

Abstract
Acinetobacter baumannii is an important nosocomial pathogen in civilian intensive care units. Recently the incidence has increased in wounded military personnel. Morphine is documented in numerous animal studies to be immunosuppressive and to sensitize to infection. The hypotheses were tested that morphine, administered for analgesia in the battlefield, predisposes to Acinetobacter infection, and that the opioid may have an additive or synergistic effect with trauma. To test these hypotheses, an intraperitoneal infection model was established in mice using several Acinetobacter strains. Morphine administered for 48 hr by implantation of a slow-release morphine pellet increased mortality compared to animals receiving a placebo pellet, an effect that was blocked by the mu-opioid receptor antagonist, naltrexone. Acinetobacter burdens in the blood, spleens, livers, and lungs of morphine-treated mice, were significantly higher than those in placebo-treated animals, confirming that mortality was due to potentiated growth of the bacteria. There were also elevated levels of pro-inflammatory cytokines in morphine-treated versus placebo-treated mice. Morphine caused a reduction in the total number of cells in the peritoneal cavity, a decrease in the percentage and total numbers of neutrophils, and a decrease in the total number of macrophages. Morphine treatment also suppressed levels of the neutrophil-inducing molecules, IL-17A and KC/CXCL1. However, IL-17A−/− mice given morphine were not sensitized to Acintobacter infection to a greater degree than similarly treated wild-type mice. Trauma alone did not sensitize to Acinetobacter infection, and there was no additive effect between morphine and trauma. These results support the hypothesis that morphine potentiates Acinetobacter infection.
Introduction
Acinetobacter baumannii is an emerging Gram-negative pathogen primarily associated with nosocomial infections, especially in intensive care units (Towner 2009; Vincent et al., 2009; Kang et al., 2010; Boucher et al., 2009; Scott 2004). Recently, an increased incidence of multiply-drug resistant (MDR) Acinetobacter infections have been associated with war-related trauma in military personnel (Petersen et al., 2007, Scott et al., 2007; Scott 2004). Acinetobacter most frequently causes wound infections, pneumonia, bacteremia and urinary tract infections. Bacteremia in particular has emerged as a rising problem in military and civilian populations. Between 2002 and 2004 there was a significant increase in multiply drug resistant A. baumannii isolated from blood of military personnel returning from combat in Iraq and Afghanistan (Scott 2004). In a large study of civilian hospitals in the U.S., A. baumannii ranked as the tenth most common isolate in nosocomial bloodstream infections during the period 1995–2002, accounting for 1.3% of ICU bacteremias with a mortality rate over 34 percent (Wisplinghoff et al, 2004). This organism has emerged as a major nosocomial pathogen due to its ability to survive for long periods of time in the hospital environment, and because of development of MDR organisms, making treatment difficult and leading to a high mortality rate (Towner 2009; Wendt et al., 1997; Dijkshoorn et al., 2007; Scott et al., 2007). Epidemiologic studies have shown that MDR Acinetobacter infections are associated with longer hospital stays, with a positive correlation between longer ICU stays and mortality (Kang et al., 2010; Boucher et al., 2009). While the antibiotic resistance patterns of A. baumannii have been widely studied, the host-immune mechanisms required to combat the pathogen are understudied. Several models of Acinetobacter pneumonia have been developed to examine innate immune parameters and/or antibiotic resistance during infection (van Faassen et al., 2007; Bernabeu-Wittel et al., 2005; Renckens et al., 2006; Russo et al., 2008; Joly-Guillou et al., 1997). To date, there are no studies in the literature documenting the innate immune responses to systemic, intraperitoneal infection with A. baumannii, which can be considered a model for sepsis.
Morphine is commonly given to injured soldiers on the battlefield and it is also used in ICUs. There is a clinical literature linking opioid use to increased infections (Hussey and Katz 1950; Louria et al., 1967; Rogers et al., 2005). There is a strong intersection between intravenous drug abuse and HIV infection (Centers for Disease Control and Prevention, 2006) (Horsburgh et al., 1989; McCoy et al., 1998). One explanation for the association of infections with opioid use is self-infection with dirty needles. It is also possible that morphine itself may sensitize to infection. In fact, morphine has been shown in laboratory studies to sensitize to several pathogens including Salmonella typhimurium (MacFarlane et al., 2000), Klebsiella pneumoniae (Tubaro et al., 1983), Toxoplasma gondii (Chao et al., 1990), and Streptococcus pneumonia (Wang et al., 2008; Ma et al., 2010), as well as Candida albicans (Tubaro et al., 1983), Leishmania donovani (Singh and Singal 2007; Singal et al., 2002), and herpes simplex virus-1 (Lioy et al., 2006; Mojadadi et al., 2009). Morphine has also been shown to suppress immune function when given in vivo or when added in vitro to leukocytes, affecting a variety of endpoints including, among others, Natural Killer cell activity (Shavit et al., 1986; Weber and Pert 1989), macrophage phagocytosis (Casellas et al., 1991; Szabo et al., 1993), and antibody production (Bussiere et al., 1993; Vassou et al., 2008). An anticipated consequence of immunosuppression would be increased sensitization to infection.
The increased incidence of Acinetobacter infections in the battlefield arena has frequently occurred in severely wounded soldiers. There is a robust literature showing that trauma and surgery can lead to immunosuppression (McCarter et al., 1998; Monroy et al., 2007; Angele and Chaudry 2005; Kimura et al., 2010). The current study tested the hypothesis that morphine sensitizes to systemic A. baumannii infection and investigated whether morphine and trauma had additive effects. It was found that mice implanted with slow release morphine pellets followed by intraperitoneal infection with A. baumannii, had increased bacterial burdens in blood and organs, greater pro-inflammatory cytokine levels, and increased mortality in comparison to placebo treated controls. Trauma did not increase morphine-induced sensitization to Acinetobacter infection. The rapidity of infection suggested that host defense was dependent upon innate immune factors. Morphine was shown to suppress the percentage and total number of cells in the peritoneal cavity 12 hours post-infection. The role of IL-17A, a cytokine that induces a chemokine, KC/CXCL1, that is active in recruiting neutrophils, was investigated in morphine treated mice.
Materials and Methods
Animals
Specific pathogen-free, female or male, six to eight week-old C57BL/6J and C3HeB/FeJ mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice with a genetic disruption of the IL-17A gene, (IL-17a −/− knockout (KO)) mice, were obtained from Dr. Jay Kolls (LSU School of Medicine), with the permission of Dr. Yoishiro Iwakura (The Institute of Medical Science, the University of Tokyo, Japan), who originally developed these animals (Nakae et al., 2002). All IL-17a −/− and age-matched wild type (WT) littermates were bred and maintained in the Central Animal Facility of Temple University. All animals were allowed to acclimate for at least 1 week before use. Rodent chow (Purina, St. Louis) and fresh water were available ad libitum. Animals were housed in the Central Animal Facility. All experiments were carried out with the approval of the Institutional Animal Care and Use Committee at Temple University.
Reagents
Opioid agonists and antagonists were provided by The National Institute on Drug Abuse (NIDA; Rockville, MD). These included 25 and 75 mg extended release morphine pellets, 30 mg naltrexone pellets, and placebo pellets.
Acinetobacter baumannii
Clinical strains of A. baumannii: 4502, 5798, and 6143 were provided by Colonel David Craft (Walter Reed Army Institute of Research, Silver Spring, MD). Organisms were stored by freezing at −80°C in DMSO. To grow organisms for in vitro or in vivo experimentation, a sterile loop was touched to the frozen stock and used to streak out two blood agar plates. Plates were incubated at 37°C overnight. Ten isolated colonies were picked and inoculated into a 50 ml conical tube (Becton-Dickinson; Franklin Lakes, NJ), containing 10 ml of Brain-Heart Infusion (BHI) media. The tube was incubated at 37°C with rotary agitation (250 rpm) for 3.5 hours. The top 5 ml was drawn off and used to inoculate a 200 ml Erlenmeyer flask containing 45 ml of BHI. This flask was incubated at 37°C with agitation (200 rpm) on a G24 Environmental Incubator Shaker (New Brunswick Scientific Co., New Brunswick, NJ) for 2.5 hours to produce late log-phase organisms. After 2.5 hours, the flask was placed on ice to retard further growth of the bacteria. To estimate the number of organisms/ml for inoculation into mice, an appropriate dilution of the desired culture was made in 10% paraformaldehyde and counted in a Petroff-Hausser counter. The culture was diluted with 0.9% sterile, pyrogen-free saline (Hospira; Lake Forest, IL) in an endotoxin-free, sterile vial to obtain the desired concentration of organisms/ml. Viable counts on blood agar were used to determine the actual number of colony-forming units (CFUs) per ml, and to calculate the precise inoculum injected. For in vivo inoculation, mice were injected intraperitoneally (i.p.) with the desired dose using a 26 gauge needle in a 200μl volume. Doses of bacteria were chosen which would permit survival of animals to indicated time points under different experimental conditions.
Pellet Implantation Surgery
Mice were anesthetized with isoflurane and an area of the back was shaved. A 1-cm incision was made in the skin, and mice were implanted subcutaneously (s.c.) between the scapula with either a 25 mg (C57BL/6 mice) or a 75 mg (C3HeB/FeJ) morphine extended release pellet, a 30 mg naltrexone pellet, a placebo pellet or a morphine plus a naltrexone pellet. The difference in dose of morphine used for the two mouse strains is due to their different sensitivities to the opioid. Surgical staples were used to close the wound, and mice were allowed to recover under a heat lamp. Morphine pellets are used routinely by pharmacologists to yield a continuous supply of the drug without having animals experience episodes of withdrawal (Bryant et al., 1988a; Cheney and Goldstein 1971; Cerletti et al., 1976;). The dose of morphine in the blood using the 75 mg pellets is in the physiologic range measured in humans given morphine for analgesia, about 0.6 to 2.0 μg/ml (Bryant et al., 1988b; Feng et al., 2006). The 30 mg naltrexone pellet is standard and dispenses a sufficient amount of drug to antagonize the morphine in both the 75 and 25 mg pellets.
Survival Studies
Susceptibility to infection was assessed by mortality. Mortality was scored for 7 days. The LD50values were previously determined (Breslow et al. 2011).
Necropsy
Mice were anesthetized with 100 μl of a 50 mg/ml solution of sodium pentobarbital injected intramuscularly (i.m.). Blood was collected via cardiac puncture using a 22 gauge needle, into heparinized 3 ml syringes. 0.1 ml of blood or an appropriate dilution was plated on Levine Eosin Methylene Blue (EMB) agar plates and incubated at 37°C overnight. The number of colony forming units (CFU) were counted and expressed as CFU/0.1ml blood. For plasma collection, heparinized blood was centrifuged at 12,000 x g for 10 min at 4°C, and the plasma layer removed to 1.5 ml Eppendorf tubes and frozen at −80°C until further use to determine levels of cytokines and chemokines. Mice were euthanized via cervical dislocation, and if desired, peritoneal exudate fluid (PEF) was collected by lavage with Hanks Balanced Salt Solution and frozen at −80°C until further use, also to determine levels of cytokines and chemokines. Organs were removed aseptically from individual animals and homogenized in 3 ml of ice-cold phosphate-buffered saline (PBS) in 14 ml round-bottom tubes (BD; Franklin Lakes, NJ), using a Tekmar Tissuemizer® (Tekmar, Cincinnati, Ohio). Homogenates were serially diluted in sterile water and spread onto EMB agar plates and incubated at 37°C overnight. The number of CFU was counted and the results expressed as CFU/0.1gm tissue or CFU/0.1ml blood. The limit of detection in undiluted samples was 30 CFU/0.1gm organ or 1 CFU/0.1ml blood.
Trauma model
The method used to administer trauma to the mice has been previously described (Mackrell et al., 2001). Briefly, the animals were anesthetized with inhaled isoflurane. The right femur of each mouse was fractured as follows. A 1.0 cm incision was made over the medial aspect of the right thigh. The femur was cleared of overlying connective tissue and fractured with sterile scissors. After hemostasis was achieved, the wound was closed with nonabsorbable sutures. The mouse was then bled 40% of total blood volume via a standard retro-orbital approach with a heparinized microcapillary tube. While under anaesthesia, if desired, mice were implanted with a morphine or a placebo pellet. Animals were infected with Acinetobacter 48 hrs after the surgery.
Cytokine Measurement by Cytometric Bead Array (CBA)
Levels of IL-6, IL-10, MCP-1, IFN-γ, TNF-α and IL-12p70 in plasma were analyzed simultaneously in a multiplex assay using the Mouse Inflammatory Cytometric Bead Array (CBA) kit (BD Biosciences, San Jose, CA). The assay was carried out according to the manufacturer’s instructions. Briefly, capture beads for the various cytokines and chemokines were mixed together to form a bead suspension. Fifty μl aliquots of the bead suspension was then incubated with 50 μl of sample plasma, and 50 μl of anti-murine phycoerythrin (PE) detection reagent for 2 hours at room temperature in the dark. One ml of wash buffer was added to each assay tube and centrifuged at 200 x g for 10 minutes. Supernatants were discarded and 200 μl of wash buffer was added to resuspend the bead pellet. For each sample, 300 events per capture bead were gated and analyzed on a BD FACScan flow cytometer calibrated with set-up beads (provided by the manufacturer) for optimization of instrument settings at specific fluorescent intensities. Cytokine levels were quantified by comparison with standard curves generated by flow cytometric analysis of cytokine standards incubated with mixed capture beads and anti-murine PE detection antibody.
Peritoneal Lavage
Mice were euthanized by cervical dislocation and the skin was pulled away to expose the peritoneum. A 1 ml volume of ice-cold Mg++Ca++ free Hank’s Balanced Salt Solution (Invitrogen, Carlsbad, CA) was injected intraperitoneally. Peritoneal exudate fluid (PEF) was collected using a 22 gauge needle into a 1.5 ml Eppendorf tube. PEF was centrifuged at 12,000 x g for 10 min at 4°C. Supernatants were collected and frozen at -80°C until further use.
ELISA
Levels of IL-17 and KC/CXCL1 were assessed in PEF or cell culture supernatants by a sandwich ELISA. IL-17 and KC/CXCL1 antibodies and reagents were purchased from R&D Systems (Minneapolis, MN) as IL-17 and KC/CXCL1 Duoset ELISA Development kits. The assay was carried out according to the manufacturer’s instructions. Briefly, a 96-well Costar (Corning, NY) plate was coated with the desired concentration of capture antibody, and incubated overnight at room temperature. The solution was aspirated and washed with 300 μl Wash Buffer three times. Reagent Diluent (1% bovine serum albumin in PBS; 300 μl) was added and incubated for 1h when the plate was washed three times. 100 μl of diluted sample or standards were added to appropriate wells and the plate was covered and allowed to incubate for 2h at room temperature. The plate was washed three times and 100 μl Detection Antibody was added to each well. The plate was covered, allowed to incubate for 2h at room temperature, and followed by three washes. One hundred microliters of a working concentration of Steptavidin-horseradish peroxidase (HRP) solution was added to each well, the plate was covered and incubated for 20 minutes in the dark at room temperature. Three more washes were performed and 100 μl of Color Reagent Solution containing a 1:1 mixture of H2O2 and tetramethylbenzidine was added to each well and the plate incubated 20 minutes in the dark at room temperature. Finally, 50 μl of Stop Solution containing 2N H2SO4 was added to all wells and the absorbance was read at 450nm on an OMEGA microplate reader (BMG Labtech, Inc., Cary, NC). Levels of cytokines or chemokines were quantified in plasma or in PEF using standard curves generated within each experiment of the assay. Samples were run in duplicate.
Flow Cytometry
Peritoneal exudate cells were adjusted to contain 106 cells in 100 μl PBS containing 2% bovine serum albumin (FACS Buffer). Samples were blocked to prevent non-specific antibody binding via the Fc receptor using rat anti-mouse CD16/CD32 (Mouse Fc Block; BD; Franklin Lakes, NJ), by adding 2 μl Fc Block to each sample and incubating for 15 minutes at 4°C. The cells were incubated for 30 minutes at 4°C with pre-titrated, optimal concentration of 2 μl PE-F4/80 (eBioscience, San Diego, CA), FITC-Ly6G, and APC-CD-11b (BD Biosciences, San Diego, CA). After the incubation, cells were washed twice with 500 μl of FACS Buffer and the cells were centrifuged at 250 x g for 5 minutes after each wash. After the last wash, 200 μl of FACS buffer was added followed by 200μl of 4% paraformaldehyde in 0.9% saline to fix the cells. Data was collected from 50,000 gated live cells on a BD FACSCalibur flow cytometer (BD; Franklin Lakes, NJ). Data were analyzed using FloJo software (Tree Star; Ashland, OR).
Statistical Analyses
Data were analyzed using SAS V9.1 (Cary, NC) by John G. Gaughan, Ph.D. at the Biostatistics Consulting Center at Temple University. The dependent variables, cell counts, marker levels, etc. were treated as continuous variables for all analyses. Means, standard deviations, and number of observations were presented for each variable. The experimental unit was each individual animal or culture samples. The experiments used a factorial design with each animal evaluated at individual time points. The null hypothesis was that there would be no difference between or within treatment groups over time. Prior to analysis, data were tested for normality using the Shapiro-Wilk test. If the data were significantly non-normal, a ‘normalized-rank’ transformation was applied to the data. The rank-transformed data were analyzed using a mixed-model ANOVA with or without repeated measures followed by multiple comparisons to detect significant differences between means (treatment groups and times). Multiple pair-wise comparisons (treatment groups and times) were not adjusted for Type 1 error. Two-group experiments were analyzed using t-tests and Wilcoxon rank sums test for non-normal distributed dependent variables. Survival studies were carried out using Kaplan-Meier Product Limit estimation. Between group differences were tested using the log rank test. A p-value of 0.05 was used for statistical significance in all studies.
Results
Effect of Morphine on Acinetobacter infection
The hypothesis was tested that morphine would sensitize to Acinetobacter infection. To test this premise, mice of two different strains were implanted with morphine pellets, challenged 48 hr later with sub-lethal doses of one of two strains of Acinetobacter, and scored for survival. As shown in Table 1, deaths occurred only in mice that had received morphine. Both strains of Acinetobacter resulted in mortality of morphine treated animals. When the results are considered in aggregate, mice that had received a morphine pellet were significantly sensitized to infection, with 14/27 dead, whereas all placebo treated mice survived (0/23 dead). To further investigate this observation, the study was repeated in both mouse strains using the opioid-receptor antagonist, naltrexone. Groups of mice received a morphine pellet, a placebo pellet, a naltrexone pellet, or both a naltrexone and morphine pellet 48 hours before i.p. challenge with A. baumannii, strain 5798. As shown in Table 2, morphine resulted in 100% mortality in C57BL/6 mice and 40% mortality in C3HeB/FeJ mice, whereas all placebo and naltrexone treated animals survived. Naltrexone blocked the morphine-mediated sensitization to infection, showing that sensitization is opioid-receptor dependent.
Table 1.
Effect of morphine on mortality of mice challenged i.p. with A. baumannii.
| Bacterial Straina | Mouse Strain and Dose | Morphine Dead/Total | Placebo Dead/Total |
|---|---|---|---|
| 5798 | C57BL/6 (0.1 LD50) | 4/6 | 0/4 |
| C3HeB/FeJ (0.2 LD50) | 5/8 | 0/8 | |
| 6143 | C57BL/6 (0.1 LD50) | 3/5 | 0/3 |
| C3HeB/FeJ (0.1 LD50) | 2/8 | 0/8 | |
| Total | 14/27 | 0/23 |
Mice were implanted with pellets 48 hours before i.p. challenge with A. baumannii.
Table 2.
Naltrexone blocks increased mortality in Acinetobacter-infected mice induced by morphine.
| Treatmenta | C3HeB/FeJb Dead/Total |
C57BL/6c Dead/Total |
|---|---|---|
| Morphine | 4/10 * | 6/6 ** |
| Placebo | 0/5 | 0/5 |
| Naltrexone | 0/5 | 0/5 |
| Morphine + Naltrexone | 0/10 | 0/10 |
Mice were implanted with pellets 48 hours before i.p. challenge with A. baumannii, strain 5798.
The challenge dose for C3HeB/FeJ mice was 0.02 LD50 (9.0 × 105 CFU).
The challenge dose for C57BL/6J mice was 0.12 LD50 (9.0 × 105 CFU).
p<0.05 for morphine vs placebo, naltrexone, or morphine + naltrexone;
p<0.0001 for morphine vs placebo, naltrexone, or morphine + naltrexone
To examine the mechanism of morphine mediated sensitization to infection, studies investigated whether morphine had any direct effect on the growth or viability of Acinetobacter in vitro. As shown in Figure 1, the presence of morphine in the culture medium had no effect on the growth of the bacteria, as measured either by optical density of the culture medium (Figure 1A) or by viability, as measured by colony counts (Figure 1B).
Figure 1. Morphine does not affect the growth or viability of A. baumannii, in vitro.


A. baumannii, strain 5798, was cultured in Brain Heart Infusion (BHI) in the presence or absence of a 6-log10 fold dose range of morphine and evaluated spectrophotometrically for growth (Panel A) or plated on BA plates to determine viability by CFU (Panel B). There was no statistically significant difference in growth rate of morphine treated cultures as compared to control cultures.
Morphine mediated increase in Acinetobacter burdens
Survival data showing sensitization to infection by morphine were corroborated by assessing bacterial burdens in the organs and blood of infected mice that received the drug, as compared to animals given placebo. All animals were challenged i.p. with A. baumannii, 48 hours after morphine pellet implantation. Based on results obtained from kinetic studies of bacterial burdens carried out previously (Breslow et al. 2011), animals were sacrificed at 12 hours after Acinetobacter infection, at which time lungs, livers, spleens, and blood were collected for necropsy. Experiments were carried out in both C57BL/6 and C3HeB/FeJ mice. As shown in Figure 2A, morphine treated C57BL/6 mice had a statistically significant increase in bacterial burdens over placebo-treated mice in lungs, spleen, and blood, with a strong trend towards significance in liver. Similar results were observed in C3HeB/FeJ mice (Figure 2B), where a statistically significant increase in bacterial burdens in morphine-treated mice was observed in lungs, livers, and blood with a trend toward statistical significance in the spleen. These data show that morphine potentiates Acinetobacter bacterial growth and burdens in the organs and blood of both C57BL/6 and C3HeB/FeJ mice.
Figure 2. Morphine potentiates bacterial burdens in organs and blood of two strains of mice.


Mice were implanted with either a morphine pellet or a placebo pellet 48 hours before i.p. infection. Panel A: C57BL/6 mice (0.1 LD50 dose, A. baumannii, strain 5798, 8.2 × 105 CFU) or Panel B: C3HeB/FeJ mice (0.2 LD50 dose, A. baumannii, strain 4502, 7.2 × 105 CFU). Organs and blood were harvested at 12 hours post-infection for necropsy. Points represent individual animals. Lines are median values for bacterial burdens. Symbols on the X-axis represent animals whose Acinetobacter burden was below the detectable limit of 30 CFU/0.1gram tissue or 1 CFU/0.1ml blood.
Morphine mediated increase in inflammatory mediators
Studies were carried out to test the effect of morphine on levels of inflammatory cytokines and chemokines in plasma following Acinetobacter infection. C57BL/6 or C3HeB/FeJ mice were implanted with either a morphine pellet or a placebo pellet and were challenged i.p. 48 hours after pellet implantation with a sub-lethal dose of A. baumannii strain, 5798. At 8 hours post-infection, plasma was collected and analyzed using a multiplex Cytometric Bead Array (CBA) assay for mouse pro-inflammatory cytokines. Treatment of C57BL/6 mice with morphine before infection resulted in increased levels of all cytokines tested except IFN-γ(Figure 3A). In C3HeB/FeJ mice (Figure 3B), morphine treatment caused a significant increase only in IL-10, IL-12p70, IFN-γ, and TNF-α. C3HeB/FeJ and C57BL/6 mice differed in their MCP-1 response after morphine, with the former showing suppression, and the latter an increase. Additionally, no effect of morphine on IL-6 production was observed in C3HeB/FeJ mice.
Figure 3. Morphine increases levels of proinflammatory cytokines and chemokines induced by infection.


Mice were implanted with either a morphine pellet or a placebo pellet 48 hours before i.p. infection with a 0.1 LD50 dose (C57BL/6 mice; A. baumannii, strain 5798, 6.5 × 105 CFU; Panel A) or a 0.4 LD50 dose (C3HeB/FeJ mice; A. baumannii, strain 4502, 1.7 × 106 CFU; Panel B) of A. baumannii. Plasma was collected at 8 hours post-infection for analysis by Cytometric Bead Array. Points represent individual animals. Lines are median values for cytokine levels.
Morphine mediated suppression of neutrophil responses, IL-17, and KC/CXCL1 to Acinetobacter infection
Due to the rapidity of the Acinetobacter infection in morphine-treated mice, it was hypothesized that the effect of the drug was likely to be on neutrophil recruitment or on levels of chemokines that recruit neutrophils. This hypothesis was tested by quantitating the neutrophil response to Acinetobacter in morphine, as compared to placebo treated animals. Strain 4502 was selected for these studies, as previously this strain was used to examine the effect of Acinetobacter infection without morphine on neutrophils and IL-17 levels (Breslow et al. 2011).
At 12 hours post-infection, peritoneal exudate cells (PECs) were collected and stained for Gr-1 (neutrophil marker), F4/80 (macrophage/monocyte marker), and CD11b (also called Mac-1; neutrophil and macrophage marker). Morphine suppressed the total numbers of cells present in the peritoneal cavity in comparison to placebo treated controls (Figure 4A). Morphine also suppressed both the percentage (Figure 4B) and total numbers of neutrophils (Figure 4C), as well as the total numbers of macrophages (Figure 4E) within the peritoneal cavity at this same time point. There was no effect observed on the percentage of macrophages in the peritoneal cavity (Figure 4D). These results demonstrate that morphine suppressed the numbers of neutrophils and macrophages responding to the site of infection at 12 hours post-infection, suggesting that morphine may interfere with the chemotaxis of cells to the infected site.
Figure 4. Effect of morphine on peritoneal cell numbers following infection.

C3HeB/FeJ mice were implanted with morphine pellets or placebo pellets 48 hours before i.p. infection with a 0.4 LD50 (1.7 × 106 CFU) dose of A. baumannii, strain 4502. Peritoneal cells were collected at 12 hours post-infection for analysis by flow cytometry for total numbers of cells (Panel A), percentage of neutrophils (Panel B: Gr-1+CD11b+) and total numbers of neutrophils (Panel C), and for percentage of macrophages (Panel D: F4/80+CD11b+) and total numbers of macrophages (Panel E). Points represent individual animals. Lines are median values. Data are pooled results of 2 experiments.
KC/CXCL1 is the rodent homologue of human IL-8 and is recognized as one of the major chemokines that recruit neutrophils. It has been shown that IL-17 induces KC/CXCL1 (Witowski et al., 2004). The impact of morphine on this circuit during Acinetobacter infection was examined by measuring levels of IL-17 and KC/CXCL1. C3HeB/FeJ mice were implanted with either a morphine pellet or a placebo pellet 48 hours before i.p. infection with A. baumannii, strain 4502. At 6 and 12 hours following infection, mice were euthanized and peritoneal exudate fluid was harvested for analysis by ELISA. As shown in Figure 5, morphine pellets caused a significant reduction in the amount of IL-17 and KC/CXCL1 produced in the peritoneal cavity at 12 hours post-infection. No difference was seen at the 6 hour time point. The morphine mediated suppression of both IL-17 and KC/CXCL1 at 12h post-infection suggests that the opioid may decrease the numbers of neutrophils responding to infection by suppressing the levels of this cytokine and this chemokine.
Figure 5. Effect of morphine delivered by extended release pellet on production of IL-17 and KC/CXCL1.

C3HeB/FeJ mice were implanted with a morphine pellet or a placebo pellet 48h before infection with a 1.2–2.4 LD50 (5.1 × 106 – 1.0 × 107 CFU) dose of A. baumannii, strain 4502. Peritoneal exudate fluid was harvested at indicated times following infection and analyzed by ELISA for IL-17 (Panel A) or KC/CXCL1 concentrations (Panel B).
The effect of genetic deletion of IL-17 was assessed in animals given a morphine pellet (Fig. 6). The absence of IL-17 in mice given morphine did not alter the effect of the opioid on bacterial burdens in lung, liver or spleen. There was no statistically significant difference between the wild-type and IL-17 KO animals.
Figure 6. Lack of IL-17 does not increase bacterial burden in morphine treated mice.

Wild-type and IL-17A KO C57BL/6 mice were implanted with slow-release morphine pellets. After 48 hrs, all mice received an ip inoculation of 0.3 LD50 of A.baumannii, strain 4502 (5.9 × 104 CFU. At 12 hrs post inoculation, all mice were euthanized and lungs, livers and spleens were harvested for necropsy. Horizontal lines indicate median values for data points. IL-17A KO vs. WT = not significant.
Effect of trauma alone or with morphine on Acinetobacter infection
The effect of trauma on Acinetobacter infection was assessed using a standard model employing a femur break and partial hemorrhage. As shown in Table 3, trauma alone did not sensitize to Acinetobacter infection in two strains of mice challenged with two different strains of Acinetobacter. In addition, infection was tested at three different times after the trauma, immediately, and one or seven days post trauma. These studies were extended to test the hypothesis that morphine plus trauma would have an added or synergistic effect in sensitizing to Acinetobacter infection. As shown in Fig. 7, there was no significant difference in Acinetobacter burdens in organs of animals given morphine alone or morphine plus trauma.
Table 3.
Trauma does not sensitize mice to Acinetobacter infection.
| Acinetobacter baumannii Strain | Mouse Strain | Dose | Time of Challenge Post Trauma | Survivors/ Total |
|---|---|---|---|---|
| 5798 | C3HeB/FeJ | 9.4 × 105 CFU (0.02 LD50) | 1 day | Control 10/10 Trauma 10/10 |
| 5798 | C57BL/6J | 8.2 × 105 CFU (0.11 LD50) | 7 days | Control 10/10 Trauma 10/10 |
| 4502 | C3HeB/FeJ | 4.8 × 104 CFU (0.15 LD50) | Immediately | Control 10/10 Trauma 10/10 |
Figure 7. Trauma does not increase bacterial burden in morphine treated mice.
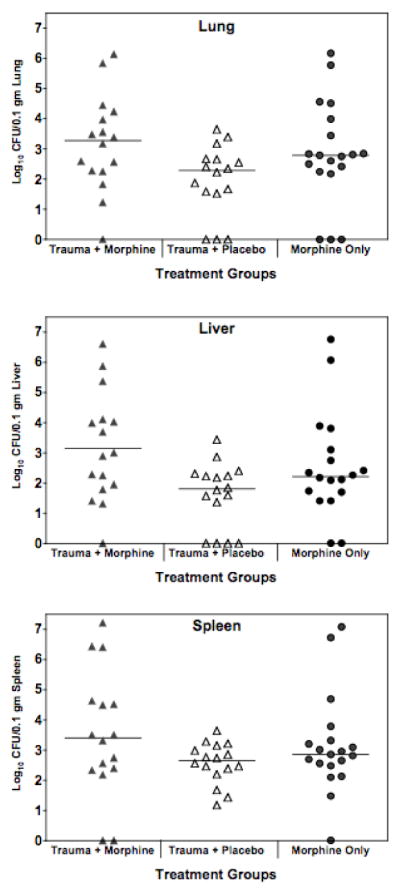
C3HeB/FeJ female mice received trauma, followed immediately by implantation of either a slow-release morphine pellet or a placebo pellet. After 48 hrs, all mice received an ip inoculation of 0.18 to 0.25 LD50 A.baumannii, strain 4502 (7.63 × 105 to 1.06 × 106 CFU). At 12 hours post infection, mice were euthanized and organs harvested for necropsy. Horizontal lines indicate median values for data points. Data are pooled from 2 experiments. Trauma + morphine vs morphine alone = Not significant
Discussion
These studies addressed a possible link between morphine administration, trauma, and the increased incidence of infection with MDR A. baumannii both in hospital ICUs and on military bases. Morphine is well documented to induce immunosuppression and is routinely administered on the battlefield for wounds. Trauma and surgery are also known to lead to immunosuppression (Kimura et al., 2010), to development of sepsis and multiple organ dysfunction (Roumen et al., 1993), and to sensitization to infection (Mack et al., 1997). Soldiers suffering wounds from improvised explosive devices can expect to receive morphine, have hemorrhagic loss of blood, and subsequent surgery, all conditions that could lead to depressed immune responses and increased susceptibility to bacterial infection. The results presented in this paper show that treatment of two strains of mice with morphine for 48 hr resulted in increased susceptibility to two different strains of A. baumannii, as measured by mortality, bacterial burdens in organs and blood, and increased pro-inflammatory cytokines in plasma. Traumatic injury caused no further modulation of infection in comparison to animals that were treated with morphine alone. The studies suggest that the mechanism by which morphine sensitizes to infection is through a decrease in the number of neutrophils mobilized in response to Acinetobacter infection. Results of survival studies using morphine treated mice showed that the opioid sensitized to Acinetobacter infection through a mu-opioid receptor mediated mechanism, as naltrexone blocked the effect of the drug. This result is consonant with the literature where the implantation of slow-release morphine pellets has been shown to sensitize to other microbes. MacFarlane et al reported that morphine pellets resulted in dramatic sensitization of mice orally challenged with Salmonella typhimurium, which correlated with increased numbers of bacteria recovered in the spleens, livers, Peyer’s patches, and mesenteric lymph nodes following infection (MacFarlane et al., 2000). Wang et al showed that morphine pellets increased the number of Streptococcus pneumoniae recovered in the spleen at 4 hours post-infection, and in the blood, lung tissue, and bronchoalveolar lavage (BAF) fluid at 24 and 48 hours (Wang et al., 2005). In addition to the morphine pellet implantation paradigm, morphine injections have also been shown to increase the burdens of other infectious organisms recovered from animals, including Candida albicans and Klebsiella pneumonia (Tubaro et al., 1983), Plasmodium berghei (Singh et al., 1993), Leishmania donovani (Singh and Singal 2007; Singal et al., 2002), and Listeria monocytogenes (Asakura et al., 2006). While the effect of morphine on Acinetobacter infection was measurable, it was not as robust as observed in some of the other infection models such as Salmonella and S. pneumoniae.
In the present Acinetobacter studies, organisms were inoculated by the intraperitoneal route, which resulted in colonization and growth of these organisms in distant organs, including the lungs, liver and spleen of morphine treated animals. Organisms were also consistently cultured from the blood of mice given morphine. In a previous study exploring the intraperitoneal inoculation model in greater depth (Breslow et al, 2011), higher inoculating doses of Acinetobacter were used, and a persistent Acinetobacter septic state was observed in blood of animals not given morphine. Thus, it is our interpretation that the organisms inoculated intraperitoneally traveled via the bloodstream to the lung and other organs, simulating sepsis that is a documented outcome in humans infected with Acinetobacter (Scott, 2004; Wisplinghoff et al, 2004). In limited studies using intratracheal inoculation of mice (unpublished studies), it was found that Acinetobacter seeded to distant organs and could be cultured from the blood. Thus, this organism has the capacity to spread systemically, regardless of whether the infection is initially mucosal or nonmucosal. Other investigators have also shown that intranasally inoculated animals have seeding of these bacteria to distant organs (Van Faasen et al, 2007).
Morphine was also shown in the present studies to alter the levels of pro-inflammatory cytokines following Acinetobacter infection. Morphine treated mice had elevated levels of IL-10, IL-12p70, and TNF-α in comparison to placebo controls. Interestingly, MCP-1 was increased in morphine treated C57BL/6 mice, but was decreased in the C3HeB/FeJ strain. The reason for this mouse strain difference is not known. The generally increased levels of pro-inflammatory cytokines is probably a reflection of the increased Acinetobacter burdens in the organs of morphine treated animals, resulting in a more robust innate immune response mounted by the host in order to eliminate the replicating pathogen. As far as we are aware, there are no publications reporting measurement of cytokine levels in humans infected with Acinetobacter. The differences we observed in the response of mouse strains to this organism could be an indication of possible variation in human responses during infection. The observation that morphine treatment enhances the levels of pro-inflammatory cytokines in infection of mice has precedent. In studies in which C3HeB/FeJ mice were implanted with morphine pellets and challenged with Salmonella, mRNA levels for IL-12p40, IFNγ, TNF-α, and iNOS were increased at 40 hours post-infection in the Peyer’s patches in comparison to placebo treated mice, and this enhancement could be reversed by the addition of naltrexone (MacFarlane et al., 2000). In another study the chronic treatment of mice with slow-release pellets, followed by intranasal challenge with S. pneumoniae caused a decrease in lung and BAF levels of TNF-α, IL-1, IL-6, MIP-2, and KC/CXCL1 at an early time point of 4 hours post-infection, but by 24 and 48 hours post-infection, the levels of all of these molecules were increased in comparison to placebo treated controls (Wang et al., 2005).
In regard to the mechanism of morphine action, a direct impact on the growth or viability of A. baumannii strains was ruled out. No direct effect of morphine sulfate was found when it was added to in vitro cultures of A. baumannii over a 106-fold dose range, as measured by optical density and viability at various times over a 24 hour period. The rapidity of infection suggested that protective immunity lies within the innate arm of the immune response. The current studies support this hypothesis as morphine was shown to result in a statistically significant decrease in the percentage and number of neutrophils recovered from the peritoneal cavity of infected mice. While the percentage of macrophages did not change, the reduced number of total cells recovered from morphine treated mice resulted in a significant decrease in the total number of macrophages recovered. The observed decrease in macrophage numbers may be a result of morphine-mediated apoptosis. Morphine has been shown to increase apoptosis of a macrophage cell line, and increases apoptosis of macrophages harvested from morphine-treated mice (Yin et al., 1999; Singhal et al., 2002). The observation of a decrease in neutrophils is in agreement with the effect of morphine on neutrophils following S. pneumoniae infection in the lung and BAF at 4, 6, and 24 hours post-infection (Ma et al., 2010; Wang et al., 2005). In addition, morphine injections have been shown to decrease the number of neutrophils (Shirzad et al., 2009) and to decrease neutrophil chemotaxis in a wound incision model (Clark et al., 2007; Martin et al., 2010; Martin et al., 2010). In the absence of infection, opioids have been shown to decrease the response of human monocytes to chemokines by a mechanism of heterologous desensitization between opioid and chemokine receptors (Szabo et al., 2003; Grimm et al., 1998). Morphine pellet implantation has also been shown to decrease leukocyte rolling and adhesion to the arteriole and venule walls of the murine vasculature, an effect that could be reversed by treatment with the antagonist naltrexone (Ni et al., 2000). Thus, morphine treatment may impede neutrophil recruitment in one of several ways, directly by interfering with chemokine signaling, and indirectly by preventing egress of the cells from the vessels into the peritoneal cavity. The present studies are the first documentation of a morphine-mediated decrease in neutrophil recruitment following systemic infection with A. baumannii, and they fit with the observation that depletion of neutrophils with antibodies also sensitizes to this organism (Breslow et al. 2011).
The hypothesis was pursued that a mechanism by which morphine might decrease neutrophil influx was by decreasing chemokine levels. As Acinetobacter was previously shown to induce high levels of IL-17 and KC/CXCL1 (Breslow et al. 2011), the effect of morphine on these proteins was assessed. It was found that morphine pellets suppressed the levels of IL-17 and KC/CXCL1 produced in PEF of C3HeB/FeJ at 12 hours post-infection, while no difference was observed at 6 hrs. Similar experiments were not carried out in C57BL/6 mice but would have been desirable. The 12 hr time point correlated with the time of morphine-mediated exacerbation of the Acinetobacter burdens. Similar results have been observed in a pneumonia model of S. pneumoniae infection, where implantation of morphine pellets 24 hours before intranasal infection with S. pneumoniae resulted in suppression of IL-17 (Ma et al., 2010) and KC/CXCL1 (Wang et al., 2005) in the lung tissue at 6 hours post-infection, a time that corresponded with a decrease in neutrophils and an increase in the number of S. pneumoniae recovered in the lungs and blood. Thus, the studies on the effect of morphine on two different bacterial infections in regard to the IL-17 circuit yielded similar results. Interestingly, however, IL-17 appears not to be sufficient for protection against Acinetobacter, as bacterial burdens in organs of IL-17 C57BL/6 KO mice were not different from those of wild-type mice given morphine. Similar results were previously obtained when IL-17 was tested as a factor in progression of Acinetobacter infection given by the intraperitoneal route to animals not treated with morphine (Breslow et al. 2011). It was hypothesized that deletion of a single chemokine that signals PMN recruitment may not be sufficient to give strong sensitization to Acinetobacter due to by-pass pathways using other chemokines (Breslow et al. 2011). While the current studies did not address the source of the IL-17, the rapid appearance of this cytokine within 4 hrs of Acinetobacter infection, suggests that a cell in the innate immune system is poised to produce it upon initial contact with the pathogen, rather than via induction of Th17 cells (Matsuzaki and Umamura, 2007). Indeed, γ/δT cells have been shown to produce IL-17 in the early hours following challenge with E. coli (Shibata et al., 2007), Mycobacterium tuberculosis (Lockhart et al., 2006), and Listeria monocytogenes (Riol-Blanco et al., 2010; Meeks et al., 2009). Similarly, Natural Killer T cells (NKT) have been shown to rapidly produce IL-17 upon stimulation (Rachitskaya et al., 2008). Both of these cell types are found at mucosal surfaces, including the peritoneal cavity, where we observed the presence of IL-17 producing cells.
The present studies also investigated, in a limited set of experiments, the effect that traumatic injury may play as a cofactor in sensitizing morphine treated mice to Acinetobacter infection. As many of the Acinetobacter infected military personnel also suffered wounds and trauma, these are important variables to examine when investigating increased incidence of infection during combat. The literature suggests that traumatic injury causes immunomodulation that may play a role in sensitizing to infection and sepsis (Lenz et al., 2007; Moore and Moore 1995). Studies suggest that macrophage responses are dampened one day following trauma and then rebound with excessive inflammatory responses at 7 days following injury (McCarter et al., 1998; Monroy et al., 2007; Mackrell et al., 2001). In the present studies trauma alone had no effect on Acinetobacter infection when the inoculation was given immediately after the trauma, one day later or 7 days later. To test the effect of morphine plus trauma, animals were infected 48 hr after morphine pellet implantation, femur break and partial hemorrhage. Using this time frame, no additional effect of the trauma was found on sensitization to Acinetobacter infection. The possibility cannot be ruled out that use of other time frames with regard to morphine, trauma, and time of challenge might yield a positive interaction.
The immunosuppressive effects of opioids have been well documented in the literature. The current studies add to this evidence by demonstrating that morphine sensitizes to systemic A. baumannii infection. These results suggest that clinicians should take a proactive approach in preventing A. baumannii infection in patients receiving opioid analgesics.
Acknowledgments
These studies were supported by USAMRMC grant W81XWH-06-1-0147, NIDA grant DA13429, and NIDA Training Grant T32DA07237.
We thank Col. Craft for providing the Acinetobacter strains. We are grateful to Dr. Iwakura for giving us permission to obtain the IL-17a−/− mice, and to Dr. Kolls for supplying us with breeding stock of the IL-17a −/− mice.
Footnotes
Disclosure: There is no financial or other relationship that might lead to a conflict of interest in regard to the data being reported.
Reference List
- Angele MK, Chaudry IH. Surgical trauma and immunosuppression: pathophysiology and potential immunomodulatory approaches. Langenbecks Arch Surg. 2005;390:333–341. doi: 10.1007/s00423-005-0557-4. [DOI] [PubMed] [Google Scholar]
- Asakura H, Kawamoto K, Igimi S, Yamamoto S, Makino S. Enhancement of mice susceptibility to infection with Listeria monocytogenes by the treatment of morphine. Microbiol Immunol. 2006;50:543–547. doi: 10.1111/j.1348-0421.2006.tb03824.x. [DOI] [PubMed] [Google Scholar]
- Bernabeu-Wittel M, Pichardo C, Garcia-Curiel A, Pachon-Ibanez ME, Ibanez-Martinez J, Jimenez-Mejias ME, Pachon J. Pharmacokinetic/pharmacodynamic assessment of the in-vivo efficacy of imipenem alone or in combination with amikacin for the treatment of experimental multiresistant Acinetobacter baumannii pneumonia. Clin Microbiol Infect. 2005;11:319–325. doi: 10.1111/j.1469-0691.2005.01095.x. [DOI] [PubMed] [Google Scholar]
- Boucher HW, Talbo GH, Bradly JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Breslow JM, Meissler JJ, Hartzell RH, Spence PB, Truant A, Gaughan J, Eisenstein TK. Innate immune responses to systemic Acinetobacter baumannii infection in mice: Neutrophils, but not IL-17, mediate host resistance. Infect Immun. 2011;79:3317–3327. doi: 10.1128/IAI.00069-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant HU, Bernton EW, Holaday JW. Morphine pellet-induced immunomodulation in mice: temporal relationships. J Pharmacol Exp Ther. 1988;245:913–920. [PubMed] [Google Scholar]
- Bryant HU, Yoburn BC, Inturrisi CE, Bernton EW, Holaday JW. Morphine-induced immunomodulation is not related to serum morphine concentrations Eur. J Pharmacol. 1988;149:165–169. doi: 10.1016/0014-2999(88)90057-x. [DOI] [PubMed] [Google Scholar]
- Bussiere JL, Adler MW, Rogers TJ, Eisenstein TK. Cytokine reversal of morphine-induced suppression of the antibody response. J Pharmacol Exp Ther. 1993;264:591–597. [PubMed] [Google Scholar]
- Casellas AM, Guardiola H, Renaud FL. Inhibition by opioids of phagocytosis in peritoneal macrophages. Neuropeptides. 1991;18:35–40. doi: 10.1016/0143-4179(91)90161-b. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Cases of HIV Infection and AIDS in the United States and Dependent Areas, 2005. US Dept. of Health and Human Services; Atlanta: 2006. HIV/AIDS Surveillance Report. [Google Scholar]
- Cerletti C, Keinath SH, Reidenberg MM, Adler MW. Chronic morphine administration: Plasma levels and withdrawal syndrome in rats. Pharmacol Biochem Behav. 1976;4:323–327. doi: 10.1016/0091-3057(76)90249-5. [DOI] [PubMed] [Google Scholar]
- Chao CC, Sharp BM, Pomeroy C, Filice GA, Peterson PK. Lethality of morphine in mice infected with Toxoplasma gondii. J Pharmacol Exp Ther. 1990;252:605–609. [PubMed] [Google Scholar]
- Cheney DL, Goldstein A. Tolerance to opioid narcotics: time course and reversibility of physical dependence in mice. Nature. 1971;232:477–478. doi: 10.1038/232477a0. [DOI] [PubMed] [Google Scholar]
- Clark JD, Shi X, Li X, Qiao Y, Liang DY, Angst MS, Yeomans DC. Morphine reduces local cytokine expresssion and neutrophil infiltration after incision. Molecular Pain. 2007;3:28–40. doi: 10.1186/1744-8069-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkshoorn L, Nemec A, Seifert H. An increasing threat in hospitals: Multidrug-resistant Acinetobacter baumannii. Nature Rev Microbio. 2007;5:939–951. doi: 10.1038/nrmicro1789. [DOI] [PubMed] [Google Scholar]
- Feng P, Rahim RT, Cowan A, Liu-Chen L-Y, Peng X, Gaughan J, Meissler JJ, Jr, Adler MW, Eisenstein TK. Effects of mu, kappa or delta opioids administered by pellet or pump on oral Salmonella infection and gastrointestinal transit. Eur J Pharmacol. 2006;534:250–257. doi: 10.1016/j.ejphar.2006.01.048. [DOI] [PubMed] [Google Scholar]
- Grimm MC, Ben-Baruch A, Taub DD, Howard OMZ, Resau JH, Wang JM, Ali H, Richardson R, Snyderman R, Oppenheim JJ. Opiates transdeactivate chemokine receptors: δ and μ opiate receptor-mediated heterologous desensitization. J Exp Med. 1998;188:317–325. doi: 10.1084/jem.188.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horsburgh CR, Anderson RA, Boyko EJ. Increased incidence of infections in intravenous drug use. Infect Control Hosp Epidemiol. 1989;10:211–215. doi: 10.1086/646004. [DOI] [PubMed] [Google Scholar]
- Hussey HH, Katz S. Infections resulting from narcotic addiction. Am J Med. 1950;9:186–193. doi: 10.1016/0002-9343(50)90021-0. [DOI] [PubMed] [Google Scholar]
- Joly-Guillou M-L, Wolff M, Pocidalo J-J, Walker F, Carbon C. Use of a new mouse model of Acinetobacter baumannii pneumonia to evaluate the post antibioic effect of imipenem. Antimicrob Agents Chemo. 1997;41:345–351. doi: 10.1128/aac.41.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang G, Hartzell JD, Howard R, Wood-Morris RN, Johnson MD, Fraser S, Wintrob A, Wortmann G. Mortality associated with Acinetobacter baumannii complex bacteremia among patients with war-related trauma. Infect Control Hosp Epidemiol. 2010;31:92–94. doi: 10.1086/649220. [DOI] [PubMed] [Google Scholar]
- Kimura F, Shimizu H, Yoshidome H, Ohtsuka M, Miyazaki M. Immunosuppression following surgical and traumatic injury. Surg Today. 2010;40:793–808. doi: 10.1007/s00595-010-4323-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Lioy D, Sheridan PA, Hurley SD, Walton JR, Martin AM, Olschowka JA, Moynihan JA. Acute morphine exposure potentiates the development of HSV-1-induced encephalitis. J Neuroimmunol. 2006;172:9–17. doi: 10.1016/j.jneuroim.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Lockhart EL, Green AM, Flynn JL. IL-17 production is dominated by γδ T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- Louria DB, Hensle T, Rose J. The major medical complications of heroin addiction. Ann Int Med. 1967;67:1–22. doi: 10.7326/0003-4819-67-1-1. [DOI] [PubMed] [Google Scholar]
- Ma J, Wang J, Wan J, Charboneau R, Chang Y, Barke RA, Roy S. Morphine disrupts interleukin-23 (IL-23)/IL-17-mediated pulmonary mucosal host defense against Streptococcus pneumoniae infection. Infect Immun. 2010;78:830–837. doi: 10.1128/IAI.00914-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki G, Umemura M. Interleukin-17 as an effector molecule of innate and acquired immunity against infections. Microbiol Immunol. 2007;51:1139–1147. doi: 10.1111/j.1348-0421.2007.tb04008.x. [DOI] [PubMed] [Google Scholar]
- MacFarlane AS, Peng X, Meissler JJ, Jr, Rogers TJ, Geller EB, Adler MW, Eisenstein TK. Morphine increases susceptibility to oral Salmonella typhimurium infection. J Infect Dis. 2000;181:1350–1358. doi: 10.1086/315403. [DOI] [PubMed] [Google Scholar]
- Mack VE, McCarter MD, Naama HA, Calvano SE, Daly JM. Candida infection following severe trauma exacerbates Th2 cytokines and increases mortality. J Surg Res. 1997;69:399–407. doi: 10.1006/jsre.1997.5093. [DOI] [PubMed] [Google Scholar]
- Mackrell PJ, Daly JM, Mestre JR, Stapleton PP, Howe LR, Subbaramaiah K, Dannerberg AJ. Elevated expression of cyclooxygenase-2 contributes to immune dysfunction in a murine model of trauma. Surgery. 2001;130:826–833. doi: 10.1067/msy.2001.116669. [DOI] [PubMed] [Google Scholar]
- Martin JL, Koodie L, Krishnan AG, Charboneau R, Barke RA, Roy S. Chronic morphine administration delays wound healing by inhibiting immune cell recruitment to the wound site. Amer J Path. 2010;176 doi: 10.2353/ajpath.2010.090457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarter MD, Mack VE, Daly JM, Naama HA, Calvano SE. Trauma-induced alterations in macrophage function. Surgery. 1998;123:96–101. [PubMed] [Google Scholar]
- McCoy CB, Metsch LR, Chitwood DD, Shapshak P, Comerford ST. Parenteral transmission of HIV among injection drug users: assessing the frequency of multiperson use of needles, syringes, cookers, cotton, and water. JAIDS. 1998;18(Suppl 1):S25–S29. doi: 10.1097/00042560-199802001-00006. [DOI] [PubMed] [Google Scholar]
- Meeks KD, Sieve AN, Kolls JK, Ghilardi N, Berg RE. IL-23 is required for protection against systemic infection with Listeria monocytogenes. J Immunol. 2009;183:8026–8034. doi: 10.4049/jimmunol.0901588. [DOI] [PubMed] [Google Scholar]
- Mojadadi S, Jamali A, Khansarinejad B, Soleimanjahi H, Bamdad T. Acute morphine adminstration reduces cell-mediated immunity and induces reactivation of latent herpes simplex virus type 1 in BALB/c mice. Cellular and Molecular Immunology. 2009;6:111–116. doi: 10.1038/cmi.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy MA, Opperman KK, Pucciarelli M, Yerrum S, Berg DA, Daly JM. The PPARγ ligand 15D-PGJ2 modulates macrophage activation after injury in a murine trauma model. Shock. 2007;28:186–191. doi: 10.1097/shk.0b013e3180310982. [DOI] [PubMed] [Google Scholar]
- Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin N Amer. 1995;75:257–277. doi: 10.1016/s0039-6109(16)46587-4. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impared in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Ni X, Gritman KR, Eisenstein TK, Adler MW, Arfors KE, Tuma RF. Morphine attenuates leukocyte/endothelial interactions. Microvasc Res. 2000;60:121–130. doi: 10.1006/mvre.2000.2253. [DOI] [PubMed] [Google Scholar]
- Petersen K, Riddle MS, Danko JR, Blazes DL, Hayden R, Tasker SA, Dunne JR. Trauma-related infections in battlefield casualties from Iraq. Ann Surg. 2007;245:803–811. doi: 10.1097/01.sla.0000251707.32332.c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachitskaya AV, Hansen AM, Horai R, Li Z, Villasmil R, Luger D, Nussenblatt RB, Caspi RR. Cutting edge: NKT cells constitutively express IL-23 receptor and RORγt and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J Immunol. 2008;180:5167–5171. doi: 10.4049/jimmunol.180.8.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renckens R, Roelofs JJ, Knapp S, deVos AF, Florquin S, van der Poll T. The acute-phase response and serum amyloid A inhibit the inflammatory response to Acinetobacter baumannii pneumonia. J Infect Dis. 2006;193:187–195. doi: 10.1086/498876. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Lazarevic V, Awasthi A, Mitsdoerffer M, Wilson BS, Croxford A, Waisman A, Kuchroo VK, Glimcher LH, Oukka M. IL-23 receptor regulated unconventional IL-17-producing T cells that control bacterial infection. J Immunol. 2010;184:1710–1720. doi: 10.4049/jimmunol.0902796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers TJ, Bednar F, Kaminsky DE, Davey PC, Meissler JJ, Jr, Eisenstein TK. Laboratory model systems of drug abuse and their relevance to HIV infection and dementia. In: Gendelman HE, Grant I, Everall IP, Lipton SA, Swindells S, editors. The Neurology of AIDS. Oxford: Oxford University Press; 2005. pp. 310–320. [Google Scholar]
- Roumen RM, Hendriks T, van der Ven-Jongekrijg J, Nieuwenhiujzen GAP, Sauerwein RW, van der Meer JW, Goris RJA. Cytokine patterns in patients after major surgery, hemorrhagic shock, and severe blunt trauma. Ann Surg. 1993;6:769–776. doi: 10.1097/00000658-199312000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo TA, Beanan JM, Olson R, MacDonald U, Luke NR, Gill SR, Campagnari AA. Rat pneumonia and soft-tissue infection models for the study of Acinetobacter baumannii biology. Infect Immun. 2008;76:3577–3586. doi: 10.1128/IAI.00269-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P, Deye G, Srinivasan A, Murray C, Moran K, Hulten E, Fishbain J, Craft D, Riddell S, Lindler L, Mancuso J, Milstrey E, Bautista CT, Patel J, Ewell A, Hamilton T, Geddy C, Tenney M, Christopher G, Peterson K, Endy T, Petruccelli B. An outbreak of multidrug-resistant Acinetobacter baumannii-calcoaceticus complex infection in the US military health care system associated with military operations in Iraq. Clin Infect Dis. 2007;44:1577–1584. doi: 10.1086/518170. [DOI] [PubMed] [Google Scholar]
- Scott PT. Acinetobacter baumannii infections among patients at military medical facilities treating injured U.S. service members. 2002–2004 Morbidity and Mortality Weekly Report. 2004;53:1064–1065. [PubMed] [Google Scholar]
- Shavit Y, Terman GW, Lewis JW, Zane CJ, Gale RP, Liebeskind JC. Effects of footshock stress and morphine on natural killer lymphocytes in rats: studies of tolerance and cross-tolerance. Brain Res. 1986;372:382–385. doi: 10.1016/0006-8993(86)91149-2. [DOI] [PubMed] [Google Scholar]
- Shibata K, Yamata H, Hara H, Kishihara K, Yoshikai Y. Resident Vγ1+ γδ T cells control early infiltration of neutrophils after Escherichia coli infection via IL-17 production. J Immunol. 2007;178:4466–4472. doi: 10.4049/jimmunol.178.7.4466. [DOI] [PubMed] [Google Scholar]
- Shirzad H, Shahrani M, Rafieian-Kopaei M. Comparison of morphine and tramadol effects on phagocytic activity of mice peritoneal phagocytes in vivo. Int Immunopharm. 2009;9:968–970. doi: 10.1016/j.intimp.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Singal P, Kinhikar AG, Singh S, Singh PP. Neuroimmunomodulatory effects of morphine in Leishmania donovani-infected hamsters. Neuroimmunomod. 2002;10:261–269. doi: 10.1159/000069970. [DOI] [PubMed] [Google Scholar]
- Singh PP, Singal P. Morphine-induced neuroimmunomodulation in murine visceral leishmaniasis: the role(s) of cytokines and nitric oxide. J Neuroimm Pharmacol. 2007;2:338–351. doi: 10.1007/s11481-007-9094-y. [DOI] [PubMed] [Google Scholar]
- Singh PP, Singh S, Dutta GP, Srimal RC. Immunomodulation by morphine in Plasmodium berghei-infected mice. Life Sci. 1993;54:331–339. doi: 10.1016/0024-3205(94)00789-6. [DOI] [PubMed] [Google Scholar]
- Singhal PC, Bhaskaran M, Patel J, Patel K, Kasinath BS, Duraisamy S, Franki N, Reddy K, Kapasi AA. Role of P38 mitogen-activated protein kinase phophorylation and Fas-Fas ligand interaction in morphine-induced macrophage apoptosis. J Immunol. 2002;168:4025–4033. doi: 10.4049/jimmunol.168.8.4025. [DOI] [PubMed] [Google Scholar]
- Szabo I, Rojavin M, Bussiere JL, Eisenstein TK, Adler MW, Rogers TJ. Suppression of peritoneal macrophage phagocytosis of Candida albicans by opioids. J Pharmacol Exp Ther. 1993;267:703–706. [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen L-Y, Bednar F, Henderson EE, Howard OMZ, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–1082. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- Towner KJ. Acinetobacter: an old friend but a new enemy. J Hosp Infect. 2009;73:355–363. doi: 10.1016/j.jhin.2009.03.032. [DOI] [PubMed] [Google Scholar]
- Tubaro E, Borelli G, Croce C, Cavallo G, Santiangeli C. Effect of morphine on resistance to infection. J Infec Dis. 1983;148:656–666. doi: 10.1093/infdis/148.4.656. [DOI] [PubMed] [Google Scholar]
- Van Faassen H, KuoLee R, Harris G, Zhao X, Conlan JW, Chen W. Neutrophils play an important role in host resistance to respiratory infection with Acinetobacter baumannii in mice. Infect Immun. 2007;75:5597–5608. doi: 10.1128/IAI.00762-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassou D, Badogeorgou E, Kampa M, Dimitriou H, Hatzoglou A, Castanas E. Opioids modulate constitutive B-lymphocyte secretion. Int Immunopharm. 2008;8:634–644. doi: 10.1016/j.intimp.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Vincent JL, Bello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. International study of the prevalence and outcomes of infection in Intensive Care Units. JAMA. 2009;302:2323–2329. doi: 10.1001/jama.2009.1754. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Charboneau R, Roy S. Morphine impairs host innate immune response and increases susceptibility to Streptococcus pneumoniae lung infection. J Immunol. 2005;174:426–434. doi: 10.4049/jimmunol.174.1.426. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. Morphine induces defects in early response of alveolar macrophages to Streptococcus pneumoniae by modulating TLR9-NFkB signaling. J Immunol. 2008;180:3594–3600. doi: 10.4049/jimmunol.180.5.3594. [DOI] [PubMed] [Google Scholar]
- Weber RJ, Pert A. The periaqueductal gray matter mediates opiate-induced immunosuppression. Science. 1989;245:188–190. doi: 10.1126/science.2749256. [DOI] [PubMed] [Google Scholar]
- Wendt C, Dietze B, Dietz E, Ruden H. Survival of Acinetobacter baumannii on dry surfaces. J Clin Microbiol. 1997;35:1394–1397. doi: 10.1128/jcm.35.6.1394-1397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent M, Seifert H, Wenzel RP, Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- Witowski J, Ksiazek K, Jorres A. Interleukin-17: A mediator of inflammatory responses. Cell Mol Life Sci. 2004;61:567–572. doi: 10.1007/s00018-003-3228-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin D, Mufson A, Wang R, Shi Y. Fas-mediated cell death promoted by opioids. Nature. 1999;397:218. doi: 10.1038/16612. [DOI] [PubMed] [Google Scholar]