or CCK-8 vehicle given ip (0.1% BSA, saline). Food intake was measured 30 min after animals (n = 17) received ip injections of CCK-8 or vehicle and access to food at the onset of the dark cycle. *P < 0.05 vs. CCK-8.
Fig. 3.
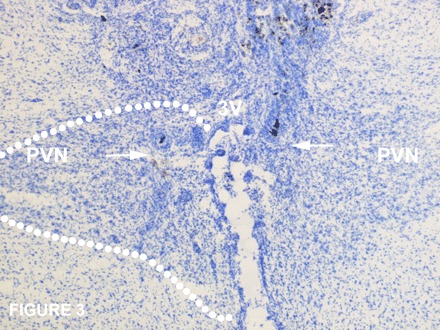
Representative photomicrograph taken of injection sites within the PVN. The arrow represents the ventral most portion of the injection sites on both sides of the PVN.
Fig. 4.
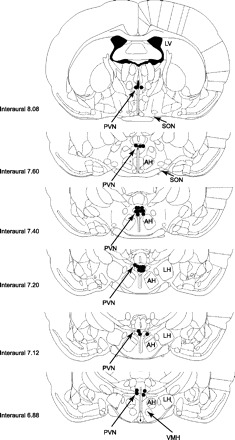
Histological analysis of injection sites within the PVN. Distribution of injection sites from animals that had guide cannulas directed at the PVN. Data from an animal were excluded if its injection site extended beyond the boundary of the PVN.
Effect of 4V SHU9119 on 30-min food intake.
Our aim was to determine whether doses of SHU9119 found ineffective at stimulating 30-min food intake in the PVN were effective at stimulating 30-min food intake in nuclei outside the PVN. To test this aim, we examined the effects of 4V administration of SHU9119 at doses (0.2, 0.4 nmol) that were 1.5- to 2.5-fold higher than the total subthreshold dose of SHU9119 given bilaterally into the PVN (0.08 nmol/side or 0.16 nmol total). SHU9119 administration (0.2, 0.4 nmol) into the 4V was ineffective at stimulating 30-min food intake (P = NS; Table 1).
Table 1.
Effects of SHU9119 administration into the 4V on 30-min food intake
| Vehicle | 0.2 nmol | 0.4 nmol | |
|---|---|---|---|
| 30-min food intake | 4.2±0.27 | 4.3±0.25 | 4.8±0.44 |
Values are means ± SE. Rats received SHU9119 injections 75 min before access to food and start of the dark cycle (n =19/dose). Food intake was measured at 30 min following the start of the dark cycle. Each animal received each treatment at 48-h intervals.
MC4R expression in pPVN cells that project to the NTS.
To determine whether components of the MC4R signaling pathway exist in projections from the pPVN to the NTS, we initially injected retrograde fluorescently labeled (Alexa 488) tracer (CTB) into the cmNTS and waited 4 days for retrograde transport of the CTB to the PVN (10). pPVN cells that contained the CTB (representing pPVN neurons that had direct projections to the hindbrain) were collected by LCM (Fig. 5, A–C) and analyzed for MC4R mRNA content by quantitative RT-PCR. Analysis of oxytocin, and CRH mRNA expression, peptides found in neurons that project from the pPVN to the NTS (39, 40) were also analyzed as a positive control to confirm collection of pPVN neurons. Neurons in the SCN (CTB−) were also collected by LCM and analyzed similarly, as a control nucleus that was expected to express relatively low levels of each of these transcripts (24, 31). In addition, unlabeled cells from the pPVN were collected and screened for MC4R mRNA, oxytocin mRNA, and CRH mRNA.
Fig. 5.
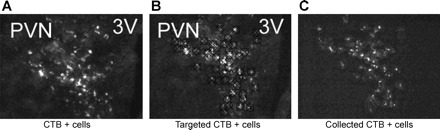
Representative image visualized using the Arcturus AutoPix Fluorescent LCM System of parvocellular PVN (pPVN) neurons labeled with Alexa 488-conjugated cholera toxin subunit B (CTB) that project to the nucleus of solitary tract (NTS). A: Alexa 488-conjugated CTB-labeled cells in pPVN neurons as visualized on the Arcturus AutoPix Fluorescent LCM System. B: Alexa 488-conjugated CTB-labeled cells targeted for collection (before collection). C: Alexa 488-conjugated CTB-labeled cells collected for analysis.
CTB+ cells in the pPVN that project to the NTS contained nearly five-, six-, and eightfold greater levels of mRNAs for MC4R (Fig. 6A), oxytocin (Fig. 6B), and CRH (Fig. 6B), respectively, compared with the SCN (P < 0.01). There were no significant differences in MC4R, oxytocin, and CRH mRNA between CTB-negative cells from the pPVN and CTB+ cells in the pPVN (P = NS; data not shown), consistent with the earlier studies reporting the pPVN to be enriched in these transcripts (27, 31, 37, 39). These findings provide evidence that pPVN neurons that directly project to the NTS contain MC4R mRNA, and mRNA for oxytocin and CRH.
Fig. 6.
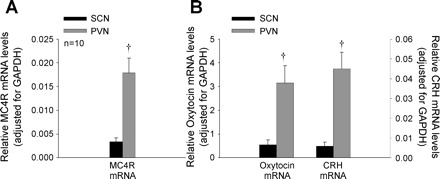
Relative mRNA levels of MC4R, oxytocin, and corticotropin-releasing hormone (CRH) on pPVN neurons that project to the NTS. Data represent means ± SE. A: relative MC4R mRNA levels following analysis of Alexa 488-conjugated CTB-labeled pPVN neurons by quantitative RT-PCR, with the data normalized to GAPDH mRNA levels (44). Relative MC4R mRNA levels in CTB-labeled pPVN neurons were compared with levels in suprachiasmatic nucleus (SCN) as negative control. B: relative oxytocin- and CRH-mRNA following analysis of Alexa 488-conjugated CTB-labeled pPVN neurons by quantitative RT-PCR, with the data normalized to GAPDH mRNA levels. Relative oxytocin- and CRH-mRNA levels were compared with levels in SCN as a negative control. †P < 0.01 vs. SCN.
DISCUSSION
As a first step to clarify the role of melanocortin signaling in the satiety response to CCK, we sought to extend earlier findings performed in MC4R−/− mice (20, 48). We showed here that suppression of satiety in mice required higher doses of CCK-8 in the absence of melanocortin signaling. CCK-8 administration (ip) at doses of 0.75 and 2.5 nmol/kg lbm was ineffective in reducing 30-min food intake in MC4R−/− mice, whereas the same doses of ip CCK-8 reduced food intake by 24 and 29%, respectively, in wild-type mice. In this study, only the 7.5-nmol/kg lbm dose of CCK-8 was effective in the MC4R−/− mice, resulting in a 38% suppression of 30-min food intake, comparable to the 42% suppression produced by this dose in wild-type mice.
Moreover, here we show that administration of SHU9119 into the PVN decreased the effectiveness of CCK-8-induced satiety in rats. Finding this effect now in rats as well as mice lends credence to the possible existence of a physiological mechanism in which endogenous melanocortin signaling through melanocortin-receptive neurons in the PVN contributes to the magnitude of the satiety responses to CCK that is released from the intestines during meals. Furthermore, based on their expression of MC4R mRNA, pPVN neurons that project to the region of the cmNTS are strong candidates for an anatomical link between melanocortin circuitry in the hypothalamus and hindbrain nuclei sensitive to peripheral CCK. While not ruling out the contribution of other hypothalamic circuits or the contribution of PVN projections to hindbrain areas outside the NTS [dorsal motor nucleus of the vagus (DMV), area postrema (AP)] labeled during the CTB injection process, these data support the hypothesis that melanocortin-sensitive neurons in the PVN play a physiological role to regulate the satiety effects of CCK. The present findings highlight the contribution of melanocortin signaling in the PVN toward regulating the magnitude of the satiety effects of CCK-8, but the satiety response to CCK-8 is undoubtedly also regulated by melanocortin-dependent pathways in other brain regions and/or melanocortin-independent mechanisms.
Understanding how melanocortins influence food intake potentially has important therapeutic ramifications. Farooqi and O'Rahilly suggested that mutations in the MC4R gene underlie the most common form of monogenic human obesity (21). Consistent with previous reports (2, 20, 46, 53), the present findings strengthen the emerging hypothesis that melanocortin signaling decreases meal size through increasing the CNS responsivity to satiety signals such as CCK in both rats and mice, and raise the possibly that this may represent a generalized mechanism for regulating food intake in mammals. Furthermore, the adipocyte hormone leptin also reduces food intake via a mechanism that involves increased sensitivity to satiety signals (6, 17, 18) as well as activation of hypothalamic melanocortin-containing neurons that project to the PVN (43). Thus, the mechanism linking changes of leptin signaling to proportionate changes in the response to satiety signals may involve regulation of melanocortin signaling in the PVN.
The observation that CCK-8 decreased food intake in wild-type mice at 0.75 and 2.5 nmol/kg lbm, but failed to suppress food intake at these doses in MC4R−/− mice that are devoid of MC4Rs, is consistent with the findings reported by Fan et al. (20), yet appears somewhat at variance with the findings reported by Vaughan et al. (48). Differences in diets and doses of CCK-8 between laboratories possibly could explain this disparity.
Previous studies showed that rebound hyperphagia following a fast resulted in either no difference in food intake between the MC4R−/− and wild-type mice (48) or resulted in a hyperphagia in the MC4R−/− mice compared with wild-type control mice (20, 23). This response, therefore, appears to be a poorly understood phenomenon. Our findings reveal that the MC4R−/− mice consumed nearly 26% less food over a 30-min period compared with wild-type mice after a 24-h fast, but these findings do not provide a definitive resolution to the conflicting literature. It is possible that a hyperphagic response in the MC4R−/− mice would be difficult to detect in 30 min because of the relatively small amount of food normally consumed in that time. Moreover, if the animals are eating continuously during that period, the amount of food consumed may not have exceeded 0.5–0.6 g of chow under any treatment. The present studies were not designed to address this particular issue and thus future work is required to better understand this response in MC4R−/− mice.
Although the response to other satiety-inducing stimuli in the MC4R−/− mice was not examined in the current studies, others reported intact or even heightened responses to bombesin and PYY(3–36) in these animals (20, 48). Thus, MC4R−/− mice appear to be responsive to other satiety-inducing agents and these effects appear to be relatively specific to CCK-8.
The possibility that all or a portion of the inhibition of food intake observed at the highest dose of CCK-8 (7.5 nmol/kg lbm) in both wild-type and MC4R−/− mice could be secondary to discomfort or malaise (34) cannot be ruled out. Comparable doses of CCK-8 have been reported to inhibit food intake (36) and activate neurons in the area postrema (13, 22), which has a leaky blood-brain barrier (52), contains CCK-1 receptors (22, 29), and is associated with nausea and vomiting (11).
Although injections of the Alexa 488-conjugated CTB fluorescent tracer (0.5 μl) potentially diffused to areas surrounding the NTS (DMV, AP), any particular labeled PVN neurons likely projected to the NTS, DMV, AP, or immediate surrounding area that was positioned to uptake the CTB fluorescent tracer. The findings support the conclusion that some of the labeled PVN neurons project to the NTS and that this is consistent with literature showing that PVN in this area has direct projections to the NTS, and that the NTS has a rich innervation of oxytocin and CRH neurons from the PVN (37, 39, 40).
The key findings of this study support a mechanism by which endogenous melanocortin signaling to the PVN contributes to the normal satiety response to CCK through activation of a descending PVN-NTS projection. The PVN contains a population of MC4R-expressing cells (31) which receives endogenous melanocortin signaling through proopiomelanocortin (POMC) projections from the ARC (15). In addition, MC4Rs are expressed by pPVN neurons that project to the cmNTS, providing an anatomical link between hypothalamic melanocortin circuitry and hindbrain nuclei sensitive to peripheral CCK. Moreover, PVN administration of MTII and α-MSH reduces food intake at doses that do not cause an aversive response (51); this indicates that MC4R activation in the PVN is sufficient to inhibit food intake. Restoration of MC4R into the PVN of mice that do not express MC4R attenuates their hyperphagia and obesity (5). Thus, the findings presented here fit well with the emergent hypothesis that melanocortin signaling through MC4Rs in the PVN may be a critical component of CNS circuitry that regulates the magnitude of normal satiety responses to CCK.
The findings reported here may be relevant to understanding the CNS mechanism by which leptin reduces food intake because the melanocortin signaling in the hypothalamus is implicated as a link in the transduction of hypothalamic leptin action to the activation of descending neuronal pathways to the NTS, where meal size is regulated. It has been established that leptin activates POMC neurons in the ARC (42), some of which have projections to the PVN (13, 16) while others project to the NTS (32, 53). Electrophysiological studies indicate that individual neurons in the pPVN respond similarly to both α-MSH and MTII, and the specificity of these effects was verified by blockade by AGRP and SHU9119, respectively (13). Benoit et al. (7) reported that administration of either the selective MC4R agonist R027–3225 or MTII into the third ventricle produced a similar pattern of neuronal activation in the PVN, consistent with a role for MC4R in the actions of these ligands within the PVN. Evidence that POMC projections from the ARC also directly innervate the NTS (32, 53) raises the possibility that other melanocortin circuits, in addition to those originating from the PVN, may also play a role in regulating the hindbrain response to satiety signals as well as the interaction of these inputs with leptin signaling to the CNS.
The observation in the present study that SHU9119 administration into the PVN did not completely block the food intake response to CCK-8 to the levels seen in control animals can most likely be explained by the fact that melanocortin signaling through MC4R in other brain nuclei such as the NTS is also important in regulating the satiety response to CCK (20, 46, 53). Furthermore, the dose of SHU9119 that was bilaterally injected into the PVN was unlikely to have accessed all MC4Rs in the entire PVN; some PVN MC4Rs were likely unaffected or not reached by the injected compounds. In addition, the regulation of CCK-induced satiety mechanisms undoubtedly involves melanocortin-independent pathways that would not have been directly affected by these interventions. However, these effects are unlikely to be explained by the MC3/MC4R antagonist acting in areas outside the PVN, including the hindbrain, which also contains MC4Rs, as administration of SH9119 administered into the 4V at doses higher than delivered to the PVN failed to stimulate food intake.
If melanocortin signaling through pPVN neurons links the ability of 3V leptin to interact with CCK-sensitive neurons at the level of the hindbrain, oxytocin (10, 25), CRH (27), thyrotropin-releasing hormone (TRH) (1), and gastrin-releasing peptide (GRP) (26) neurons in the pPVN would be logical candidates to mediate this effect. TRH neurons and GRP neurons were reported to be activated after leptin administration into the 3V (1, 26). We reported previously that 3V leptin treatment increased CRH mRNA content in rat PVN (41). Moreover, 3V infusion of a CRH receptor antagonist blocked the effect of leptin to reduce food intake (47) and body weight. Recent findings link MC4R expression to CRH neurons in the pPVN (27), some of which have known direct projections to the NTS and DMV as well as spinal cord (39).
Perspectives
These data are consistent with the hypothesis that melanocortin signaling regulates the satiety effects of CCK and that under defined experimental conditions, MC4R neurons in the PVN contribute to the satiety response to a pharmacological dose of CCK in rats. Furthermore, these data indicate that the satiety effects of CCK-8 are also regulated by melanocortin-dependent mechanisms in mice. The present findings highlight the contribution of melanocortin signaling in the PVN toward regulating the magnitude of the satiety effects of CCK-8, but the satiety response to CCK-8 is undoubtedly also regulated by melanocortin-dependent pathways in other brain regions and/or melanocortin-independent mechanisms. The present findings also provide evidence that MC4R mRNA is expressed by pPVN neurons that project to the cmNTS, providing a functional circuit to link hypothalamic melanocortin-responsive neurons that are sensitive to peripheral CCK-8 and putatively influence meal size. Since the hypothalamic melanocortin system is implicated as a key mediator of leptin's anorexic action in the brain, the present work suggests that melanocortin-sensitive neurons that project to the NTS regions from the medial parvocellular region of the PVN contribute to leptin's ability to enhance the satiety effect of CCK.
GRANTS
This material is based on work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs including the VA Career Development Program, Merit Review Research Program, Research Enhancement Award Program, and Career Scientist Program. D. G. Baskin is the recipient of a Department of Veterans Affairs Senior Research Career Scientist Award at the VA Puget Sound Health Care System. The research was also supported by the biomedical research core programs, particularly the Cellular and Molecular Imaging Core of the National Institutes of Health (NIH) Diabetes Endocrinology Research Center and NIH CNRU at the University of Washington. Grant support for the research was provided by a Pilot and Feasibility grant from the University of Washington CNRU and NIH Grants DK-17047, P30 DK-035816, P30 DK-017047–31, P30 DK-017047–31689, P30 DK-035816, and PO1 DK-068384.
Acknowledgments
The authors thank the Clinical Nutrition Research Unit CNRU biostatistician B. Fish at the University of Washington and the technical support of B. Thatcher.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E, Flier JS. Role of leptin in the neuroendocrine response to fasting. Nature 382: 250–252, 1996 [DOI] [PubMed] [Google Scholar]
- 2.Aja SM, Sahandy S, Ladenheim EE, Schwartz GJ, Moran TH. Intracerebroventricular CART peptide reduces food intake and alters motor behavior at a hindbrain site. Am J Physiol Regul Integr Comp Physiol 281: R1862–R1867, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Azzara AV, Schuss B, Hong S, Chua SC, Schwartz GJ. Melanocortin 4 receptor (MC4R) knockout mice have increased food intake and meal size, and decreased sensitivity to post-oral nutrient stimulation. Appetite 44: 332, 2005 [Google Scholar]
- 4.Azzara AV, Sokolnicki JP, Schwartz GJ. Central melanocortin receptor agonist reduces spontaneous and scheduled meal size but does not augment duodenal preload-induced feeding inhibition. Physiol Behav 77: 411–416, 2002 [DOI] [PubMed] [Google Scholar]
- 5.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123: 493–505, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Barrachina MD, Martinez V, Wang L, Wei JY, Tache Y. Synergistic interaction between leptin and cholecystokinin to reduce short-term food intake in lean mice. Proc Natl Acad Sci USA 94: 10455–10460, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benoit SC, Schwartz MW, Lachey JL, Hagan MM, Rushing PA, Blake KA, Yagaloff KA, Kurylko G, Franco L, Danhoo W, Seeley RJ. A novel selective melanocortin-4 receptor agonist reduces food intake in rats and mice without producing aversive consequences. J Neurosci 20: 3442–3448, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blevins JE, Dixon KD, Hernandez EJ, Barrett JA, Gietzen DW. Effects of threonine injections in the lateral hypothalamus on intake of amino acid imbalanced diets in rats. Brain Res 879: 65–72, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Blevins JE, Eakin TJ, Murphy JA, Schwartz MW, Baskin DG. Oxytocin innervation of caudal brain stem nuclei activated by cholecystokinin. Brain Res 993: 30–41, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Blevins JE, Schwartz MW, Baskin DG. Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol 287: R87–R96, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Borison HL. History and status of the area postrema. Fed Proc 43: 2937–2940, 1984 [PubMed] [Google Scholar]
- 12.Cheung CC, Clifton DK, Steiner RA. Proopiomelanocortin neurons are direct targets for leptin in the hypothalamus. Endocrinology 138: 4489–4492, 1997 [DOI] [PubMed] [Google Scholar]
- 13.Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron 24: 155–163, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Ebihara K, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Satoh N, Tamaki M, Yoshioka T, Hayase M, Matsuoka N, Aizawa-Abe M, Yoshimasa Y, Nakao K. Involvement of agouti-related protein, an endogenous antagonist of hypothalamic melanocortin receptor, in leptin action. Diabetes 48: 2028–2033, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol 402: 442–459, 1998 [PubMed] [Google Scholar]
- 16.Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA 95: 741–746, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emond M, Ladenheim EE, Schwartz GJ, Moran TH. Leptin amplifies the feeding inhibition and neural activation arising from a gastric nutrient preload. Physiol Behav 72: 123–128, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Emond M, Schwartz GJ, Ladenheim EE, Moran TH. Central leptin modulates behavioral and neural responsivity to CCK. Am J Physiol Regul Integr Comp Physiol 276: R1545–R1549, 1999 [DOI] [PubMed] [Google Scholar]
- 19.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385: 165–168, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin-mediated suppression of feeding involves the brain stem melanocortin system. Nat Neurosci 7: 335–336, 2004 [DOI] [PubMed] [Google Scholar]
- 21.Farooqi IS, O'Rahilly S. Monogenic human obesity syndromes. Recent Prog Horm Res 59: 409–424, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Hill DR, Campbell NJ, Shaw TM, Woodruff GN. Autoradiographic localization and biochemical characterization of peripheral type CCK receptors in rat CNS using highly selective nonpeptide CCK antagonists. J Neurosci 7: 2967–2976, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88: 131–141, 1997 [DOI] [PubMed] [Google Scholar]
- 24.Jing X, Ratty AK, Murphy D. Ontogeny of the vasopressin and oxytocin mRNAs in the mouse hypothalamus. Neurosci Res 30: 343–349, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Kirchgessner AL, Sclafani A, Nilaver G. Histochemical identification of a PVN-hindbrain feeding pathway. Physiol Behav 42: 529–543, 1988 [DOI] [PubMed] [Google Scholar]
- 26.Ladenheim EE, Behles R, Bi S, Moran TH. Gastrin-releasing peptide mRNA expression in the hypothalamic paraventricular nucleus is altered by melanocortin receptor stimulation and food deprivation. Endocrinology In press. [DOI] [PMC free article] [PubMed]
- 27.Lu XY, Barsh GS, Akil H, Watson SJ. Interaction between alpha-melanocyte-stimulating hormone and corticotropin-releasing hormone in the regulation of feeding and hypothalamo-pituitary-adrenal responses. J Neurosci 23: 7863–7872, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsh DJ, Hollopeter G, Huszar D, Laufer R, Yagaloff KA, Fisher SL, Burn P, Palmiter RD. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet 21: 119–122, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Moran TH, Robinson PH, Goldrich MS, McHugh PR. Two brain cholecystokinin receptors: implications for behavioral actions. Brain Res 362: 175–179, 1986 [DOI] [PubMed] [Google Scholar]
- 30.Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, Baskin DG, Schwartz MW. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest 115: 703–710, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mountjoy KG, Mortrud MT, Low MJ, Simerly RB, Cone RD. Localization of the melanocortin-4 receptor (MC4-R) in neuroendocrine and autonomic control circuits in the brain. Mol Endocrinol 8: 1298–1308, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Palkovits M, Mezey E, Eskay RL. Pro-opiomelanocortin-derived peptides (ACTH/beta-endorphin/alpha-MSH) in brain stem baroreceptor areas of the rat. Brain Res 436: 323–338, 1987 [DOI] [PubMed] [Google Scholar]
- 33.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. (2nd ed.). Orlando, FL: Academic, 1986
- 34.Perez C, Sclafani A. Cholecystokinin conditions flavor preferences in rats. Am J Physiol Regul Integr Comp Physiol 260: R179–R185, 1991 [DOI] [PubMed] [Google Scholar]
- 35.Raposinho PD, Castillo E, d'Alleves V, Broqua P, Pralong FP, Aubert ML. Chronic blockade of the melanocortin 4 receptor subtype leads to obesity independently of neuropeptide Y action, with no adverse effects on the gonadotropic and somatotropic axes. Endocrinology 141: 4419–4427, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Reidelberger RD, Hernandez J, Fritzsch B, Hulce M. Abdominal vagal mediation of the satiety effects of CCK in rats. Am J Physiol Regul Integr Comp Physiol 286: R1005–R1012, 2004 [DOI] [PubMed] [Google Scholar]
- 37.Rinaman L. Oxytocinergic inputs to the nucleus of the solitary tract and dorsal motor nucleus of the vagus in neonatal rats. J Comp Neurol 399: 101–109, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 139: 4428–4431, 1998 [DOI] [PubMed] [Google Scholar]
- 39.Sawchenko PE. Evidence for differential regulation of corticotropin-releasing factor and vasopressin immunoreactivities in parvocellular neurosecretory and autonomic-related projections of the paraventricular nucleus. Brain Res 437: 253–263, 1987 [DOI] [PubMed] [Google Scholar]
- 40.Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. J Comp Neurol 205: 260–272, 1982 [DOI] [PubMed] [Google Scholar]
- 41.Schwartz MW, Seeley RJ, Campfield LA, Burn P, Baskin DG. Identification of targets of leptin action in rat hypothalamus. J Clin Invest 98: 1101–1106, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes 46: 2119–2123, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW. Melanocortin receptors in leptin effects. Nature 390: 349, 1997 [DOI] [PubMed] [Google Scholar]
- 44.Segal JP, Stallings NR, Lee CE, Zhao L, Socci N, Viale A, Harris TM, Soares MB, Childs G, Elmquist JK, Parker KL, Friedman JM. Use of laser-capture microdissection for the identification of marker genes for the ventromedial hypothalamic nucleus. J Neurosci 25: 4181–4188, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanley BG, Lanthier D, Leibowitz SF. Multiple brain sites sensitive to feeding stimulation by opiod agonists: a cannula-mapping study. Pharmacol Biochem Behav 31: 825–832, 1988 [DOI] [PubMed] [Google Scholar]
- 46.Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. Endocrinology 146: 3739–3747, 2005 [DOI] [PubMed] [Google Scholar]
- 47.Uehara Y, Shimazu T, Ohtani K, Sato N, Mori M. Hypothalamic corticotropin-releasing hormone is a mediator of the anorexigenic effect of leptin. Diabetes 47: 890–893, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Vaughan CH, Haskell-Luevano C, Andreasen A, Rowland NE. Effects of oral preload, CCK or bombesin administration on short term food intake of melanocortin 4-receptor knockout (MC4RKO) mice. Peptides 27: 3226–3233, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Williams DL, Grill HJ, Weiss SM, Baird JP, Kaplan JM. Behavioral processes underlying the intake suppressive effects of melanocortin 3/4 receptor activation in the rat. Psychopharmacology (Berl) 161: 47–53, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Williams DL, Kaplan JM, Grill HJ. The role of the dorsal vagal complex and the vagus nerve in feeding effects of melanocortin-3/4 receptor stimulation. Endocrinology 141: 1332–1337, 2000 [DOI] [PubMed] [Google Scholar]
- 51.Wirth MM, Olszewski PK, Yu C, Levine AS, Giraudo SQ. Paraventricular hypothalamic alpha-melanocyte-stimulating hormone and MTII reduce feeding without causing aversive effects. Peptides 22: 129–134, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Wsniewski H, Olszewski J. Vascular permeability in the area postrema and hypothalamus. A study using iodinated radioactive albumin. Neurology 13: 885–894, 1963 [DOI] [PubMed] [Google Scholar]
- 53.Zheng H, Patterson LM, Pfifer CB, Berthoud HR. Brain stem melanocortinergic modulation of meal size and identification of hypothalamic POMC projections. Am J Physiol Regul Integr Comp Physiol 289: R247–R258, 2005 [DOI] [PubMed] [Google Scholar]